

Abstract
CREB binding protein (CBP) is a transcriptional coactivator with histone acetyltransferase activity. Our prior study suggested that CBP might be a key target of presenilins in the regulation of memory formation and neuronal survival. To elucidate the role of CBP in the adult brain, we generated conditional knock-out (cKO) mice in which CBP is completely inactivated in excitatory neurons of the postnatal forebrain. Histological analysis revealed normal neuronal morphology and absence of age-dependent neuronal degeneration in the CBP cKO cerebral cortex. CBP cKO mice exhibited robust impairment in the formation of spatial, associative, and object-recognition memory. In addition to impaired long-term memory, CBP cKO mice also displayed deficits in short-term associative and object-recognition memory. Administration of a histone deacetylase inhibitor, trichostatin A, rescued the reduction of acetylated histones in the CBP cKO cortex but failed to rescue either short- or long-term memory deficits, suggesting that the memory impairment may not be caused by general reduction of histone acetyltransferase activity in CBP cKO mice. Further microarray and Western analysis showed decreased expression of calcium-calmodulin-dependent kinase isoforms and NMDA and AMPA receptor subunits in the cerebral cortex of CBP cKO mice. Collectively, these findings suggest a crucial role for CBP in the formation of both short- and long-term memory.
Introduction
Mutations in the presenilin genes are the major cause of familial forms of Alzheimer's disease. Previous genetic analyses have demonstrated that presenilins play essential roles in synaptic function, memory, and neuronal survival (Yu et al., 2001; Beglopoulos et al., 2004; Saura et al., 2004; Zhang et al., 2009). Expression of CREB (cAMP responsive element binding protein) target genes and CREB-binding protein (CBP) is reduced in the absence of presenilins (Saura et al., 2004). Furthermore, the CBP promoter contains a putative RBP-Jκ (recombination signal-binding protein 1 for J-κ) binding site, raising the possibility that presenilins may regulate transcription of CBP and CREB target genes through γ-secretase-mediated production of Notch intracellular domains, which are translocated to the nucleus and relieve transcriptional suppression by RBP-Jκ (Fortini, 2002).
CBP is a transcriptional coactivator with histone acetyltransferase (HAT) activity (Goodman and Smolik, 2000) and plays essential roles during embryonic development (Tanaka et al., 1997; Oike et al., 1999; Kung et al., 2000). Several lines of CBP mutant mice with reduced CBP or HAT function have been reported to exhibit defects in long-term memory and synaptic plasticity, and administration of histone deacetylase (HDAC) inhibitors rescues these defects (Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005). However, it was unclear whether CBP is required for neuronal survival in the aging brain. To date, only partial loss of function CBP mutants, such as CBP+/−, transgenic (Tg) mice expressing truncated forms of CBP lacking the HAT domain, or knock-in mice expressing triple mutations in the CBP KIX domain, have been reported (Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005, 2006).
CBP has been implicated in a number of neurodegenerative diseases, including Huntington's disease (Ferrante et al., 2003; Taylor et al., 2003; Bates et al., 2006; Rouaux et al., 2007; Pallos et al., 2008) and amyotrophic lateral sclerosis (Rouaux et al., 2003, 2007). For example, deletion of one copy of CBP enhances polyglutamine-induced neurodegeneration in C. elegans (Bates et al., 2006), and upregulation of CBP rescues polyglutamine-induced neurodegeneration in Drosophila (Taylor et al., 2003). To test whether CBP is involved in presenilin-dependent memory and neuronal survival in the aging cerebral cortex, we generated CBP conditional knock-out (cKO) mice using the same Cre line that was used for the generation of presenilin cKO mice (Yu et al., 2001; Saura et al., 2004). The advantage of this approach is that CBP or presenilins are inactivated in similar spatial and temporal patterns in the adult cerebral cortex, permitting accurate comparison of the phenotypes. CBP cKO mice also provide a unique genetic model system, compared with the previously reported partial loss-of-function mutants, to uncover the full effects of complete loss of CBP function in memory formation and neuronal survival.
Materials and Methods
Animals.
