

Abstract
TGF-β1 and its target gene encoding plasminogen activator inhibitor-1 (PAI-1) are major regulators of capillary outgrowth, vessel maturation and angiogenic network stability. The increasing realization of the complexity of PAI-1 action in the vascular system requires analysis of specific signaling events that impact its expression in a physiologically-relevant cell system. PAI-1 was required for tubular differentiation and maintenance of cellular survival in complex gels since targeted disruption of PAI-1 synthesis or activity with antisense constructs or function-blocking antibodies resulted in network regression. Indeed, serum-deprivation-induced apoptosis of tubulogenic T2 cells was concentration-dependently inhibited by addition of a stable PAI-1 mutant protein consistent with the established pro-survival role of PAI-1 in vascular endothelial cells. PAI-1 induction and ERK pathway activation in response to TGF-β1 was attenuated by EGFR signaling blockade (with AG1478) or preincubation with the MMP/ADAM inhibitor GM6001. The combination of AG1478 + GM6001 completely ablated both responses suggesting that EGFR transactivation is important in PAI-1 gene control and may, at least partially, involve ligand shedding. TGF-β1-stimulated PAI-1 induction was preceded, in fact, by EGFR phosphorylation on Y845 (a src kinase target residue). EGFR1 knockdown with lentiviral shRNA constructs, moreover, effectively decreased (by >75%) TGF-β1-stimulated PAI-1 expression whereas infection with control (i.e. GFP) viruses had no effect. TGF-β1 failed to induce PAI-1 synthesis in EGFR-deficient fibroblasts while introduction of a wild-type EGFR1 construct in EGFR−/− cells rescued the PAI-1 response to TGF-β1 confirming, at a genetic level, the targeted knockdown data. The continued clarification of novel cooperative signaling cascades that impact expression of important angiogenic genes (e.g. PAI-1) may provide therapeutically useful targets to manage the pathophysiology of human neoplastic and vascular diseases.
Keywords: SERPINE1, PAI-1, angiogenesis, TGF-β1, EGFR, apoptosis, tubulogenesis
Introduction
TGF-β signaling in endothelial cells utilizes heteromeric receptor complexes consisting of TGF-βRII and the activin receptor-like kinases ALK1/ALK5.1-6 ALK1/ALK5 receptor signaling balances angiogenic stimulatory (ALK1) and vessel maturation (ALK5) programs by impacting control of the relevant downstream cascades and their transcriptional targets.7-10 Indeed, binding of TGF-β to ALK1 receptor complexes initiaties phosphorylation of Smad1/5 as part of the activation phase of angiogenesis resulting in Id1 expression and endothelial proliferation/ migration. ALK5 engagement, in contrast, promotes capillary maturation while inhibiting endothelial cell proliferation, migration and network regression.2,5,6 Although recent reports suggest that the physiologic role of ALK5 in the endothelium may be limited,11 ALK5-dependent plasminogen activator inhibitor type-1 (PAI-1) expression is a critical element in neovessel stability. PAI-1 maintains an angiogenic scaffold, stabilizes nascent capillary structure by inhibiting downstream matrix metalloproteinase (MMP) activation and collagen degradation while negatively regulating (endothelial) cell proliferation.12-18
TGF-β1-mediated angiogenesis in murine microvessel endothelial cells requires epidermal growth factor receptor (EGFR) transactivation via TGF-β1-induced autocrine secretion of the EGFR ligand TGF-α; EGFR/TGF-α interactions, in turn, activate the PI3K and MEK/ERK pathways promoting endothelial cell survival.19 EGFR recruitment appears to be part of a kinase receptor cross-talk network initiated by TGF-β120 involving both ligand-dependent and independent mechanisms.19,21,22 Since PAI-1 is a major TGF-β1 response gene and a critical regulator of endothelial apoptosis/vessel regression, it was important to determine if PAI-1 induction in TGF-β1-stimulated human endothelial cells similarly required EGFR participation.
Materials and Methods
Cell culture
Human microvessel endothelial cells (HMEC-1), as well as the EC-1, T2, RK and HaCaT II-4 cell lines, were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Transfection of EC-1 cells with the PAI-1 antisense construct Rc/CMVIAP provided for selection of the stable PAI-1 functionally-null 4HH line.23 For tubulogenesis assays, cells were seeded either directly on hydrated Matrigel-coated surfaces (2-D culture) or suspended in a more complex Vitrogen-supplemented Matrigel 3-D environment. EGFR+/+ and EGFR−/− mouse embryonic fibroblasts (MEFs) were provided by Dr. Jennifer R. Grandis (University of Pittsburgh Medical Center). Serum-deprived cells were stimulated with EGF (10 ng/ml) or 4 mM HCl-activated TGF-β1 (1–10 ng/ml) for the times indicated. Pretreatment with the EGFR inhibitor AG1478, the MEK inhibitors U0126 and PD98059 or the MMP/ADAM inhibitor GM6001 is described in the text. AG1478 was used at 1.2 to 2.5 μM where its action is specific for EGFR1. Much higher concentrations of AG1478 are required for inhibition of the kinase activities of erb-B2 (IC50 > 100 μM), the PDGFR(IC50 > 100 μM) and p210Bcr-Abl (IC50 > 50 μM) (Calbiochem datasheet 658552, revised 2008). Apoptotic cells were scored using DAPI to identify condensed chromatin and fragmented nuclear bodies. The recombinant PAI-1 active mutant 14-1b (t½ = 145 hr) and PAI-1 neutralizing antibodies were used at the final concentrations indicated.
