

Abstract
E2F transcription factors (E2F1-8) are best known for their role in cell proliferation, although it is clear that they regulate many other biological processes through the transcriptional modulation of distinct target genes. However, the specific set of genes regulated by each E2F remains to be characterized. To gain insight into the molecular pathways regulated by E2F2, we have analyzed the proteome of antigen receptor–activated T cells lacking E2F2. We report that loss of E2F2 results in a deregulated Aryl-hydrocarbon-receptor pathway. Proliferating E2F2−/− T lymphocytes expressed significantly higher levels of Aip, Ahr, and Arnt relative to wild-type (WT)1 controls. The mechanism for increased levels of Aip appears straightforward, involving direct regulation of the Aip gene promoter by E2F2. Although the Ahr and Arnt promoters also bind E2F2, their regulation appears to be more complex. Nevertheless, exposure to the environmental xenobiotic 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), a well-known exogenous ligand of the Ahr pathway, led to overexpression of the Ahr target gene Cyp1a1, and to increased sensitivity to TCDD-triggered apoptosis in E2F2−/− T cells compared with WT controls. These results suggest that E2F2 modulates cellular sensitivity to xenobiotic signals through the negative regulation of the Ahr pathway.
E2F transcription factors (E2F1-8) are critical regulators of the cell cycle, in connection with the retinoblastoma family of proteins. They were initially recognized for their ability to regulate the G1/S transition of the cell cycle by controlling the transcription of genes involved in DNA replication, nucleotide biosynthesis, and cell cycle progression (1). However, subsequent studies identified E2F targets involved not only in G1/S entry and progression, but also in mitosis, apoptosis, DNA repair, chromosome organization, carboxylic acid and amino acid metabolism, or differentiation (2, 3), implying that the role of E2F transcription factors in cellular physiology is more complex than it was originally thought to be. The list of functions in which E2Fs are involved is probably not complete. For instance, a role for E2F in development has been proposed recently in Xenopus and zebrafish (4, 5), and there is also evidence for their role in processes such as endocycle regulation and plant cell size control (6, 7), or in autophagy (8).
Phenotypic characterization of mice carrying targeted mutations for E2F family members has confirmed the complexity of the regulatory network controlled by E2F transcription factors (2, 9). These analyses have shown that each E2F member plays a unique role in mouse physiology, although there is a certain degree of functional compensation among them. Moreover, conflicting data have been gathered in the analysis of these mice, which has led to propose that the function of each individual E2F member may be modulated differently, depending on the cellular context (2). This seems to be the case with E2F2. Its positive role in cell-cycle progression has been demonstrated for certain cell types, such as fibroblasts and hematopoietic progenitors (10–12). Conversely, E2F2 has also been shown to regulate negatively the proliferation of T cells through the transcriptional repression of genes involved in DNA replication and cell-cycle progression (13–15). It has also been shown that it plays a role in apoptosis induction (9) or in neurogenic differentiation (16). The identification of the genes that are regulated in each particular cellular context by E2F2 could help explain the different roles played by this transcription factor.
Global gene expression analyses at the mRNA and protein level have become extremely useful to investigate gene expression signatures that are specific for a given physiological or pathological condition (17–21). Functional genomic analyses with DNA microarrays have contributed greatly to the identification of gene signatures that are regulated upon ectopic expression or functional inactivation of E2F transcription factors (15, 22–24). Transcriptomic studies are generally performed on the basis that there should be a good correlation between the RNA level and the protein level. This assumption, however, is not always true, probably due to technical differences between both techniques, and because the level of mRNA change may not be directly translated into a change of protein level (25, 26). Thus, both types of molecules should be examined to obtain a more complete picture of the cellular process under study (27).
We have previously performed transcriptomic and proteomic analyses with quiescent T lymphocytes lacking E2F2, and have provided evidence demonstrating the aberrantly activated state of these cells before stimulation, which could account for their hyperproliferative phenotype (15, 28). To deepen our understanding on the mechanism by which E2F2 regulates T cell physiology, we analyzed the proteome of E2F2-deficient T lymphocytes that have initiated their proliferative cycle after activation through the T-cell receptor (TCR). We found that the mediators of the Ahr pathway are aberrantly expressed in activated E2F2−/− T cells compared with WT cells, which renders them highly sensitive to apoptosis upon exposure to environmental xenobiotics, such as TCDD. These results underscore the importance of expression proteomics combined with gene knockout technology in defining the functional role of specific genes.
MATERIALS AND METHODS
Mouse Strains and MEF Preparation
E2F2−/− and WT mice were maintained in a C57Bl6:129Sv background on a normal light/dark cycle in cages with microisolator lids, and were genotyped by standard PCR technology, as previously described (13). All procedures were approved by the Animal Care and Use Committee of the University of the Basque Country.
Harvest, Purification, Flow Cytometry and Culture of T Lymphocytes
Lymph nodes from 4- to 6-week-old E2F2−/− and WT mice were obtained and lymphocytes collected. For cell surface staining, antibodies conjugated to FITC (CD44, CD69), to phycoerithrin (CD4), or to PerCP (CD8) were used (BD Biosciences). Cells were analyzed on a FACSCalibur (BD Biosciences) flow cytometer.
Lymphocytes from three individual mice of each genotype were extracted and T lymphocyte purification was performed by pulling down and discarding B-lymphocytes using biotinylated anti-B220 antibody (BD Biosciences) and M-280 streptavidin Dynabeads (Dynal Biotech, Oslo, Norway). T lymphocyte purification was checked by flow cytometry as previously described (15). For analysis of TCR-mediated responses, purified T lymphocytes (106/mL) were stimulated for the indicated times with plate-immobilized antibodies against CD3 at a concentration of 1.5 μg/well (BD Biosciences).
For TCDD treatment, T lymphocytes were first activated with anti-CD3 for 36 hours. Subsequently, T lymphocyte culture media was replaced with media containing vehicle (toluene) or TCDD (Supelco, Bellefonte, PA), and incubated for the indicated times before harvest. Cells were then stained with the Annexin V apoptosis detection kit (BD Biosciences) according to the manufacturer's instructions, or subjected to RNA extraction.
RNA Isolation and RT-PCR
Total RNA was prepared from cells using TRIzol reagent (Invitrogen), according to the manufacturer's instructions, purified using RNeasy (Qiagen, Hilden, Germany), and electrophoresed on a denaturing agarose gel to examine for RNA integrity. cDNA was synthesized from 2.5 μg of total RNA using Superscript First-Strand Synthesis System for RT-PCR (Invitrogen). Real-time PCR was performed on several cDNA dilutions as well as 1x SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA) and primers at the optimized concentrations (PCR primers are shown in Table I of Supplemental Data). Reactions were performed in the Applied Biosystems 7900 Fast Real-Time PCR System, and analysis was performed with the SDS 2.2.1 software for 40 cycles (95 °C for 15 s and 60 °C for 1 min) after an initial 10-min incubation at 95 °C. Reactions were performed in triplicate, and relative amounts of cDNA were normalized to the internal control, Eef1a1 mRNA. Statistical analyses were performed using one-way analysis of variance with simple contrasts to perform comparisons among groups at a significance level of p < 0.05 with the SPSS 14.0 program.
