

Abstract
Selective acetylation of various per-O-TMS-protected carbohydrates has been accomplished. Using a protecting group exchange strategy and microwave assistance, monosaccharides (glucose, galactose and mannose) can be selectively acetylated producing either the 6-O-monoacetate or 1,6-O-diacetylated species. This new class of molecules can be deprotected without migration of the acetyl groups providing useful synthetic intermediates. To demonstrate the scope of the reaction, the methodology was successfully extended to TMS-protected ceramide.
With the current interest in glycochemistry and glycobiology growing, regioselectively functionalized carbohydrates are targets that garner significant attention. Carbohydrate molecules contain a number of hydroxyl units that are difficult to manipulate selectively due to similar reactivity profiles. Hence, methodologies that achieve efficient protection and selective modification of monosaccharides are important synthetic tools in organic chemistry.
One of the most fundamental and yet significant transformations in carbohydrate chemistry is the acetylation of monosaccharides. However, there has been little success in the selective acetylation of unprotected sugars, owing largely to the insolubility of carbohydrates in organic solvents. Enzymatic acylation1 has been shown to be useful for the preparation of selectively acetylated pyranosides,2 furanosides,3 sialosides,4 oligosaccharides and natural glycosides.5 This technology has also been extended to more complex carbohydrate structures, such as the selective acetylation of ginsenosides6 and diterpenoid glycosides,7 the production of gangliosides8 and combinatorial arrays,9 and has been shown to be effective in ionic liquids.10 Yet until now, a non-enzymatic method for the selective acetylation of unfunctionalized carbohydrates with minimal synthetic manipulation has not been available.
Non-enzymatic acetylations of carbohydrates typically utilize alkyl glycosides as substrates. Acetylation of the hydroxyl at C-6 can be achieved preferentially on alkyl glycosides, as conventional wisdom would predict.11 However, unprotected carbohydrates reportedly exhibit anomalous behavior. For example, Kurahashi, Mizutani and Yoshida found that the secondary hydroxyls in glycosides were preferentially acetylated over the primary 6-OH using acetic anhydride and DMAP, presumably due to intramolecular hydrogen bonding.12 Kattnig and Albert showed that selectivity of acetylation was also influenced by the counterion and that, while acetyl chloride reacted primarily at the C-6 hydroxyl, acetic anhydride yielded an inseparable mixture of acetylated products.13 With both of these approaches, it is difficult to remove the alkyl functionality at the anomeric position. Hence, the products of these reactions have yet to find wide utility as synthetic precursors.
As opposed to using either alkyl glycosides or unfunctionalized sugars, we explored the possibility of directly exchanging a protecting group with an acetyl group. Acetylation of a protected sugar would provide increased solubility in organic solvents, loss of intramolecular hydrogen bonding in the substrate and the ability to orthogonally deprotect and functionalize the product. We regularly utilize per-O-TMS-protected pyranosides and believed these would prove promising substrates, especially since acetic anhydride had been shown to acetylate per-O-TMS-protected N-acetylgalactosamine.14
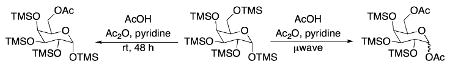
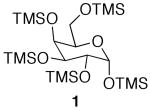
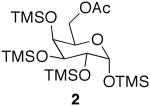
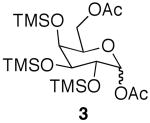
The initial substrate we chose to investigate was per-O-trimethylsilylgalactose (1), which was prepared in a single step from galactose.15 The galactoside was stirred with 2 equiv of acetic acid for 2 days, and while TLC showed less than 50% reaction completion, 35% of 6-O-monoacetate 2 and 2% of 1,6-O-diacetate 3 were isolated cleanly via column chromatography (Scheme 1). When the amount of acid was increased to 4 equiv, the substrate was completely consumed after 48 h, yielding 90% of the monoacetylated product and 7% of diacetate 3.16
Scheme 1.

Acetylation of per-O-TMS-galactoside
While encouraged by the results, a major drawback to the methodology was the long reaction period. In order to expedite the reaction time, the reaction was subjected to microwave irradiation. The reaction progression was monitored via TLC, and showed complete consumption of starting material after three 25 min cycles, as opposed to 48 h at rt (Scheme 1). Interestingly, the microwave assisted reaction produced a larger amount of diacetate 3 than was produced at room temperature. This led us to investigate whether the diacetylated product could be produced as the major product in the selective acetylation reaction. Indeed, when 4 equiv of acetic acid were used along with microwave assistance, diacetate 3 was the major product.