Northern blotting
Cytoplasmic RNA was isolated and separated by electrophoresis on denaturing 1% agarose/2.2 M formaldehyde gels prior to transfer to nitrocellulose. Input RNA content was confirmed by quantitation of ribosomal RNA in each sample. Hybridization was at high stringency in the presence of 10% dextran sulfate, 50% formamide and 4x saline/sodium citrate buffer (SSC). Blots were incubated for 4 hr at 42 °C then subsequently hybridized with a 32P-labeled EcoRI-HindIII fragment of PAI-1 cDNA (specific activity 1–2 × 108 cpm/μg DNA) for 48 hr at 40 °C. The recombinant pBluescript SK(–) phagemid pRPAISSI-3, containing a 3.0 kb EcoRI/SstII-flanked cDNA insert encoding PAI-1, was used for isolation of the pRPAImr1-4 probe used for hybridization. Briefly, pRPAISS1-3 was digested with EcoRI/HindIII at 37 °C for 1 hr and fragments separated in 1% agarose gels. After staining with ethidium bromide, bands representing the PAI-1 cDNA insert were excisesd and electroeluted. This insert fragment (pRPAImr1-4) was labeled with 32P-dCTP by random priming. Following hybridization, membranes were washed for 20 minutes in 2 × SSC/0.1% SDS (twice) and then in 1 × SSC/0.1% SDS, all at 55 °C. Blots were exposed to X-Omat film with intensifying screens and autoradiographs scanned to quantitate relative PAI-1 mRNA abundance.
Construction and transfection of an antisense PAI-1 expression vector
pBluescript containing a full-length PAI-1 cDNA was digested with EcoR1 and HindIII to generate a 2.6 kb insert (representing nucleotides −118 to +2572). Agarose-gel-purified DNA was blunt-ended with Klenow fragment/dNTPs using a fill-in reaction. NotI linkers were ligated, the fragments digested with NotI and purified by agarose-gel electrophoresis. Flanked inserts were ligated to NotI-digested calf intestinal phosphatase-treated gel-purified Rc/CMV expression vector DNA and subsequently transformed into competent INVαF’ Escherichia coli. Plasmid DNA was isolated from ampicillin-resistant colonies. Restriction enzyme digestion and Southern-blot analysis of Oncor membrane blots of electrophoretically separated endonuclease-digested DNA, using a 726 bp PstI/ApaI-digested cDNA fragment labeled with [32P]dCTP, confirmed antisense (Rc/CMVIAP) insert orientation. Transfected cells were stably selected with G418.
Western blotting
Cells were disrupted in 4% SDS/PBS for 10 minutes, lysates vortexed briefly, boiled for 5 minutes then centrifuged at 14,000 × g for 15 minutes. Aliquots (containing 30 μg cellular protein) were electrophoretically-separated, transferred to nitrocellulose, membranes blocked in 5% milk in 0.05% Triton-X 100/PBS, incubated overnight with specific antibodies to PAI-1, pERK1/2 and ERK2, actin, p38, EGFR, pSMAD2Ser465/467 and EGFRpY845 in blocking buffer and washed three times in 0.05% Triton-X 100/PBS prior to incubation with secondary antibodies. Immunoreactive proteins were visualized with ECL reagent and quantitated by densitometry. Statistical analyses of data utilized the t test to derive p values.
Fluorescence microscopy
For immunocytochemical localization of activated (pY845) EGFR and PAI-1, formalin-fixed cells were washed 3 times with Ca+2/Mg+2-free PBS, permeabilized in 0.1% Triton X-100 in PBS for 10 minutes followed by 3 PBS washes and a 2% BSA block for 20 minutes. Cells were incubated with diluted antibodies to pY845 EGFR or PAI-1 for 1 hour at room temperature, washed 3 times, and incubated with appropriate secondary antibodies for 45 minutes. Following a final series of PBS rinses, coverslips were mounted with VectaShield reagent containing DAPI (to visualize nuclei) (Vector Laboratories).
EGFR “rescue” and knockdown
EGFR−/− MEFs were infected (MOI = 50–100) with adenoviruses bearing either control (GFP) or wild-type human EGFR1 expression constructs24 in low-serum-containing medium. Stimulation with TGF-β1 (0.1 ng/ml) was for 4 hours prior to cell extraction. Adenovirus infectivity was >90% (assessed by GFP fluorescence). HaCaT II-4 cells were similarly infected with EGFR shRNA-bearing lentiviruses (Sigma).
Results
Branching morphogenesis in response to TGF-β1 requires EGFR and MEK signaling
T2 cells cultured on dried Matrigel surfaces formed contact-inhibited monolayers (Fig. 1A) unlike the highly - differentiated honeycomb - like tubular structures when plated onto thin, hydrated Matrigel undercoatings (Fig. 1B). In contrast, branched complexes with sprouting foci typified network construction on thick (Fig. 1C) as compared to thin (Fig. 1B) hydrated gels; the branching phenotype was most highly-developed in a 3-D collagen I-supplemented Matrigel environment (Fig. 1D).
Figure 1.