Table I. T-cell subset composition in E2F2+/+ and E2F2−/− lymph nodes.
| E2F2+/+ (n = 6) | E2F2−/− (n = 6) | |
|---|---|---|
| CD4+ | 33.9% ± 0.8 | 34.8% ± 2.2 |
| CD8+ | 20.8% ± 2.5 | 22.0% ± 3.4 |
| CD4+/CD69+ | 0.4% ± 0.2 | 0.2% ± 0.1 |
| CD8+/CD69+ | 0.4% ± 0.2 | 0.24% ± 0.1 |
| CD4+/CD44hi | 8.7% ± 0.8 | 9.0% ± 0.7 |
| CD8+/CD44hi | 4.8% ± 0.7 | 6.1% ± 0.4 |
Protein Sample Preparation
For total protein extraction, activated T lymphocytes (95% pure) were incubated in lysis buffer (7 mol/L urea, 2 mol/L thiourea, 4% CHAPS, 1% dithioerythritol, 0.4% IPG buffer pH 4–7) and thoroughly vortexed and incubated for 45 min at room temperature. Samples were centrifuged at 13,000 g for 15 min and supernatants were collected. Protein was quantified with 2D Quant Kit (GE Healthcare) and 2D Clean-Up kit (GE Healthcare) was applied to the samples for removal of interfering substances. Protein pellets were then resuspended in extraction lysis buffer, incubated for one hour under agitation, and stored at −20 °C.
2-DE Analysis
Two independent experiments were performed using three different biological replicates per genotype and experiment. Six gels were run simultaneously in each experiment, three for each genotype, each corresponding to a different mouse sample. IEF was performed on IPG strips (pH 4–7, 18 cm) on an IPGphor unit (GE Healthcare) and 150 μg of sample were loaded in each gel. Strips were actively rehydrated at 50 V for 15 hours. The IEF protocol was as follows: two hours at 300 V, 300–3,000 V linear gradient for one hour, two hours at 5,000 V, 5,000–8,000 V linear gradient for two hours, and two hours at 8,000 V up to a total of 40,000 Vh. After the first dimension, the strips were incubated in equilibration solution (6 mol/L urea, 2% SDS, 40 mmol/L Tris- HCl pH 8.8, 10% glycerol) containing 2% DTT for 15 minutes, and then were transferred to equilibration solution containing 2.5% iodoacetamide. Separation in the second dimension was performed using 12.5% self-cast acrylamide gels in an Ettan Dalt system (GE Healthcare).
Protein Visualization and Image Analysis
For fluorescent staining, gels were immersed in 40% ethanol and 10% acetic acid for 30 minutes followed by overnight staining with SYPRO Ruby (Bio-Rad). Gels were washed in a solution containing 10% ethanol and 7% acetic acid for 30 minutes, then protein patterns were digitized using a Molecular Imager FX scanner and the Quantity One 4.1–0 software (Bio-Rad). Gel image analysis was performed with the Imagemaster Platinum software (GE Healthcare). Gel patterns from each independent analysis were matched together and the relative volume of each spot (%V) in the two gel sets (control and knock-out) were compared using Student's t test at a significance level of p < 0.05. Gels were stored in deionized water at 4 °C. Silver staining was performed (29) to identify points of interest on the gels.
Protein Identification by MS
Differentially expressed spots were excised from stained gels and trypsin in-gel digestion performed following the method described by Shevchenko et al. (30) with minor modifications. LC-MS/MS spectra were acquired using a Q-Tof micro mass spectrometer (Waters, Manchester, UK) interfaced with a CapLC System (Waters). An aliquot (8 μL) of each sample was loaded onto a Symmetry 300 C18 NanoEase Trap precolumn (Waters) and washed with 0.1% formic acid for 5 min at a flow rate of 20 μL/min. The precolumn was connected to a XBridge BEH130 C18, 75 μm x 150 mm, 3.5 μm (Waters) equilibrated in 5% acetonitrile and 0.1% formic acid and peptides were eluted at 300 nL/min with a 30-minute linear gradient of 10–60% acetonitrile directly onto a NanoEase Emitter (Waters). Data-dependent MS/MS acquisitions were performed on precursors with charge states of 2, 3, or 4 over a survey m/z range of 400–1,500. Collision energies were varied as a function of the m/z and charge state of each peptide. To avoid redundancy of MS/MS peptides that had already undergone collision-induced dissociation, dynamic peak exclusion was incorporated for 45 seconds. Obtained spectra were processed using default processing parameters by Virtual Expert Mass Spectrometrist version 2006 (31) and searched using MASCOT 2.2 (Matrix Science, London, UK) against SwissProt 57.8 (509019 sequences; 178948533 residues) database restricted to mouse.
For protein identification, the following parameters were adopted: carbamidomethylation of cysteines as fixed modification, oxidation of methionines as variable modification, 50 ppm of peptide mass tolerance, 0.1 Da fragment mass tolerance, and one missed cleavage was allowed. The cut-off score used to filter peptide identifications was the Mascot identity score.
Western Blotting
Cells were lysed by five minutes of boiling in loading buffer containing 50 mmol/L Tris pH 6.8, 5% glycerol, 1.67% β-mercaptoethanol, 1.67% SDS. Twenty micrograms of protein were loaded per lane, fractionated by SDS-PAGE in 10% or 12% polyacrylamide gels, and transferred onto nitrocellulose membranes (Bio-Rad). Antibodies against the following proteins were used: Ahr (ab2770, Abcam), Aip (ab468, Abcam), β-Actin (A5441, Sigma-Aldrich) Arnt (ab2771, Abcam), Crkl (ab49930, Abcam), Cyp1a1 (sc-25304, Santa Cruz Biotechnology), E2F2 (sc-2281, Santa Cruz Biotechnology), Gsta1 (ab53940, Abcam), Lamb1 (ab20396, Abcam), Immunocomplexes were visualized with horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin G antibodies (GE Healthcare), followed by chemiluminiscence detection (ECL, GE Healthcare) with a ChemiDoc camera (Bio-Rad).
Chromatin Immunoprecipitation Analyses
The chromatin immunoprecipitation was performed as previously described (15). T lymphocytes were crosslinked by addition of formaldehyde to 1.1% final concentration. Crosslinking was allowed to proceed at room temperature for 10 minutes and was stopped with glycine. After cell lysis, nuclei were collected by centrifugation, resuspended in 50 mmol/L Tris-Cl pH 8, 10 mmol/L EDTA pH 8.0, 1% SDS, 1 mmol/L PMSF and protease inhibitor mixture. Chromatin was sonicated to an average length of 300–400 bp (Misonix sonicator). After centrifugation, the supernatant was diluted 10-fold in chromatin immunoprecipitation (ChIP) dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mmol/L EDTA, 16.7 mmol/L Tris-Cl pH 8.0, 167 mmol/L NaCl, 1 mmol/L PMSF and protease inhibitor mixture) and pre-cleared with 50% slurry of protein A-Sepharose (GE Healthcare), blocked with salmon sperm DNA and BSA at 4 °C for three hours. Samples of 100–120 μg of precleared chromatin were incubated overnight at 4 °C with 4–5 μg of anti-E2F2 (sc-633) or anti-SV40Tag (sc-147) antibodies (Santa Cruz Biotechnology). Next, samples were incubated with protein A-Sepharose at 4 °C for two hours. Immune complexes were recovered and washed. The elution of the immune complexes was performed with a buffer containing 0.1 mol/L NaHCO3 and 1% SDS. Crosslinking was reversed by addition of NaCl to a final concentration of 200 mmol/L, and overnight incubation at 65 °C. Proteins were digested with 80 μg of proteinase K at 42 °C for two hours, and DNA was extracted with phenol-chloroform and ethanol precipitation.
DNA was amplified with primers complementary to the Aip, Ahr, Arnt, and Ahrr promoters (PCR primers are shown in Table II of Suppl. Data). PCR reaction conditions were as follows: 30 seconds at 94 °C, 30 seconds at 60 °C, and 45 seconds at 72 °C.
Table II. List of the proteins identified in experiment 1 and 2.