While selective acetylation at the 6-position was noteworthy in that it produced 6-O-acetyl-monosaccharides non-enzymatically, there have been no reported preparations of unfunctionalized 1,6-di-O-acetyl-monosaccharides, to the best of our knowledge. Hence, these novel molecules could prove useful as synthetic intermediates, either as the TMS-protected silyl ethers or the deprotected free sugar variant. Moreover, since anomeric acetates can be removed selectively,17 the 1-O-acetate is essentially orthogonal to the 6-O-acetate.
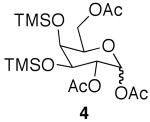
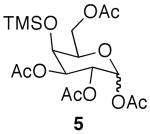
It is worth noting that use of microwave conditions also produced selectively acetylated triacetates (4) and tetraacetates (5) (Table 1, entry 4), and while they are minor products, the ease of separation allows access to ample quantities of material upon scale-up. Compounds 4 and 5 are useful synthetic intermediates, as galactosides similar to 4 have been used in the synthesis of glycans, such as Sialyl Lewis X.18
Table 1. Selective acetylation of per-O-TMS-protected monosaccharides.
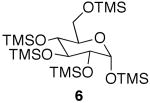
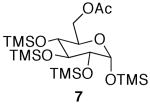
| entry | TMS-monosaccharide | conditions | equiv AcOH | monoacetate | diacetate | triacetate | tetraacetate |
|---|---|---|---|---|---|---|---|
 |
 |
 |
 |
 |
|||
| 1 | galactose | rt, 48 h | 2 | 35% | 2% | -a | -a |
| 2 | galactose | rt, 48 h | 4 | 90% | 7% | -a | -a |
| 3 | galactose | μwave, 3 × 25 min | 2 | 72% | 23% | -a | -a |
| 4 | galactose | μwave, 3 × 25 min | 4 | 22% | 52% | <5% | <5% |
 |
 |
 |
|||||
| 5 | glucose | rt, 48 h | 2 | 45% | 7% | -a | -a |
| 6 | glucose | rt, 48 h | 4 | 66% | 22% | -a | -a |
| 7 | glucose | μwave, 3 × 25 min | 2 | 42% | 28% | 10%b | -a |
 |
 |
 |
 |
 |
|||
| 8 | mannose | rt, 48 h | 2 | 70% | 16% | -a | -a |
| 9 | mannose | μwave, 3 × 25 min | 2 | 50% | 27% | 6% | 3% |
 |
 |
||||||
| 10 | N-acetylglucosamine | rt, 48 h | 2 | -a | 81% | -a | -a |
No product isolated.
Mixture of inseparable triacetates isolated.
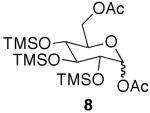
Similar reactivity was observed in the acetylation of per-O-TMS glucose (6).15 The reaction progression was slow at rt and, when 2 equiv of acid were used, the reaction did not proceed to completion (Table 1, entry 5). In contrast, TLC analysis showed complete substrate consumption when using 4 equiv of acetic acid after 48 h yielding 66% of 6-O-acetylglucoside 7 and 22% of 1,6-di-O-acetyl-glucoside 8 (Table 1, entry 6).16
These results indicate that the first site of acetylation of the glucoside is the primary C-6 position to afford monoacetate 7, which is further acetylated at the anomeric position to yield diacetate 8 as a mixture of anomers. The reaction rate was increased by use of microwave irradiation and triacetylated glucosides were isolated (Table 1, entry 7). With glucose, however, selectivity was not observed and an inseparable mixture of triacetates was obtained.
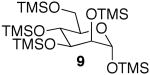
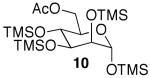
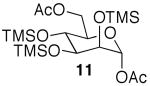
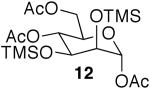
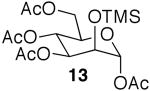
The promising reactivities of the galactose and glucose systems led to the extension of the technology to mannose. When per-O-TMS-mannose (9)15 was treated with 2 equiv of acetic acid, the reaction progressed more rapidly than either of the per-O-silylated galactose or glucose analogs at rt (Table 1, entry 8). The main product was monoacetate 10, which was isolated in 70% yield; and diacetate 11 was isolated in 16% yield. Under microwave irradiation, the main products were again 10 and 11, but, the microwave conditions also yielded selectively acetylated triacetate 12 and tetraacetate 13 in small quantities (Table 1, entry 9). These molecules are likely to be important analogs for the preparation of highmannose oligosaccharides.