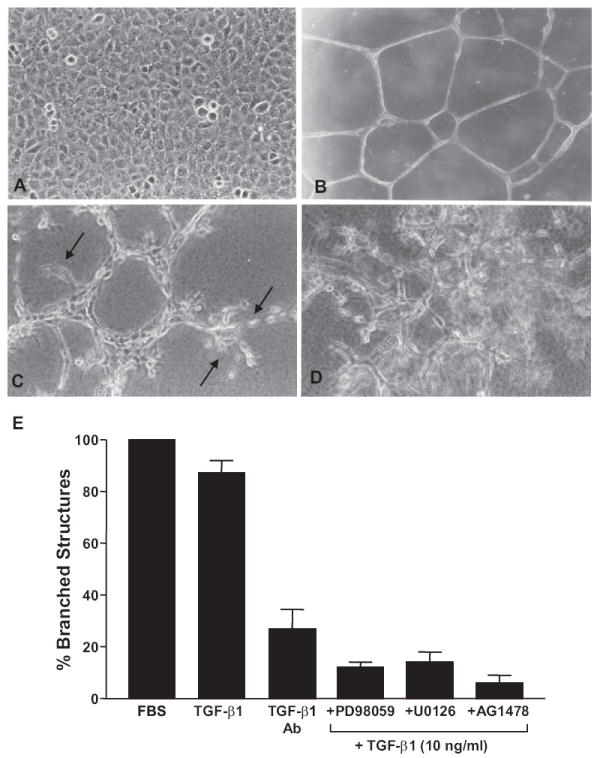
Tubular differentiation of T2 cells in Matrigel culture requires TGF-β1 signaling. T2 cells cultured on dried Matrigel surfaces established contact-inhibited monolayers (A) contrasting with the development of smooth branched tubular networks when seeded onto thin hydrated Matrigel coatings. (B) Cells cultured on thick, hydrated, Matrigel underlays constructed tubular aggregates with extensive sprouting (arrows) evident at terminal foci.(C) Sprout formation and network complexity was most highly developed in 3-D suspension culture in a combination Virtogen:Matrigel support scaffold. (D) Addition of TGF-β1 alone yielded an almost equivalent tubular differentiation response on thin Matrigel matrices compared to FBS supplementation. (E) Incubation with pan-TGF-β antibodies capable of neutralizing TGF-β1, β2, β3 and β5 largely inhibited this response indicating that network formation in the Matrigel system was largely TGF-β-dependent. Pharmacologic blockade of MEK/ERK and EGFR activity with PD98059/U0126 or AG1478, respectively, effectively inhibited TGF-β1-initiated tubular differentiation. Data plotted (in E) is the mean ± SD of triplicate experiments using a calibrated ocular grid to quantify the number of network branchpoints (e.g. B) on thin Matrigel coatings.
TGF-β1 is a major tubulogenic factor, at least for murine 1G11 endothelial cells, and EGFR/MEK activity is essential for capillary differentiation in collagen I gels.19 Addition of serum (10% final concentration) at the time of plating onto hydrated Matrigel-coated surfaces resulted in optimal tubular differentiation of T2 cells. Replacement of serum with TGF-β1 (10 ng/ml) in the 3-D suspension culture model promoted tubular network formation almost as effectively as serum (Fig. 1E). Since Matrigel contains approximately 2 ng/ml TGF-β1, the role of endogenous TGF-β1 as a potential tubulogenic effector in the current in vitro model was assessed even though Matrigel was a minor component in the Vitrogen: Matrigel system. Addition of pan-TGF-β neutralizing antibodies at the time of initiation of 3-D suspension culture markedly reduced the incidence of branched structures formed by T2 cells (Fig. 1E). TGF-β1 signaling was clearly important in tubular differentiation as inclusion of the MEK inhibitors PD98059 or U0126 or the EGFR tyrphostin inhibitor AG1478 significantly reduced tubular network formation (Fig. 1E) consistent with previous findings.19
PAI-1 is required for in vitro tubulogenesis
PAI-1 is the major upstream negative regulator of plasminogen-dependent capillary regression in collagen gel culture.16 PAI-1 transcripts, in fact, were significantly elevated in T2 cells cultured on Matrigel and maintained at high levels (approximately 18-fold relative to quiescent monolayers) in formed tubular structures (i.e. 24 hours after initial seeding) (Fig. 2A). Since it was not possible to introduce PAI-1 neutralizing antibodies reliably in a 3-D culture system, a genetic approach was used to manipulate PAI-1 expression prior to initiation of tubular differentiation in more complex gels. Cells engineered to constitutively express PAI-1 antisense transcripts, resulting in complete ablation of PAI-1 synthesis (i.e. 4HH) (Figs. 2B, C), failed to form stable, highly-branched, tubular networks in suspension culture typical of wild-type cells (Figs. 2D, E). Tubular remnants were evident, however, suggesting abortive network formation with an inability to stabilize these primitive structures (Fig. 2E).
Figure 2.
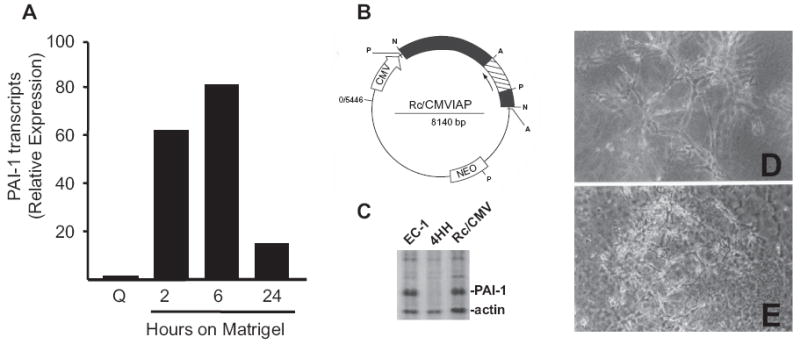
Targeted disruption of PAI-1 expression inhibits formation of stable tubular networks in 3-D Vitrogen:Matrigel suspension culture. PAI-1 transcripts are rapidly up-regulated within 2 hours after plating T2 cells onto thin, hydrated, Matrigel coatings and are maximal at 6 hours corresponding with rapid migration into primitive tube precursor structures. (A) Tubular networks are well-developed by 24 hours at which time PAI-1 mRNA levels remain 18-fold elevated over quiescent (Q) T2 monolayer cultures. Transfection of the Rc/CMVIAP PAI-1 antisense vector (B) into EC-1 cells resulted in derivation of the stable, PAI-1 functionally-null, 4HH cell line. Gel electrophoresis of the 35S-methionine-labeled, matrix-enriched, protein fraction of EC-1, 4HH and empty vector (Rc/CMV)-transfectants confirmed that 4HH cells were, in fact, PAI-1-deficient. (C) Unlike parental and T2 cells that formed stable complex tubular networks in 3-D culture (D), 4HH cells failed to construct stable differentiated structures and effectively degraded the supporting matrix scaffold (E).