Entry and protein name are obtained from SwissProt 57.8 (509019 sequences; 178948533 residues). Average Ratio: E2F2/WT of normalized spot volumes * Protein identified with a single peptide (additional information on this peptide in supplementary Figure 1 and supplementary Table III and IV). Representative 2-DE images of experiments 1 and 2 are provided in supplementary Figures 2 and 3.
| Experiment | Spot | Uniprot number | Entry name | Protein name | Average ratio | Std. dev. | Mascot Score | N peptides | Seq. coverage |
|---|---|---|---|---|---|---|---|---|---|
| Experiment 1 | 1 | O08915 | AIP_MOUSE | AH receptor-interacting protein | 1.56 | 0.15 | 128 | 7 | 0.25 |
| 2 | P56480 | ATPB_MOUSE | ATP synthase subunit beta, mitochondrial | 1.37 | 0.12 | 1037 | 16 | 0.42 | |
| 3 | P38647 | GRP75_MOUSE | Stress-70 protein, mitochondrial | 1.50 | 0.10 | 776 | 17 | 0.34 | |
| 4 | Q9D1Q6 | ERP44_MOUSE | Endoplasmic reticulum resident protein ERp44 | 1.37 | 0.06 | 215 | 5 | 0.15 | |
| 5 | Q8CAQ8 | IMMT_MOUSE | Mitochondrial inner membrane protein | 1.30 | 0.15 | 27 | 2 | 0.03 | |
| 6 | P29758 | OAT_OUSE | Ornithine aminotransferase, mitochondrial | 1.44 | 0.14 | 273 | 8 | 0.21 | |
| 7 | P62137 | PP1A_MOUSE | Serine/threonine-protein phosphatase PP1-alpha catalytic subunit | 1.21 | 0.05 | 256 | 9 | 0.28 | |
| 7 | P62806 | H4_MOUSE | Histone H4 | 1.21 | 0.05 | 153 | 3 | 0.29 | |
| 7 | Q02257 | PLAK_MOUSE | Junction plakoglobin | 1.21 | 0.05 | 51 | 2 | 0.03 | |
| 7 | O70456 | 1433S_MOUSE | 14-3-3 protein sigma | 1.21 | 0.05 | 31 | 2 | 0.08 | |
| 8 | P20108 | PRDX3_MOUSE | Thioredoxin-dependent peroxide reductase, mitochondrial | 1.20 | 0.10 | 85 | 2 | 0.09 | |
| 9 | O35955 | PSB10_MOUSE | Proteasome subunit beta type-10 | 1.20 | 0.10 | 47 | 4 | 0.15 | |
| 10 | P22935 | RABP2_MOUSE | Cellular retinoic acid-binding protein 2 | 1.44 | 0.17 | 108 | 4 | 0.28 | |
| 11 | Q99JB2 | STML2_MOUSE | Stomatin-like protein 2 | 1.46 | 0.24 | 459 | 8 | 0.31 | |
| 12 | Q9WUP7 | UCHL5_MOUSE | Ubiquitin carboxyl-terminal hydrolase isozyme L5 | 1.24 | 0.11 | 190 | 6 | 0.20 | |
| 13 | P47941 | CRKL_MOUSE | Crk-like protein | 0.46 | 0.04 | 65 | 3 | 0.14 | |
| 14 | Q05816 | FABP5_MOUSE | Fatty acid-binding protein, epidermal | 0.69 | 0.06 | 37 | 2 | 0.17 | |
| 15 | Q8BH04* | PCKGM_MOUSE | Phosphoenolpyruvate carboxykinase [GTP], mitochondrial | 0.35 | 0.06 | 43 | 1 | 0.02 | |
| Experiment 2 | 1 | O08915 | AIP_MOUSE | AH receptor-interacting protein | 1.95 | 0.14 | 99 | 3 | 0.21 |
| 2 | P08030 | APT_MOUSE | Adenine phosphoribosyltransferase | 1.57 | 0.24 | 209 | 3 | 0.10 | |
| 3 | P57776 | EF1D_MOUSE | Elongation factor 1-delta | 1.32 | 0.05 | 204 | 5 | 0.25 | |
| 4 | Q9D8Y0 | EFHD2_MOUSE | EF-hand domain-containing protein D2 | 1.53 | 0.03 | 233 | 4 | 0.22 | |
| 5 | P30416 | FKBP4_MOUSE | FK506-binding protein 4 | 2.29 | 0.22 | 215 | 5 | 0.11 | |
| 5 | Q8JZK9 | HMCS1_MOUSE | Hydroxymethylglutaryl-CoA synthase, cytoplasmic | 2.29 | 0.22 | 105 | 2 | 0.14 | |
| 6 | Q9WUK2 | IF4H_MOUSE | Eukaryotic translation initiation factor 4H | 1.45 | 0.10 | 81 | 7 | 0.21 | |
| 7 | P63242 | IF5A1_MOUSE | Eukaryotic translation initiation factor 5A-1 | 2.12 | 0.26 | 140 | 5 | 0.30 | |
| 8 | Q9D1J3 | SARNP_MOUSE | SAP domain-containing ribonucleoprotein | 1.90 | 0.21 | 175 | 3 | 0.27 | |
| 8 | Q8K2T1 | NMRL1_MOUSE | NmrA-like family domain-containing protein 1 | 1.90 | 0.21 | 129 | 4 | 0.16 | |
| 9 | Q61990 | PCBP2_MOUSE | Poly(rC)-binding protein 2 | 1.39 | 0.10 | 150 | 3 | 0.28 | |
| 9 | Q8BXL7 | ARFRP_MOUSE | Actin-related protein 2 | 1.39 | 0.10 | 29 | 2 | 0.06 | |
| 10 | P70296 | PEBP1_MOUSE | Phosphatidylethanolamine-binding protein 1 | 1.27 | 0.14 | 147 | 5 | 0.21 | |
| 11 | P67778 | PHB_MOUSE | Prohibitin | 1.21 | 0.06 | 252 | 8 | 0.31 | |
| 12 | P34022 | RANG_MOUSE | Ran-specific GTPase-activating protein | 1.25 | 0.08 | 100 | 2 | 0.10 | |
| 13 | Q9CQV8 | 1433B_MOUSE | 14-3-3 protein beta/alpha | 0.55 | 0.12 | 56 | 2 | 0.09 | |
| 14 | P63101 | 1433Z_MOUSE | 14-3-3 protein zeta/delta | 0.52 | 0.03 | 141 | 3 | 0.15 | |
| 15 | P61164 | ACTZ_MOUSE | Alpha-centractin | 0.48 | 0.09 | 29 | 2 | 0.08 | |
| 16 | Q8WTY4 | CPIN1_MOUSE | Anamorsin | 0.78 | 0.12 | 182 | 6 | 0.22 | |
| 17 | P47941 | CRKL_MOUSE | Crk-like protein | 0.53 | 0.07 | 85 | 5 | 0.23 | |
| 18 | Q9DCT2 | NDUS3_MOUSE | NADH dehydrogenase [ubiquinone] iron-sulfur protein 3, mitochondrial | 0.68 | 0.06 | 56 | 2 | 0.09 | |
| 19 | P62814 | VATB2_MOUSE | V-type proton ATPase subunit B, brain isoform | 0.72 | 0.04 | 90 | 3 | 0.08 |
Quantification of immunoprecipitate-enriched DNA sequences was performed by real-time PCR with Power SYBR Green PCR Master Mix and the Applied Biosystems 7900 Fast Real-Time PCR System, and analysis was performed with the SDS 2.2.1 software. Samples were analyzed in triplicate. A standard curve for each promoter was constructed with five different input DNA dilutions of known concentration (X-axis) and their corresponding cycle threshold values (Y-axis). The unknown amount of E2F2-immunoprecipitated DNA was calculated from cycle threshold values through the standard curve plot.