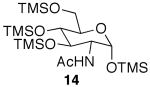
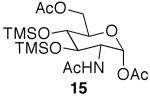
After noting the reaction's generality, attention was turned to N-acetylglucosamine (GlcNAc). Attempts to selectively acetylate per-O-TMS-protected N-acetylglucosamine (14)15 under microwave irradiation produced per-O-acetylated N-acetylglucosamine, even under shorter reaction times. Aside from being more reactive than the previous glycosides, GlcNAc compounds were very difficult to visualize by TLC, which complicated monitoring the reaction progression. However, when the substrate was stirred with 2 equiv of acetic acid at rt, 1,6-diacetyl species 15 was produced in good yields in 48 h (Table 1, entry 10). This product is poised for use in the preparation of a number of biologically important molecules, such as N-acetyllactosamine and chitobiose.
In earlier work, we have shown that per-O-TMS-protected glycosides can be readily deprotected via acidic methanolysis.19 However, there was concern that selectively acetylated TMS protected sugars might undergo acetyl cleavage or migration under the deprotection conditions. To explore this possibility, the substrates were dissolved in methanol and treated with Dowex for up to 1 h. To our delight, filtration and concentration yielded fully deprotected monosaccharides in high yield and purity (Table 2). It is important to note that while spectroscopic analysis revealed that there was no evidence of acetyl migration during deprotection, acetyl migration can occur over time. Since, there was no evidence of acetyl migration in the fully protected species, it is recommended that the acetylated intermediates be desilylated immediately prior to use, rather than stored as the deprotected sugar.
Table 2. Methanolysis of TMS-ethers with Dowex.
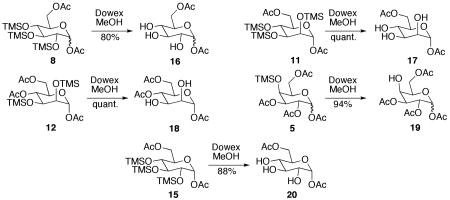 | |||
|---|---|---|---|
| substrate | product | time | yield |
| 8 | 16 | <15 min | 80% |
| 11 | 17 | <15 min | quant. |
| 12 | 18 | 15 min | quant. |
| 5 | 19 | 1 h | 94% |
| 15 | 20 | 15 min | 88% |
The synthesis of tetraacetate 19 had been previously reported by a 2-step enzymatic process, however our chemical shift and coupling constant data do not match the published NMR data.20 We are quite confident that our methodology produced 19 based upon extensive 1D and 2D NMR21 (Figure 1). Full proton assignment was accomplished using COSY and the coupling constants were indicative of an anomeric mixture. The position of the acetates was determined via HMBC NMR data, which correlated the proton signals at C-1, C-2, C-3 and C-6 with long range coupling to the carbonyl carbons.
Figure 1.

Key COSY (red) and HMBC (blue) interactions in 19
To test the selective acetylation of a molecule that was not a sugar, the methodology was extended to ceramide 21, a biologically important molecule. The ceramide was silylated cleanly using TMS-chloride and the O-TMS-protected ceramide was acetylated under microwave irradiation (Scheme 2). The reaction progressed quickly with 1 equiv of acetic acid and the reaction yielded selectively acetylated ceramide 22 in good yield along with small amounts of recovered substrate and diacetylated ceramide. Deprotection of the silyl ether proceeded without acetyl migration to yield acetylceramide 23 in 80% yield.
Scheme 2.

Selective acetylation and deprotection of ceramide
Wong and coworkers have developed a one-pot glycosylation strategy to facilitate complex oligosaccharide synthesis.22 The approach exploits the reactivity differences of various glycosyl donors on the basis of the position, number and type of protecting group; and acyl groups are generally thought to be deactivating.23 The nucleophilicity of glycosyl acceptors can also be attenuated by acyl protecting groups, as recently demonstrated in the synthesis of glycosyl ceramides.18b Moreover, Hung and coworkers have developed a regioselective one-pot protection of TMS-protected glycosides.24 The synthetic methodology presented herein nicely interfaces with these platforms providing powerful tools for accessing large arrays of glycosyl donors and acceptors for oligosaccharide synthesis.
Supplementary Material
Acknowledgments
This work was partially supported by the National Institutes of Health, NIH grant No. R01GM090262. MAW was supported by NIH fellowship F32-AI074329. The NMR spectrometers used were funded by NSF CRIF program (CHE-9808183), NSF Grants DBIO 722538, OSTI 97-24412, and NIH Grant RR11973. HRMS samples were analyzed by Dr. William Jewell at the UC Davis Campus Mass Spectrometry Facilities.