Similar to results in the T2 system, human endothelial (HMEC-1) cells formed highly branched capillary networks with clear luminal spaces when maintained on hydrated Matrigel underlays (Fig. 3A). Addition of plasminogen (Fig. 3B) or neutralizing PAI-1 antibodies in the absence of added plasminogen (Fig. 3C) significantly reduced the integrity of tubular network structure. The combined addition of PAI-1 function-blocking antibodies + plasminogen completely disrupted capillary organization with many areas of the culture reverting to a more monolayer-like growth pattern (Fig. 3D) supporting the previous conclusions that PAI-1 was an essential factor in the maintenance of endothelial tubular differentiation.
Figure 3.
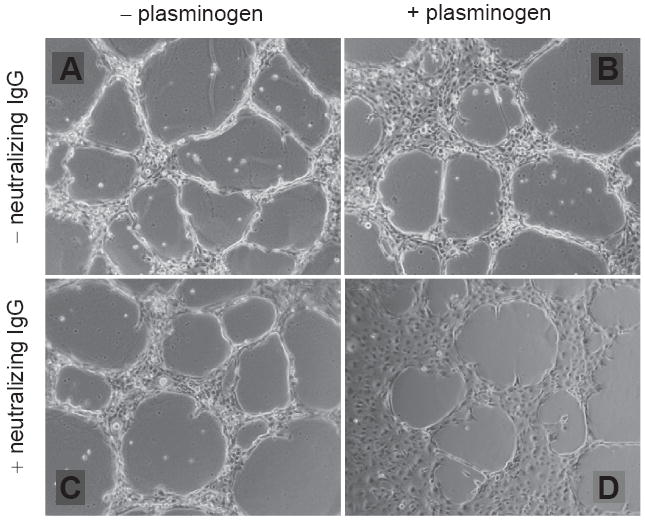
Function-blocking PAI-1 antibodies disrupt Matrigel-induced tubular network formation in human microvessel endothelial cells. HMEC-1 cells formed well-developed capillary-like structures with patent lumens within 24 hours after plating onto hydrated Matrigel coatings. (A) Network maintenance required fine regulation of the plasmin-generating cascade as addition of plasminogen (+plasminogen) (B) or PAI-1 neutralizing antibodies (+neutralizing IgG) (C) separately effectively inhibited tubular differentiation and promoted network breakdown consistent with the requirement of PAI-1 for capillary stability.15 The simultaneous addition of plasminogen and PAI-1 IgG (D) resulted in complete network disassembly with reversion to a more monolayer growth pattern.
PAI-1 protects cells from apoptosis due to serum-deprivation
PAI-1 has a prominent anti-apoptotic activity in several cell systems25-28 and, specifically, protects endothelial cells from FasL-mediated programmed cell death.29 T2 cells, which exhibit a 60%–70% apoptotic response upon serum withdrawal (Fig. 4A), were used to assess a potential survival role for PAI-1. Serum deprivation for 3-days resulted in extensive apoptosis that was dose-dependently inhibited by increasing concentrations of the active 14-1b PAI-1 mutant (t½ = 145 hr; added concomitantly with the switch to serum-free medium) (Fig. 4A). PAI-1 inhibition of serum withdrawal-induced programmed cell death was not restricted to T2 cells since 14-1b PAI-1 similarly protected newborn rat keratinocytes (RK cells) from apoptosis due to serum-deprivation (Table 1). This significant PAI-1-dependent pro-survival effect, at least in the context of serum depletion (Fig. 4B), correlated with an approximately 4-fold increase in ERK2 phosphorylation (Fig. 4C). Both the anti-apoptotic and ERK responses to exogenous PAI-1 were effectively ablated by simultaneous addition of the MEK inhibitors PD98059 or U0126 (not shown). Such signaling consequences of PAI-1 exposure are consistent with the PAI-1-dependent stimulation of JAK/STAT activation in smooth muscle cells at PAI-1 levels approximating those used here.30 These data suggest that a PAI-1:low density lipoprotein receptor-related protein (LRP) interaction, similar to that required for JAK/STAT mobilization,30 may also be involved in the downstream pERK response.
Figure 4.
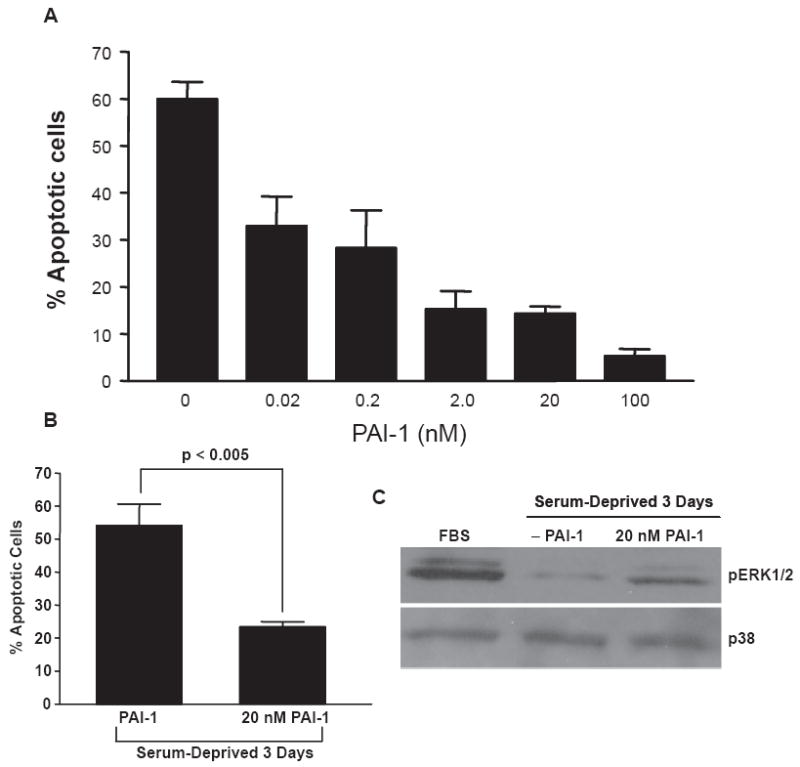
PAI-1 rescues T2 cells from apoptosis induced as a consequence of serum-deprivation. T2 cells undergo a significant apoptotic response (involving >60% of the population) following 3 days in serum-free medium. Addition of the stable 14-1b recombinant PAI-1 protein (but not BSA), in final concentrations of 0.02 to 100 nM at the time of change-over to FBS-deficient medium, protected T2 cells from apoptosis due to serum-deprivation. (A) The significant (p < 0.005) apoptotic “rescue” evident in cultures incubated in 20 nM PAI-1 (B) correlated with a PAI-1-related increase in ERK1/2 phosphorylation (C) consistent with previous observations that PAI-1 initiates signaling events in various vascular and non-vascular cell types.30 Data plotted (in A, B) is the mean ± SD of triplicate independent experiments.