RESULTS
Two Dimensional Gel Electrophoresis (2-DE) Analysis Reveals a Differential Protein Expression Pattern for Activated E2F2−/− T Lymphocytes
To gain insight into the mechanisms involved in E2F2-mediated regulation of T-cell physiology, we set out to examine the proteome profile of E2F2-deficient T cells after antigenic stimulation. We have previously shown an expansion of a subset of cells with a memory phenotype (CD44hi, CD69+) in aged E2F2−/− mice, although the percentage of these cells in young E2F2−/− mice is comparable to that in WT counterparts (13). To ensure that the lymphocyte subset composition in E2F2−/− and WT samples used in this work was comparable, only young (4- to 6-week-old) mice were used. Flow cytometry analysis showed that the fraction of CD8+ or CD4+ T lymphocytes that were CD44hi or CD69+ in WT and E2F2−/− lymph nodes was similarly low in both genotypes (Table I), indicating that their subset compositions with regard to memory cells were equivalent.
Lymph node-derived T cells obtained from WT and E2F2−/− mice were incubated for 36 hours with plate-bound anti-CD3 antibodies for their activation, which allowed cells to exit G0 and transit through G1/S (15). Protein was extracted from these cells and analyzed by 2-DE. For a reliable analysis of the gene expression pattern in these cells, two independent experiments were performed and three biological replicates were analyzed per genotype and experiment. That is, six different biological replicates per genotype were analyzed using this approach (Fig. 1). Among all 12 gels (6 WT and 6 E2F2−/− between two independent experiments), only two spots were reliably detected and observed to be different between genotypes in every case.
Fig. 1.
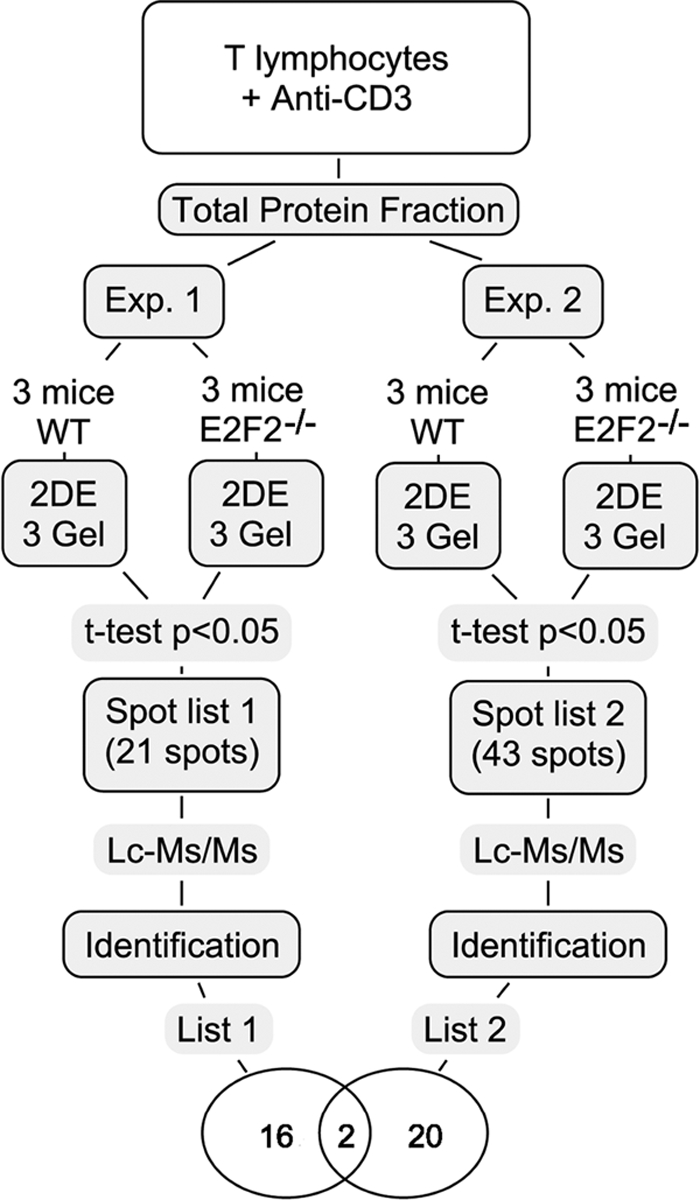
Experimental design and workflow summary. Two independent experiments were performed, and three biological replicates were used per genotype and experiment. Differentially expressed proteins with statistical significance (t test p < 0.05) were identified by LC-MS/MS, resulting two proteins commonly deregulated in both experiments: Aip and Crkl.
Many additional spots were detected in each of the two independent experiments: for experiment 1, as many as 340 spots, and for experiment 2, as many as 381 spots were detected. In experiment 1, 16 of these were increased and five decreased in E2F2−/− T cells relative to WT. In experiment 2, 29 were increased and 14 decreased in E2F2−/− T cells relative to WT. In each case, the intra-experiment change in intensity was statistically significant (t test, p < 0.05), although the spots were not observed in both independent experimental replicates.
The differentially expressed spots were chosen and digested with trypsin for subsequent LC-MS/MS analysis. Identified proteins are summarized in Table II and representative 2-DE images are shown in supplemental Figures 2 and 3. We discarded spots that failed to yield a clear identification of a single isoform. A total of 34 differentially expressed spots corresponding to 40 protein isoforms were identified, 18 of which were identified in experiment 1, and 22 in experiment 2.
Two proteins were identified with reproducibly altered expression levels in both experiments (comparing 6 WT to 6 E2F2−/− gels): Aryl-hydrocarbon receptor interacting protein (Aip), observed to be increased in E2F2−/− T cells (expression ratios in E2F2−/− T cells relative to WT were 1.95 ± 0.14 and 1.56 ± 0.15, p < 0.05), and the Crk-like protein (Crkl), observed to be decreased in E2F2−/− T cells (expression ratios in E2F2−/− T cells relative to WT were 0.53 ± 0.07 and 0.46 ± 0.04, p < 0.05).
Genes encoding the deregulated proteins were functionally classified using the DAVID Gene Functional Classification tool (http://david.abcc.ncifcrf.gov/). This analysis revealed that a high number of deregulated proteins were involved in metabolic regulation (36%). Features such as protein localization (17%) or oxidative phosphorylation (10%) were also common among them.
Consistent Deregulation of Aip and Crkl in Activated T Lymphocytes
To examine the functional consequences of aberrant protein expression in activated E2F2−/− cells, we focused on Aip and Crkl, the two proteins that appeared deregulated in both 2-DE analyses. Deregulation of these two proteins was further confirmed by Western blotting (expression ratios in E2F2−/− T cells relative to WT were 2.4 ± 0.8 in the case of Aip, and 0.4 ± 0.1 in the case of Crkl; n = 8 WT, n = 8 E2F2−/−, three independent experiments, p < 0.05), and by quantitative RT-PCR (expression ratios in E2F2−/− T cells relative to WT were 1.5 ± 0.01 in the case of Aip, and 0.8 ± 0.01 in the case of Crkl; n = 6 WT, n = 6 E2F2−/−, two independent experiments, p < 0.05) (Fig. 2).
Fig. 2.

Validation of deregulated expression of Aip and Crkl expression in activated E2F2−/− T lymphocytes. (A, C) Western analysis of Aip and Crkl expression values showing normalized ratios. The ratios Aip/β-actin and Crkl/β-actin in WT were considered as a unit. The experiment shown is representative of three independent experiments (n = 8 WT; n = 8 E2F2−/−). (B, D) Quantitative RT-PCR analysis of Aip and Crkl mRNA expression. Values were normalized against the mRNA expression levels of an irrelevant gene (eEf1a1) and given as relative values (n = 6 WT; n = 6 E2F2−/−; two independent experiments, each in triplicate).