Footnotes
Supporting Information Available Experimental procedures, data and NMR spectra have been provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Faber K, Riva S. Synthesis. 1992:895. [Google Scholar]
- 2.(a) Therisod M, Klibanov AM. J Am Chem Soc. 1986;108:5638. [Google Scholar]; (b) Therisod M, Klibanov AM. J Am Chem Soc. 1987;109:3977. [Google Scholar]; (c) Wang YF, Lalonde JJ, Momongan M, Bergbreiter DE, Wong CH. J Am Chem Soc. 1988;110:7200. [Google Scholar]; (d) Theil F, Schick H. Synthesis. 1991:533. [Google Scholar]
- 3.Hennen WJ, Sweers HM, Wang YF, Wong CH. J Org Chem. 1988;53:4939. [Google Scholar]
- 4.Kim MJ, Hennen WJ, Sweers HM, Wong CH. J Am Chem Soc. 1988;110:6481. [Google Scholar]
- 5.(a) Riva S, Chopineau J, Kieboom APG, Klibanov AM. J Am Chem Soc. 1988;110:584. [Google Scholar]; (b) Danieli B, Luisetti M, Sampognaro G, Carrea G, Riva S. J Mol Catal B: Enzym. 1997;3:193. [Google Scholar]
- 6.(a) Danieli B, Luisetti M, Riva S, Bertinotti A, Ragg E, Scaglioni L, Bombardelli E. J Org Chem. 1995;60:3637. [Google Scholar]; (b) Gebhardt S, Bihler S, Schubert-Zsilavecz M, Riva S, Monti D, Falcone L, Danieli B. Helv Chim Acta. 2002;85:1943. [Google Scholar]; (c) Teng R, Ang C, McManus D, Armstrong D, Mau S, Bacic A. Helv Chim Acta. 2004;87:1860. [Google Scholar]
- 7.Colombo G, Riva S, Danieli B. Tetrahedron. 2004;60:741. [Google Scholar]
- 8.Zhang X, Kamiya T, Otsubo N, Ishida H, Kiso M. J Carbohydr Chem. 1999;18:225. [Google Scholar]
- 9.Yu HL, Xu JH, Wang YX, Lu WY, Lin GQ. J Comb Chem. 2007;10:79. doi: 10.1021/cc7001606. [DOI] [PubMed] [Google Scholar]
- 10.Park S, Kazlauskas RJ. J Org Chem. 2001;66:8395. doi: 10.1021/jo015761e. [DOI] [PubMed] [Google Scholar]
- 11.Ishihara K, Kurihara H, Yamamoto H. J Org Chem. 1993;58:3791. [Google Scholar]
- 12.Kurahashi T, Mizutani T, Yoshida Ji. J Chem Soc, Perkin Trans I. 1999:465. [Google Scholar]
- 13.Kattnig E, Albert M. Org Lett. 2004;6:945. doi: 10.1021/ol0364935. [DOI] [PubMed] [Google Scholar]
- 14.(a) Fuchs EF, Lehmann J. Chem Ber. 1974;107:721. [Google Scholar]; (b) Auge C, David S, Gautheron C, Veyrieres A. Tetrahedron Lett. 1985;26:2439. [Google Scholar]
- 15.Bhat AS, Gervay-Hague J. Org Lett. 2001;3:2081. doi: 10.1021/ol0160405. [DOI] [PubMed] [Google Scholar]
- 16.While the α:β ratio of the anomeric acetate was found to vary based on the α:β ratio of the starting material, the reaction yielded a 2:1 α:β ratio when the substrate was composed entirely of the α-anomer.
- 17.Tiwari P, Misra AK. Tetrahedron Lett. 2006;47:3573. [Google Scholar]
- 18.Danishefsky SJ, Gervay J, Peterson JM, McDonald FE, Koseki K, Oriyama T, Griffith DA, Wong CH, Dumas DP. J Am Chem Soc. 1992;114:8329. [Google Scholar]
- 19.(a) Du W, Kulkarni SS, Gervay-Hague J. Chem Commun. 2007:2336. doi: 10.1039/b702551c. [DOI] [PubMed] [Google Scholar]; (b) Schombs MW, Park FE, Du W, Kulkarni SS, Gervay-Hague J. J Org Chem. 2010;75:4891. doi: 10.1021/jo100366v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filice M, Bavaro T, Fernandez-Lafuente R, Pregnolato M, Guisan JM, Palomo JM, Terreni M. Catal Today. 2009;140:11. [Google Scholar]
- 21.All compounds were fully characterized using 1D and 2D NMR.
- 22.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734. [Google Scholar]
- 23.(a) Douglas NL, Ley SV, Lucking U, Warrnier SL. J Chem Soc, Perkin Trans I. 1998;1:51. [Google Scholar]; (b) Crich D. Acc Chem Res. 2010 doi: 10.1021/ar100035r. ASAP. [DOI] [PubMed] [Google Scholar]
- 24.Wang CC, Lee JC, Luo SY, Kulkarni SS, Huang YW, Lee CC, Chang KL, Hung SC. Nature. 2007;446:896. doi: 10.1038/nature05730. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.