Table 1.
Addition of the stable PAI-1 mutant protein 14-1b inhibits serum deprivation-induced apoptosis in RK cells.
| Culture conditionsa | Percent apoptotic cellsb |
|---|---|
| FBS-free | 10.7 ± 1.2 |
| FBS-free + 20 nM PAI-1 | 3.3 ± 1.7 |
| FBS-free + 100 nM PAI-1 | 0.1 ± 0.1 |
RK cells were grown to 50% confluency, the conditioned medium removed, cells layers rinsed in PBS twice and serum-free DMEM ± PAI-1 14-1b (in the concentrations indicated) added for a 3-day incubation period.
Apoptotic cells were scored using DAPI to identify condensed chromatin at the nuclear periphery and fragmented nuclear bodies.
TGF-β1 stimulation of ERK phosphorylation/PAI-1 induction is inhibited by AG1478 and GM6001
Previous data implicated participation of MEK-ERK in PAI-1 expression in TGF-β1-stimulated cells.31 One mechanism for MEK-ERK mobilization in response to TGF-β1 involves transactivation of the EGFR20 which, in murine endothelial cells, is a consequence of shedding of the EGFR ligand TGF-α.19 To assess such cross-pathway involvement in TGF-β1-induced PAI-1 gene control, T2 and HMEC-1 cells were stimulated with TGF-β1 in the presence or absence of prior incubation with the EGFR inhibitor AG1478 or the broad spectrum MMP/ADAM inhibitor GM60001 or both. AG1478 and GM6001, when used separately, significantly attenuated ERK1/2 phosphorylation and PAI-1 expression in response to TGF-β1 (Figs. 5A–C). Pre-incubation with both inhibitors simultaneously completely ablated ERK1/2 phosphorylation and PAI-1 induction, even in cultures stimulated with TGF-β1 + EGF in combination (Figs. 5A, D). The pERK1/2 and PAI-1 levels in TGF-β1-stimulated dual-inhibitor-treated cultures, moreover, was not different from quiescent controls (Fig. 5A).
Figure 5.
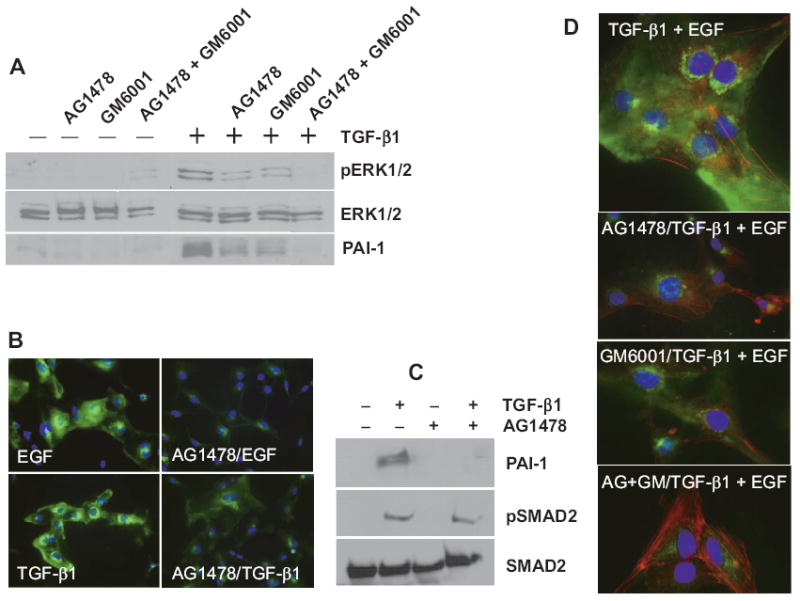
PAI-1 induction in TGF-β1-stimulated tubulogenic cells requires EGFR signaling. Pretreatment with the EGFR inhibitor AG1478 or the MMP/ADAM inhibitor GM6001 effectively attenuated both ERK1/2 phosphorylation and PAI-1 induction in TGF-β1-stimulated T2 cells; addition of both AG1478 and GM6001 simultaneously completely ablated both responses. (A) EGFR signaling was similarly required for PAI-1 expression in TGF-β1-treated HMEC-1 cultures. (B, C) While PAI-1 induction was effectively blocked by prior incubation with AG1478, SMAD2 phosphorylation was retained indicating that TGF-βR signaling was not impacted by this pharmacologic inhibitor. (C) The combination of AG1478 and GM6001 pretreatment was particularly effective at ablating ERK1/2 phosphorylation and PAI-1 in response to either singular (A–C) or dual (D) growth factor stimulation.