A search for transcription factor binding sites in the promoters of Aip and Crkl using the Consite web tool (http://asp.ii.uib.no:8090/cgi-bin/CONSITE/consite) (32) revealed an E2F site in the Aip promoter (at the −487 bp position, with an 83% similarity compared with the E2F consensus sequence), suggesting that Aip is an E2F target gene. Thus, we considered the possibility that Aip and the pathway regulated by this protein could be under E2F2 control in T lymphocytes. No such site was found in the Crkl promoter, therefore we did not pursue it further.
E2F2 Binds to the Promoter of Genes of the Ahr Pathway
Aip is a protein that interacts with and modulates the activity of Ahr/Arnt, a ligand-activated transcription factor heterodimer that binds consensus xenobiotic response element sequences, thereby regulating transcriptional activation of genes carrying these motifs (33, 34). Most of these targets are related to detoxification response phase I and II, such as Cyp1a1 and Gsta1 (35), although recent reports also suggest a role for Ahr in the regulation of cell cycle and proliferation (36–38). A physical interaction of Ahr with E2F1 has also been described, which results in inhibition of E2F1-mediated apoptotic gene expression (39).
We observed that, in addition to Aip, E2F consensus binding sites are also present in the promoters of Ahr and Arnt, based on the Consite application, with a similarity of more than 80% compared with the E2F consensus sequence (Fig. 3A). In addition, Ahrr, a factor known to repress Ahr-mediated transcriptional activation by disrupting the Ahr/Arnt heterodimer, also exhibited an E2F binding site. The E2F site found on Arnt promoter is situated within 200 bp of the transcriptional start site, a typical location for E2F binding motifs (40). E2F sites found on Aip, Ahr and Ahrr promoters are located further upstream within the core promoter, a region where approximately 10% of the E2F motifs are found (40).
Fig. 3.

E2F2 binds to the promoters of the Ahr pathway genes in quiescence or after activation. (A) Schematic representation of Aip, Ahr, Arnt, Ahrr and Chk1 promoters, indicating the position of the E2F-binding sites and the primers used for ChIP analysis. (B, C) Results of the ChIP analysis. Cell lysates from quiescent or anti-CD3 stimulated T lymphocytes were used for ChIP analysis with anti-E2F2 antibodies. Immunoprecipitated DNA was analyzed by Q-PCR using primers flanking the E2F sites of each promoter. Antibodies to large T-antigen were used as irrelevant antibody control (mock). The promoter of β-actin, a gene that has no E2F sites but is highly expressed in T lymphocytes, was used as a negative control. (B) Specificity of anti-E2F2 antibodies used in ChIP assays. Control ChIP assay of Chk1 promoter using anti-E2F2 antibodies in WT or E2F2−/− T lymphocytes. Each data point depicts the amount (ng) of immunoprecipitated DNA ± S.D. (average of three independent ChIP experiments, each analyzed in triplicate, n = 6 WT; n = 6 E2F2−/−). (C) ChIP assays of cell lysates from quiescent or proliferating T lymphocytes derived from WT mice, using antibodies anti-E2F2. Each data point depicts the amount (ng) of immunoprecipitated DNA ± S.D. (average of three independent ChIP experiments, each analyzed in triplicate, n = 9 WT). PCR amplification values for Ahr pathway promoters were compared with those of β–actin promoter by a one-way analysis of variance (p < 0.05).
To determine whether E2F2 is found bound to the promoters of these Ahr pathway genes, we performed ChIP using specific antibodies to E2F2. ChIP of E2F2 was performed both in quiescent and activated T cells, and binding was assessed by quantitative PCR. Specific primers were designed for amplifying promoter sequences surrounding E2F-binding motifs in the promoters of Ahr, Arnt, Aip, and Ahrr. Additionally, the promoter of β-actin, a non-E2F2 target gene, was used as negative control in quantitative PCR amplifications. The specificity of anti-E2F2 antibody was confirmed by comparing the binding of E2F2 protein to Chk1 promoter, a well-known E2F2-target gene (15), in E2F2+/+ and E2F2−/− T lymphocytes (Fig. 3B). ChIP analyses showed significant binding of E2F2 to the promoters of all of these members of the Ahr pathway (Fig. 3C). E2F2 is bound to the promoter of Aip in activated cells, but not in quiescent cells. By contrast, E2F2 is bound to promoters of Ahr and Arnt mostly in quiescent cells, although it is also bound to a lesser extent to the promoter of Ahr in activated cells. Finally, E2F2 proteins are also detected on Ahrr promoter, particularly in proliferating cells. These results suggest a direct regulation of the Ahr pathway by E2F2.
The Ahr Pathway is Deregulated in Activated E2F2−/− T Lymphocytes
ChIP results prompted us to examine the expression of the components of the Ahr pathway in activated E2F2−/− samples. To this end, purified T lymphocytes obtained from WT and E2F2−/− mice were stimulated with plate-bound anti-CD3, and 36 hours later mRNA and protein were extracted. Western blot and RT-PCR analyses showed that expression of Ahr and Arnt was up-regulated in activated E2F2−/− T cells compared with their WT counterparts (Fig. 4). We also showed that the expression of Ahrr was downregulated at the mRNA level (Fig. 4E). The lack of a commercially available antibody specific for Ahrr precluded an analysis of this gene at the protein level. Taken together, these results reveal an altered expression pattern for the proteins involved in Ahr pathway in the absence of E2F2, and suggest that their regulation is dependent, at least in part, upon binding of E2F2 to their promoters.
Fig. 4.

Deregulation of Ahr pathway genes in activated E2F2−/− T lymphocytes. (A, C) Western analysis of Ahr and Arnt expression. Values were normalized against β-actin. The ratios Ahr/β-actin and Arnt/β-actin in WT were considered as a unit. The experiment shown is representative of three independent experiments (n = 8 WT; n = 8 E2F2−/−). (B, D, E) Quantitative RT-PCR analysis of Ahr, Arnt and Ahrr. mRNA expression values were normalized against the mRNA expression levels of an irrelevant gene (eEf1a1) and given as relative values (n = 6 WT; n = 6 E2F2−/−; two independent experiments, each in triplicate).
Aberrant Cytosolic Accumulation of Ahr Pathway Proteins in Activated E2F2−/− T Lymphocytes
Having demonstrated an overexpression of the genes that participate in the Ahr-mediated gene regulation in activated T lymphocytes lacking E2F2, we next investigated the functional consequences of this altered expression. Cyp1a1 and Gsta1 are two proteins involved in detoxification that are transcriptionally controlled by Ahr/Arnt (41, 42) and are used as reporters of Ahr function. Expression levels of these two proteins were analyzed by Western blots to search for differences between WT and E2F2-deficient T lymphocytes. Cell extracts derived from WT and E2F2−/− T lymphocytes that had been stimulated with anti-CD3 for 36 hours were blotted and incubated with antibodies against Cyp1a1 and Gsta1. Both genes were expressed significantly in activated T lymphocytes. Surprisingly, their levels were unchanged upon E2F2 loss (Fig. 5A). These results suggested that the expression of Cyp1a1 and Gsta1 may not be under the control of the Ahr pathway in activated T cells, although several reports have shown such control (43). Alternatively, we considered the possibility that this pathway could be impaired in E2F2-deficient cells, despite the accumulation of Ahr, Arnt, and Aip proteins in these cells.
Fig. 5.

(A) Western blot analysis of Cyp1a1 and Gsta1 expression demonstrating comparable protein levels in WT and E2F2−/− T lymphocytes activated through their T-cell receptor. Expression values were normalized against β-actin. The ratios Cyp1a1/β-actin and Gsta1/β-actin in WT were considered as a unit. (B) Western blot analyzes of quiescent (0h) or proliferating (36h) T lymphocytes showing differential subcellular localization of Ahr pathway proteins upon E2F2 loss. Accumulation of Ahr pathway proteins was observed in the cytosolic fraction of E2F2 deficient T lymphocytes. LamB1 was used as nuclear marker and β-actin as loading control. The experiment shown is representative of two independent experiments (n = 6 WT; n = 6 E2F2−/−).
Translocation of Ahr to the nucleus is a necessary step for Ahr-mediated transcriptional activation of its target genes, which depends upon ligand binding to the inactive Ahr complex localized in the cytosol (44). In an effort to determine the subcellular localization of Ahr pathway proteins, we analyzed their levels in the nucleus and cytosol of activated T lymphocytes lacking E2F2. A subcellular fractionation of the cells was performed to separate the cytosolic from the non-cytosolic protein fractions. Given that Ahr can only have a cytosolic and/or nuclear localization (45), we assume that the Ahr signal from the non-cytosolic protein fraction is exclusively nuclear. β-lamin, a protein that is exclusively nuclear, was only detected in the non-cytosolic samples, confirming the purity of the fractions. Western blot analysis showed that genes coding for Ahr and Arnt are silent in quiescent T cells, regardless of the E2F2 genotype, whereas basal levels of Aip are present in resting cells. TCR-mediated activation of T lymphocytes led to the expression of Ahr and Arnt, and to increased levels of Aip, with higher expression levels being detected in E2F2-deficient cells. Remarkably, the proportion of these proteins in each subcellular fraction differed significantly in WT and E2F2−/− samples (Fig. 5B). Comparable levels of proteins were present in the nuclear compartments of either genotype, whereas a substantially higher proportion was observed in the cytosolic fraction of E2F2−/− cells relative to WT cells. These results show that lack of E2F2 leads to an accumulation of Ahr, Arnt, and Aip in the cytosol of activated T lymphocytes and not in the nucleus. Thus, the increased levels of these proteins upon E2F2 loss are not accompanied by increased levels in the compartment where the activity of the proteins take place, which is consistent with the results obtained for Cyp1a1 and Gsta1 protein expression.
Increased Apoptosis in TCDD-Treated E2F2−/− T Cells
The cytosolic accumulation of Ahr pathway proteins may prevent aberrant Ahr activity in E2F2−/− T cells. However, these accumulated proteins could be potentially activated and may therefore become biologically relevant in the presence of an appropriate stimulus. Endogenous ligands for Ahr are still unknown, but many exogenous ligands of the Ahr pathway have been described. Among these, TCDD is the best-known activator of Ahr-mediated transcriptional activity. Exposure to TCDD has been shown to induce Ahr-dependent apoptosis in activated but not quiescent T lymphocytes (46–48). To assess the biological relevance of Ahr/Arnt protein accumulation in activated E2F2−/− T cells, a TCDD-treatment was performed, and the implication of this treatment in cell survival was analyzed. Purified T cells from WT and E2F2−/− mice were activated for 36 hours with anti-CD3 and then treated for an additional 24 hours with vehicle or with TCDD, and apoptosis was determined. In WT cells, TCDD treatment triggered a slight accumulation of annexinV-positive cells, consistent with previous reports (46–48). Remarkably, apoptosis was significantly increased in TCDD-treated E2F2−/− cells (Fig. 6A), indicating that the absence of E2F2 renders T lymphocytes more susceptible to TCDD in vitro.
Fig. 6.

TCDD-mediated activation of Ahr pathway in proliferating T lymphocytes leads to an increased apoptosis in E2F2−/− T cells. T cells were activated using anti-CD3 antibody for 36 hours and subsequently treated with TCDD for the referred times. (A) Apoptosis ratio in the presence of TCDD and vehicle toluene (TOL) of activated WT and E2F2−/− T lymphocytes. (B–D) RT-PCR analysis of Cyp1a1, Bax and FasL in TCDD treated activated T cells derived from WT and E2F2−/− mice (B) Quantitative RT-PCR analysis of Cyp1a1 mRNA expression. Values were normalized against the mRNA expression levels of an irrelevant gene (eEf1a1) and given as relative values. (C, D) Quantitative RT-PCR analysis of the proapoptotic genes FasL and Bax. Values were normalized against the mRNA expression levels of an irrelevant gene (eEf1a1), and relative values obtained in wild-type samples were considered as a unit in each experimental time point (n = 9 WT; n = 9 E2F2−/−; three independent experiments).
To examine Ahr activity upon TCDD exposure in WT and E2F2−/− T cells, we measured Cyp1a1 mRNA levels in anti-CD3-stimulated WT and E2F2−/− cells treated with TCDD for several timepoints. Quantitative RT-PCR analyses were performed with specific primers for Cyp1a1 and control gene eEf1a1. A moderate increase in Cypa1a expression after TCDD treatment was detected in WT cells. Importantly, the expression of Cyp1a1 in E2F2-deficient T cells was substantially higher that in WT cells at the 12-hour timepoint, suggesting a higher Ahr activity in E2F2-devoid cells (Fig. 6B). Moreover, a significant increase of Bax mRNA expression was observed in E2F2-deficient cells exposed to TCDD by the six-hour timepoint, suggesting that the apoptotic signals are activated shortly after TCDD treatment in these cells (Fig. 6C).
It has been shown that TCDD induces apoptosis in T cells involving Fas/FasL interactions (48). We investigated whether the increased susceptibility of activated E2F2−/− T cells to TCDD was the result of an increased expression of FasL in anti-CD3 activated cells. To this end, we determined the expression of FasL in activated WT and E2F2−/− T cells exposed to TCDD or vehicle, by performing quantitative RT-PCR using mouse FasL-specific sets of primers. Remarkably, the levels of FasL mRNA were significantly higher in E2F2-deficient cells relative to WT cells (Fig. 6D), and differences in expression were observed as early as six hours after TCDD exposure, indicating an accelerated kinetic of FasL mRNA production in E2F2-deficient cells. Taken together, these results suggest that E2F2 regulates the Ahr pathway, thereby modulating cellular sensitivity to xenobiotic and/or apoptotic signals (Fig. 7).
Fig. 7.

Model of E2F2-mediated regulation of the Ahr pathway. E2F2 represses the expression of Ahr pathway components Ahr, Aip and Arnt. In the absence of E2F2, higher levels of Ahr pathway gene products accumulate in the cytosol. In response to xenobiotics, E2F2-deficient cells exhibit greater Ahr-dependent transcriptional activation, resulting in increased apoptosis.
DISCUSSION
Previous approaches to identify targets regulated by E2F2 have generally relied on large-scale analysis of mRNA expression using DNA microarrays to examine differences in response to knock-out or overexpression of E2F2 (15, 22–24). Although this approach is powerful, technical limitations make it unlikely to provide a complete picture of E2F2-regulated pathways. Proteomic analysis provides a complementary screening tool to identify candidate targets regulated by E2F2. We performed 2-DE followed by MS to identify differences in protein levels in TCR-activated primary T lymphocytes from WT and E2F2−/− mice. Despite significant experimental variation inherent to this proteomic screening method, known to suffer from a high rate of false-negative results, we identified two proteins, Aip and Crkl, which exhibit consistent protein changes in replicate comparisons. These changes were confirmed by follow-up experiments, thus emphasizing the importance of performing replicate proteomic experiments, and validating the data by orthogonal approaches.
We studied E2F2 regulation of Aip in further detail. We found that Aip protein and mRNA levels were consistently increased in TCR-activated E2F2−/− T cells relative to WT. Furthermore, E2F consensus sites are found in the Aip promoter, and ChIP reveals that E2F2 becomes bound to the Aip promoter upon TCR activation. Together the data suggests that E2F2 directly regulates the Aip promoter as an inhibitor of Aip transcription, resulting in E2F2-dependent decreases in Aip mRNA and protein in WT cells. Previous studies have demonstrated that E2F2 inhibits the expression of DNA replication and cell-cycle genes, including Mcm's, Cdc2, and survivin (15, 49). Thus, we provide here another example that challenges the classical view of E2F2 as transcriptional activator.