EGFR1 is required for TGF-β1-induced PAI-1 expression
Inhibition of PAI-1 expression as a consequence of AG1478 pre-treatment (e.g. Figs. 5A, C) necessitated assessment of EGFR activation in response to TGF-β1. This was important as AG1478 could also impact other signaling pathways, albeit at much higher concentrations than that used here (see Methods). Since TGF-β1-induced PAI-1 expression was previously shown to require src kinase activity,20,31 the effect of TGF-β1 on a src kinase target EGFR residue (i.e.Y845) was used as a measure of receptor activation. As expected, EGF stimulated EGFRY845 phosphorylation in response to growth factor addition (Fig. 6A). TGF-β1 also increased Y845 site phosphorylation (Fig. 6B) and Y845-phosphorylated EGFR internalization into prominent cytoplasmic vacuoles was evident within 15 minutes of EGF or TGF-β1 addition (Fig. 6C). The level of TGF-β1-stimulated pEGFRY845 phosphorylation was significantly reduced compared to cells exposed to EGF (cf., Figs. 6A, B) suggesting involvement of, perhaps, only a subset of EGF receptors. The existence of such fine controls on EGFR participation in TGF-βR signaling appeared highlighted, moreover, by the relatively transient window of EGFR activation contrasting with the longer term of TGF-β1-stimulated SMAD2 phosphorylation (Fig. 6B). Consistent with the marked sensitivity of the PAI-1 induction to AG1478 (Figs. 5A, C), EGFR knockdown by lentiviral-transduced shRNA constructs (Fig. 6D) similarly decreased TGF-β1-mediated PAI-1 expression (Fig. 6E). Although both the pharmacologic and shRNA findings clearly implicated EGFR involvement in TGF-β-stimulated PAI-1 induction, a genetic approach was required to specifically confirm EGFR1 involvement. Such assessments were made possible by the fact that EGFR1-null MEFs did not express PAI-1 in response to TGF-β1 stimulation (Table 2). Introduction of a wild type EGFR1 construct by adenoviral delivery in EGFR1−/− fibroblasts “rescued” the response of the PAI-1 gene to TGF-β1 (Table 2).
Figure 6.
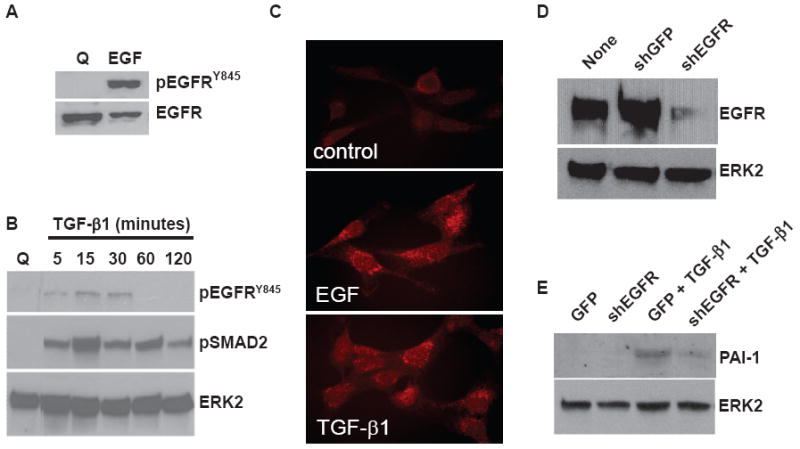
The EGFR is phosphorylated in response to TGF-β1 stimulation and required for induced PAI-1 expression. EGFR phosphorylation at the Y845 src kinase target residue was prominent in EGF- (A) and, to a lesser extent, in TGF-β (B) stimulated HMEC-1 cultures. The EGFRY845 site, however, had a relatively restricted activation window unlike SMAD2 phosphorylation which was evident within 5 minutes after TGF-β1 addition and maintained for at least 2 hours (B). TGF-β1 also stimulated EGFR internalization within 15 minutes of addition (as detected immunocytochemically with pEGFRY845 antibodies) although not to the same extent as EGFR-treated cultures (C) consistent with the differential EGFRY845 phosphorylation as detected by western blotting (A, B). EGFR knockdown by lentiviral-transduced shRNA constructs (D) effectively decreased (by >75%) TGF-β1-mediated PAI-1 expression (E).
Table 2.
“Rescue” of TGF-β1-stimulated PAI-1 induction in EGFR−/− fibroblasts engineered to re-express EGFR1.
| Cell type/treatmenta,b | PAI-1 expression level |
|---|---|
| EGFR−/−/−TGF-β1 | 1 ± 0 |
| EGFR−/−/+TGF-β1 | 1.3 ± 0.4 |
| EGFR−/−→EGFR+/+/−TGF-β1 | 2.8 ± 0.4 |
| EGFR−/−→EGFR+/+/+TGF-β1 | 11.0 ± 2.8 |
EGFR−/− MEFs were infected (MOI =50–100) with adenoviruses bearing either control (GFP) or wild-type human EGFR expression constructs in low-serum-containing medium. Indicated cell types (EGFR-null MEFs or cells engineered to re-express a wild-type EGFR) were maintained in growth factor-free medium (−TGF-β1) or stimulated with TGF-β1 (+TGF-β1; 1 ng/ml) for 5 hours.
Western blots of PAI-1 levels in cellular extracts were quantified by densitometry; data is the mean ± standard deviation of triplicate independent experiments.
Discussion
TGF-β1 activates both SMAD-dependent and independent signaling cascades32 that impact PAI-1 expression;20,24,31 their specific contributions to PAI-1 induction, however, are not well defined. TGF-β1-initiated EGFR transactivation highlights the complex cross-talk between TGF-β1 and EGFR signaling events in several cell types.19,21 The current study is the first to establish, moreover, that TGF-β1-induced PAI-1 expression in human microvessel endothelial cells, like the angiogenic response of 1G11 murine endothelial cells,19 is similarly dependent on EGFR signaling. Indeed, TGF-β1-stimulated PAI-1 induction is significantly attenuated by an EGFR pharmacologic inhibitor (AG1478) and, more specifically, by EGFR1 genetic deficiency.