Because Aip is a component of the Ahr xenobiotic response pathway, we examined the promoters of other genes in the pathway. We found that the promoters for Ahr, Arnt, and Ahrr also contain E2F consensus sites. Our analysis indicates that the role of E2F in regulating these other components of the pathway is complex. By ChIP we demonstrate that E2F2 does directly bind the promoters of each of these genes. However, our evidence demonstrates regulation of the mRNA, protein levels, and subcellular localization of Ahr and Arnt that is clearly beyond the scope of influence of promoter-bound E2F2. More study will be required to determine to what extent these reflect novel direct functions of E2F2 versus indirect responses resulting from other changes in gene expression in E2F2−/− cells. Importantly, we demonstrate that loss of E2F2 results in functional changes in the Ahr pathway response to the xenobiotic TCDD. In E2F2−/− T lymphocytes the xenobiotic TCDD triggers enhanced expression of genes regulated by the Ahr pathway and increased apoptosis relative to WT (see model in Fig. 7).
Our discovery of regulation of the Ahr pathway by E2F2 raises many interesting questions regarding the role of the Ahr pathway in producing the autoimmunity observed in E2F2−/− mice (13). Previous studies have implicated the Ahr pathway in the generation of TH17+ inflammatory cells (50, 51). Furthermore, several reports have shown that T cells in the thymus and periphery are highly sensitive to TCDD-induced apoptosis (47, 48, 52).
Many questions remain regarding the details of regulation of the Ahr xenobiotic response pathway by E2F2 and the significance of the role of this pathway in mediating the function of E2F2 in vivo. Nevertheless, led by our unbiased proteomic screening data, our results here allow us to forward the novel conclusion that E2F2 functions to regulate the Ahr pathway at multiple points.
Supplementary Material
Acknowledgments
We thank members of the Zubiaga and the Arizmendi laboratories for helpful discussions, and Nerea Osinalde and Jone Mitxelena for providing samples. We are grateful to Seth J. Field, for his critical review of the manuscript.
§ This work was supported by grants from the Spanish Ministry of Education and Science (SAF2009-12037 and OncoBIO Consolider-Ingenio 2010 Programme, CSD2007-00017, to A. M. Z.) and the Basque Government Department of Industry (Etortek-IE06-178 and Saiotek-PE06UN01 to A. M. Z. and to J. M. A.). Proteomics Core Facility-SGIKER is a member of Proteored network, and was supported by UPV/EHU, MICINN, GV/EJ, and ESF agencies. The mass spectrometry data reported in this paper have been deposited in the PRIDE database, http://www.ebi.ac.uk/pride/startsearch.do (Reference: Azkargorta).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- TCDD
- 2,3,7,8-tetrachlorodibenzodioxin
- WT
- wild-type
- TCR
- T cell receptor
- ChIP
- chromatin immunoprecipitation
- 2-DE
- two-dimensional gel electrophoresis.
REFERENCES
- 1.Dyson N. (1998) The regulation of E2F by pRBfamily proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 2.DeGregori J., Johnson D. G. (2006) Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 6, 739–748 [DOI] [PubMed] [Google Scholar]
- 3.Blais A., Dynlacht B. D. (2007) E2F-associated chromatin modifiers and cell cycle control. Curr. Opin. Cell Biol. 19, 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanaka T., Ono T., Kitamura N., Kato J. Y. (2003) Dominant negative E2F inhibits progression of the cell cycle after the midblastula transition in Xenopus. Cell Struct. Funct. 28, 515–522 [DOI] [PubMed] [Google Scholar]
- 5.Yang Q., Hu J., Ye D., Zhao C., Song S., Gong W., Tan Z., Song P. (2010) Identification and expression analysis of two zebrafish E2F5 genes during oogenesis and development. Mol. Biol. Rep. 37, 1773–1780 [DOI] [PubMed] [Google Scholar]
- 6.Lammens T., Boudolf V., Kheibarshekan L., Zalmas L. P., Gaamouche T., Maes S., Vanstraelen M., Kondorosi E., La Thangue N. B., Govaerts W., Inzé D., De Veylder L. (2008) Atypical E2F activity restrains APC/CCCS52A2 function obligatory for endocycle onset. Proc. Natl. Acad. Sci. U.S.A. 105, 14721–14726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lammens T., Li J., Leone G., De Veylder L. (2009) Atypical E2Fs: new players in the E2F transcription factor family. Trends Cell Biol. 19, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polager S., Ofir M., Ginsberg D. (2008) E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 27, 4860–4864 [DOI] [PubMed] [Google Scholar]
- 9.Iaquinta P. J., Lees J. A. (2007) Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 19, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu L., Timmers C., Maiti B., Saavedra H. I., Sang L., Chong G. T., Nuckolls F., Giangrande P., Wright F. A., Field S. J., Greenberg M. E., Orkin S., Nevins J. R., Robinson M. L., Leone G. (2001) The E2F1–3 transcription factors are essential for cellular proliferation. Nature 414, 457–462 [DOI] [PubMed] [Google Scholar]
- 11.Li F. X., Zhu J. W., Hogan C. J., DeGregori J. (2003) Defective gene expression, S phase progression, and maturation during hematopoiesis in E2F1/E2F2 mutant mice. Mol. Cell. Biol. 23, 3607–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dirlam A., Spike B. T., Macleod K. F. (2007) Deregulated E2f2 underlies cell cycle, and maturation defects in retinoblastoma null erythroblasts. Mol. Cell. Biol. 27, 8713–8728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murga M., Fernández-Capetillo O., Field S. J., Moreno B., Borlado L. R., Fujiwara Y., Balomenos D., Vicario A., Carrera A. C., Orkin S. H., Greenberg M. E., Zubiaga A. M. (2001) Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity 15, 959–970 [DOI] [PubMed] [Google Scholar]
- 14.Zhu J. W., Field S. J., Gore L., Thompson M., Yang H., Fujiwara Y., Cardiff R. D., Greenberg M., Orkin S. H., DeGregori J. (2001) E2F1 and E2F2 determine thresholds for antigen-induced T-cell proliferation and suppress tumorigenesis. Mol. Cell. Biol. 21, 8547–8564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Infante A., Laresgoiti U., Fernández-Rueda J., Fullaondo A., Galán J., Díaz-Uriarte R., Malumbres M., Field S. J., Zubiaga A. M. (2008) E2F2 represses cell cycle regulators to maintain quiescence. Cell Cycle 7, 3915–3927 [DOI] [PubMed] [Google Scholar]
- 16.Zhu Y., Jin K., Mao X. O., Greenberg D. A. (2003) Vascular endothelial growth factor promotes proliferation of cortical neuron precursors by regulating E2F expression. Faseb J. 17, 186–193 [DOI] [PubMed] [Google Scholar]
- 17.Alizadeh A., Eisen M., Davis R. E., Ma C., Sabet H., Tran T., Powell J. I., Yang L., Marti G. E., Moore D. T., Hudson J. R., Jr., Chan W. C., Greiner T., Weisenburger D., Armitage J. O., Lossos I., Levy R., Botstein D., Brown P. O., Staudt L. M. (1999) The lymphochip: a specialized cDNA microarray for the genomic-scale analysis of gene expression in normal and malignant lymphocytes. Cold Spring Harb. Symp. Quant. Biol. 64, 71–78 [DOI] [PubMed] [Google Scholar]
- 18.Iyer V. R., Eisen M. B., Ross D. T., Schuler G., Moore T., Lee J. C., Trent J. M., Staudt L. M., Hudson J., Jr., Boguski M. S., Lashkari D., Shalon D., Botstein D., Brown P. O. (1999) The transcriptional program in the response of human fibroblasts to serum. Science 283, 83–87 [DOI] [PubMed] [Google Scholar]
- 19.Bittner M., Meltzer P., Chen Y., Jiang Y., Seftor E., Hendrix M., Radmacher M., Simon R., Yakhini Z., Ben-Dor A., Sampas N., Dougherty E., Wang E., Marincola F., Gooden C., Lueders J., Glatfelter A., Pollock P., Carpten J., Gillanders E., Leja D., Dietrich K., Beaudry C., Berens M., Alberts D., Sondak V. (2000) Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2406, 536–540 [DOI] [PubMed] [Google Scholar]
- 20.Gstaiger M., Aebersold R. (2009) Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat. Rev. Genet. 10, 617–627 [DOI] [PubMed] [Google Scholar]
- 21.Ji H., Moritz R. L., Kim Y. S., Zhu H. J., Simpson R. J. (2007) Analysis of Ras-induced oncogenic transformation of NIH-3T3 cells using differential-display 2-DE proteomics. Electrophoresis 28, 1997–2008 [DOI] [PubMed] [Google Scholar]
- 22.Iglesias A., Murga M., Laresgoiti U., Skoudy A., Bernales I., Fullaondo A., Moreno B., Lloreta J., Field S. J., Real F. X., Zubiaga A. M. (2004) Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double·mutant mice. J. Clin. Invest. 113, 1398–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishida S., Huang E., Zuzan H., Spang R., Leone G., West M., Nevins J. R. (2001) Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol. 21, 4684–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Müller H., Bracken A. P., Vernell R., Moroni M. C., Christians F., Grassilli E., Prosperini E., Vigo E., Oliner J. D., Helin K. (2001) E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 15, 267–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gygi S. P., Rochon Y., Franza B. R., Aebersold R. (1999) Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Fröhlich F., Walther T. C., Mann M. (2008) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 27.Hatzimanikatis V., Lee K. H. (1999) Dynamical analysis of gene networks requires both mRNA and protein expression information. Metab. Eng. 1, 275–281 [DOI] [PubMed] [Google Scholar]
- 28.Azkargorta M., Arizmendi J. M., Elortza F., Alkorta N., Zubiaga A. M., Fullaondo A. (2006) Differential proteome profiles in E2F-2 deficient T lymphocytes. Proteomics Suppl 1, S42–S50 [DOI] [PubMed] [Google Scholar]
- 29.Kolkman A., Dirksen E. H., Slijper M., Heck A. J. (2005) Double standards in quantitative proteomics: direct comparative assessment of difference in gel electrophoresis and metabolic stable isotope labeling. Mol. Cell. Proteomics 4, 255–266 [DOI] [PubMed] [Google Scholar]
- 30.Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Mass spectrometric sequencing of proteins silverstained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 31.Matthiesen R., Trelle M. B., Højrup P., Bunkenborg J., Jensen O. N. (2005) VEMS 3.0: algorithms and computational tools for tandem mass spectrometry based identification of posttranslational modifications in proteins. J. Proteome Res. 4, 2338–2347 [DOI] [PubMed] [Google Scholar]
- 32.Sandelin A., Wasserman W. W., Lenhard B. (2004) ConSite: web based prediction of regulatory elements using cross species comparison. Nucleic Acids Res. 32, W249–W252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sogawa K., Fujii-Kuriyama Y. (1997) Ah receptor, a novel ligand-activated transcription factor. J. Biochem. 122, 1075–1079 [DOI] [PubMed] [Google Scholar]
- 34.Beischlag T. V., Morales J. L., Hollingshead B. D., Perdew G. H. (2008) The Aryl Hydrocarbon Receptor Complex and the Control of Gene Expression. Crit. Rev. Eukaryot. Gene Expr. 18, 207–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hankinson O. (1995) The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 35, 307–340 [DOI] [PubMed] [Google Scholar]
- 36.Puga A., Xia Y., Elferink C. (2002) Role of the aryl hydrocarbon receptor in cell cycle regulation. Chem.-Biol. Interact. 141, 117–130 [DOI] [PubMed] [Google Scholar]
- 37.Shimba S., Komiyama K., Moro I., Tezuka M. (2002) Overexpression of the aryl hydrocarbon receptor (AhR) accelerates the cell proliferation of A549 cells. J. Biochem. 132, 795–802 [DOI] [PubMed] [Google Scholar]
- 38.Marlowe J. L., Knudsen E. S., Schwemberger S., Puga A. (2004) The aryl hydrocarbon receptor displaces p300 from E2Fdependent promoters and represses S phasespecific gene expression. J. Biol. Chem. 279, 29013–29022 [DOI] [PubMed] [Google Scholar]
- 39.Marlowe J. L., Fan Y., Chang X., Peng L., Knudsen E. S., Xia Y., Puga A. (2008) The aryl hydrocarbon receptor binds to E2F1 and inhibits E2F1-induced apoptosis. Mol. Biol. Cell. 19, 3263–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bieda M., Xu X., Singer M. A., Green R., Farnham P. J. (2006) Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 16, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korashy H. M., ElKadi A. O. (2006) The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab. Rev. 38, 411–450 [DOI] [PubMed] [Google Scholar]
- 42.Kawajiri K., FujiiKuriyama Y. (2007) Cytochrome P450 gene regulation and physiological functions mediated by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 464, 207–212 [DOI] [PubMed] [Google Scholar]
- 43.Ko H. P., Okino S. T., Ma Q., Whitlock J. P., Jr. (1996) Dioxin induced CYP1A1 transcription in vivo: the aromatic hydrocarbon receptor mediates transactivation, enhancer promoter communication, and changes in chromatin structure. Mol. Cell. Biol. 16, 430–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Q., Whitlock J. P., Jr. (1997) A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8tetrachlorodibenzopdioxin. J. Biol. Chem. 272, 8878–8884 [PubMed] [Google Scholar]
- 45.Antenos M., Casper R. F., Brown T. J. (2002) Interaction with Nedd8, a ubiquitinlike protein, enhances the transcriptional activity of the aryl hydrocarbon receptor. J. Biol. Chem. 277, 44028–44034 [DOI] [PubMed] [Google Scholar]
- 46.Camacho I. A., Hassuneh M. R., Nagarkatti M., Nagarkatti P. S. (2001) Enhanced activation-induced cell death as a mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunotoxicity in peripheral T cells. Toxicology 165, 51–63 [DOI] [PubMed] [Google Scholar]
- 47.Camacho I. A., Nagarkatti M., Nagarkatti P. S. (2002) 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces Fas-dependent activation-induced cell death in superantigen-primed T cells. Arch. Toxicol. 76, 570–580 [DOI] [PubMed] [Google Scholar]
- 48.Singh N. P., Nagarkatti M., Nagarkatti P. (2008) Primary peripheral T cells become susceptible to 2,3,7,8tetrachlorodibenzopdioxinmediated apoptosis in vitro upon activation and in the presence of dendritic cells. Mol. Pharmacol. 73, 1722–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raj D., Liu T., Samadashwily G., Li F., Grossman D. (2008) Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis 29, 194–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura A., Naka T., Nohara K., Fujii-Kuriyama Y., Kishimoto T. (2008) Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. U.S.A. 105, 9721–9726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veldhoen M., Hirota K., Christensen J., O'Garra A., Stockinger B. (2009) Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 206, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camacho I. A., Singh N., Hegde V. L., Nagarkatti M., Nagarkatti P. S. (2005) Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells. J. Immunol. 175, 90–103 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.