TGF-β1 treatment increased EGFR phosphorylation at the Y845 src-target residue in human endothelial cells additionally implicating a src family member in EGFR transactivation in response to TGF-β1. The IC50 of the PP1-sensitive kinase, the inhibition of TGF-β1- (but not PDGF-) induced PAI-1 expression by the EGFRY845F mutant as well as a DN-Src construct20 collectively implicate EGFR/pp60c-src interactions and, in particular, the EGFRY845 pp60c-src site in PAI-1 induction.20,24 Indeed, addition of TGF-β1 to quiescent vascular smooth muscle cells resulted in a 3-fold increase in pp60c-src autophosphorylation and kinase activity.33 Similarly, TGF-β1 failed to stimulate PAI-1 expression in mouse embryonic fibroblasts (MEFs) genetically deficient in three src family kinases (i.e. c-src, c-yes-, c-fyn- null fibroblasts; SYF−/−/−) compared to identically stimulated wild-type SYF+/+/+ cells. PAI-1 synthesis was restored in SYF−/−/− MEFs engineered to re-express a wild-type pp60c-src providing proof-of-principle for involvement of this particular src kinase in the inductive response.20
Increasing evidence suggests that PAI-1 protects cells from death by modulating signaling cascades that promote cell survival or reduce apoptosis.12,13,25,27-29 In this regard TGF-β1 mediated synthesis and subsequent secretion of PAI-1 can modulate secondary signaling events (ERK or Akt activation) that prevent endothelial cell apoptosis or induce survival programs that precede and necessary for efficient morphogenesis response Pharmacological blockade of ERK1/2 and EGFR, also virtually eliminates endothelial branching induced by TGF-β1 in keeping with signaling elements necessary for TGF-β1 mediated PAI-1 induction and subsequent endothelial morphogenesis. Serum deprivation mediated apoptosis in T2 endothelial cells rescued by exogenous addition of PAI-1 likely through activation of ERK1/2 pathway.
The mechanism of MAP kinase activation in TGF-β1-stimulated cells is just becoming clear. Upon ligand binding, the TGF-βRII undergoes autophosphorylation on three tyrosines (Y259, Y336, Y424) while Y284 is a target site for src kinases.34 TGF-βRI is also subject to tyrosine phosphorylation post-receptor occupancy.35 Such phosphorylated tyrosine residues provide docking sites for recruitment of Grb2/Shc/SOS complexes with subsequent mobilization of the ras-raf-MEK-ERK cascade.20,33,34 Although ERKs are prominently activated in response to TGF-β1,20,31 the JNK and p38 MAP kinase pathways are better characterized targets of TGF-β1-initiated signaling. TGF-β1 rapidly activates JNK through MKK4 and p38 via MKK3/6 perhaps even in a cell type-specific fashion contributing to the mechanistic complexity of pathway cross-talk. Each of these kinase systems, moreover, have been implicated in a cell type-dependency of PAI-1 gene control.31,34 Should such pathways prove uniquely or, at least, preferentially utilized in specific cellular lineages, they may provide tumor type-specific targets for intervention therapy.
Acknowledgments
This work was supported by NIH grant GM57242 to PJH.
Footnotes
Disclosure
The authors report no conflicts of interest.
This is an open access article distributed under the terms of the Creative Commons Attribution License (http://www.creativecommons.org/licenses/by/2.0) which permits unrestricted use, distribution and reproduction provided the original work is properly cited.
References
- 1.Bertolino P, Deckers M, Lebrin F, ten Dijke P. Transforming growth factor-β signal transduction in angiogenesis and vascular disorders. Chest. 2005;128(6 Suppl):585S–90S. doi: 10.1378/chest.128.6_suppl.585S. [DOI] [PubMed] [Google Scholar]
- 2.Goumans MJ, Lebrin F, Valdimarsdottir G. Controlling the angiogenic switch: a balance between two distinct TGF-β receptor signaling pathways. Trends Cardiovasc Med. 2003;13(7):301–7. doi: 10.1016/s1050-1738(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 3.Byfield SD, Roberts AB. Lateral signaling enhances TGF-β response complexity. Trends Cell Biol. 2004;14(3):107–11. doi: 10.1016/j.tcb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Lebrin F, Deckers M, Bertolino P, ten Dijke P. TGF-β receptor function in the endothelium. Cardiovasc Res. 2005;65(3):599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 5.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002;21(7):1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ten Dijke P, Arthur HM. Extracellular control of TGFβ signaling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8(11):857–69. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 7.Ross S, Hill CS. How the Smads regulate transcription. Inter J Biochem Cell Biol. 2008;40(3):383–408. doi: 10.1016/j.biocel.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Ma J, Han JD, Wang N, Chen YG. Distinct regulation of gene expression in human endothelial cells by TGF-β and its receptors. Microvasc Res. 2006;71(1):12–9. doi: 10.1016/j.mvr.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Lux A, Salway F, Dressman HK, et al. Alk1 signalling analysis identifies angiogenesis related genes and reveals disparity between TGF-β and constitutively active receptor induced gene expression. BMC Cardiovasc Disord. 2006;6(13):1–17. doi: 10.1186/1471-2261-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.ten Dijke P, Miyazono K, Heldin CH. Signaling inputs converge on nuclear effectors in TGF-β signaling. Trends Biochem Sci. 2000;25(2):64–70. doi: 10.1016/s0968-0004(99)01519-4. [DOI] [PubMed] [Google Scholar]
- 11.Holderfield MT, Hughes CC. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-β in vascular morphogenesis. Circ Res. 2008;102(6):637–52. doi: 10.1161/CIRCRESAHA.107.167171. [DOI] [PubMed] [Google Scholar]
- 12.Balsara RD, Castellino FJ, Ploplis VA. A novel function of plasminogen activator inhibitor-1 in modulation of the AKT pathway in wild-type and plasminogen activator inhibitor-1-deficient endothelial cells. J Biol Chem. 2006;281(32):22527–36. doi: 10.1074/jbc.M512819200. [DOI] [PubMed] [Google Scholar]
- 13.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downsteam target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877–84. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkins-Port CE, Ye Q, Mazurkiewicz JE, Higgins PJ. TGF-β1 + EGF-initiated invasive potential in transformed human keratinocytes is coupled to a plasmin/MMP-10/MMP-1-dependent collagen remodeling axis: role for PAI-1. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-09-0043. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saunders WB, Bayless KJ, Davis GE. MMP-1 activation by serine proteases and MMP-10 induces human capillary tubular network collapse and regression in 3D collagen matrices. J Cell Sci. 2005;118(10):2325–40. doi: 10.1242/jcs.02360. [DOI] [PubMed] [Google Scholar]
- 16.Davis GE, Pintar Allen KA, Salazar R, Maxwell SA. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. J Cell Sci. 2001;114(5):917–30. doi: 10.1242/jcs.114.5.917. [DOI] [PubMed] [Google Scholar]
- 17.Wilkins-Port CE, Higgins CE, Freytag J, Higgins SP, Carlson JA, Higgins PJ. PAI-1 is a critical upstream regulator of the TGF-β1/ EGF-induced invasive phenotype in mutant p53 human cutaneous squamous cell carcinoma. J Biomed Biotech. 2007:1–8. doi: 10.1155/2007/85208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masson V, Devy L, Grignet-Debrus C, et al. Mouse aortic ring assay: a new approach of the molecular genetics of angiogenesis. Biol Proced Online. 2002;4(1):24–31. doi: 10.1251/bpo30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vinals F, Pouyssegur J. Transforming growth factor β1 (TGF-β1) promotes endothelial cell survival during angiogenesis via an autocrine mechanism implicating TGF-α signaling. Mol Cell Biology. 2001;21(21):7218–30. doi: 10.1128/MCB.21.21.7218-7230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samarakoon R, Higgins SP, Higgins CE, Higgins PJ. TGF-β1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60c-src/EGFRY845 and Rho/ROCK signaling. J Mol Cell Cardiology. 2008;44(3):527–38. doi: 10.1016/j.yjmcc.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joo CK, Kim HS, Park JY, Seomun Y, Son MJ, Kim JT. Ligand release-independent transactivation of epidermal growth factor receptor by transforming growth factor-β involves multiple signaling pathways. Oncogene. 2008;27(5):614–28. doi: 10.1038/sj.onc.1210649. [DOI] [PubMed] [Google Scholar]
- 22.Uchiyama-Tanaka Y, Matsubasr H, Mori Y, et al. Involvement of HB-EGF and EGF receptor transactivation in TGF-β-mediated fibronectin expression in mesangial cells. Kidney Int. 2002;62(3):799–808. doi: 10.1046/j.1523-1755.2002.00537.x. [DOI] [PubMed] [Google Scholar]
- 23.Higgins PJ, Ryan MP, Jelley DM. p52PAI-1 gene expression in butyrate-induced flat revertants of v-ras-transormed rat kidney cells: mechanism of induction and involvement in the morphological response. Biochem J. 1997;321(2):431–7. doi: 10.1042/bj3210431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samarakoon R, Higgins CE, Higgins SP, Higgins PJ. Differential requirement for MEK/ERK and SMAD signaling in PAI-1 and CTGF expression in response to microtubule disruption. Cell Signal. 2009;21(6):986–95. doi: 10.1016/j.cellsig.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lademann UA, Romer MU. Regulation of programmed cell death by plasminogen activator inhibitor type 1 (PAI-1) Thromb Haemost. 2008;100(6):1041–6. [PubMed] [Google Scholar]
- 26.Providence KM, Higgins SP, Mullen A, et al. SERPINE1 (PAI-1) is deposited into keratinocyte migration trails and required for optimal monolayer wound repair. Arch Dermatol Res. 2008;300(6):303–10. doi: 10.1007/s00403-008-0845-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romer MU, Larsen L, Offenberg H, Brunner N, Lademann UA. Plasminogen activator inhibitor 1 protects fibrosarcoma cells from atoposide-induced apoptosis through activation of the PI3K/AKT cell survival pathway. Neoplasia. 2008;10(10):1083–91. doi: 10.1593/neo.08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider DJ, Chen Y, Sobel BE. The effect of plasminogen activator inhibitor type 1 on apoptosis. Thromb Haemost. 2008;100(6):1037–40. [PubMed] [Google Scholar]
- 29.Bajou K, Peng H, Laug WE, et al. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14(4):324–34. doi: 10.1016/j.ccr.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Degryse B, Neels JG, Czekay RP, Aertgeerts K, Kamikubo Y, Loskutoff DJ. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem. 2004;279(21):22595–604. doi: 10.1074/jbc.M313004200. [DOI] [PubMed] [Google Scholar]
- 31.Kutz SM, Higgins CE, Samarakoon R, et al. TGF-β1-induced PAI-1 expression is E box/USF-dependent and requires EGFR signaling. Exp Cell Res. 2006;312(7):1093–105. doi: 10.1016/j.yexcr.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 32.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signaling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 33.Samarakoon R, Higgins CE, Higgins SP, Kutz SM, Higgins PJ. Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-β1-stimulated smooth muscle cells is pp60c-src/MEK-dependent. J Cell Physiol. 2005;204(1):236–46. doi: 10.1002/jcp.20279. [DOI] [PubMed] [Google Scholar]
- 34.Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res. 2009;19(1):128–39. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee MK, Pardoux C, Hall MC, et al. TGF-β activates Erk MAP kinase signaling through direct phosphorylation of ShcA. EMBO J. 2007;26(17):3957–67. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]