Generation of floxed CBP (fCBP/fCBP) mice has been described previously (Kang-Decker et al., 2004). As recommended by the Banbury Conference on genetic background in mice (Silva et al., 1997), our laboratory routinely uses B6/129 hybrid background mice for behavioral experiments, since the performance of this background is better than that of B6 or 129 (Yu et al., 2001; Saura et al., 2004, 2005; Tabuchi et al., 2009). To generate CBP cKO mice, we first crossed homozygous fCBP/fCBP mice with αCaMKII-Cre Tg mice (Yu et al., 2001) to obtain fCBP/+;CaMKII-Cre mice, which were then bred with fCBP/fCBP mice to get fCBP/fCBP;CaMKII-Cre animals. The latter were bred with fCBP/fCBP mice to get littermates for fCBP/fCBP (control) and fCBP/fCBP;CaMKII-Cre (CBP cKO) mice. The mice used for fear conditioning and object recognition were 3–8 months old. Two separate cohorts of mice (2–3 and 8–10 months) were tested in water maze. The mice were kept on 7:00 A.M.–7:00 P.M. light cycle under conditions of constant humidity and temperature (22°C). All the mice were group-housed (2–4 per house cage) throughout the experimental period and had ad libitum access to food and water. Both male and female mice were used. A total of 151 mice used for the behavioral study were handled twice a day (5 min/time) for 3 consecutive days by the experimenter (G. Chen). All of the behavioral experiments were conducted during the light phase of the cycle, between 9:00 A.M. and 6:00 P.M. Different cohorts of mice were used for different behavioral and biochemical purposes. All behavior experiments were conducted in a genotype-blind manner. The genotype was decoded only after the completion of the experiments and data analysis.
Northern blotting.
Cortices were dissected on ice and placed in TRI-Reagent (Sigma). Total RNA was then isolated from tissue homogenized in Tri-reagent. Twenty micrograms of total RNA for each sample was loaded on an RNA borate/formaldehyde gel and then transferred onto Hybond-N membrane (GE Healthcare). The membrane was then probed with a 32P-radiolabeled CBP cDNA fragment, washed, and exposed to XB-1 film (Kodak). The membrane was then reprobed with a 32P-radiolabeled GAPDH cDNA fragment to normalize loading and transfer.
Immunoblotting.
Cortices, including both the neocortex and the hippocampus, were dissected and homogenized together in cold radioimmunoprecipitation assay lysis buffer [consisting of the following (in mm): 50 Tris-HCl, pH 7.4, 150 NaCl, 1 EDTA plus 1% NP-40, and 0.5% Triton X-100] containing protease and phosphatase inhibitors (Sigma) (Saura et al., 2004, 2005). Lysates were cleared by centrifugation (12,000 rpm for 15 min). Normalized volumes of samples were resolved in 10% SDS-PAGE, transferred to nitrocellulose membrane, and immunoblotted using infrared (IR) dye-coupled secondary antibodies (goat anti-rabbit IRdye800, goat anti-rabbit IRdye680, goat anti-mouse IRdye800, goat anti-mouse IRdye680; Li-Cor). Image acquisition and data quantitation were performed using Odyssey Infrared Imaging System (Li-Cor). Primary antibodies used were as follows: anti-CBP (A22, 1:200), anti-p300 (N15, 1:200), anti-histone H3 (1:200), anti-acetylated histone H3 (1:200), anti-histone H2B (1:100), anti-acetylated histone H2B (1:100), anti-SynGAP (1:1000), anti-Synapsin (1:1000), anti-Rab3A (1:1000), and anti-rabbit β-actin (1:500) (Cell Signaling Technology); anti-glial fibrillary acidic protein (anti-GFAP; 1:500; Sigma); anti-CaMKKα (1:200), anti-CaMKKβ (1:200), anti-CaMKIγ (1:200), anti-CaMKIIβ (1:200), anti-CaMKIIγ (1:200), anti-CaMKIIδ, and anti-Valosin-containing protein (1:200) (1:200) (Santa Cruz Biotechnology); anti-GluN1 (1:100), anti-GluN2A (1:250), anti-GluA2 (1:1000), and anti-GluA3 (1:500) (Millipore Bioscience Research Reagents); anti-PSD95 (1:2000; SYSY); anti-SNAP25 (1:1000; Stressgen); and anti-mouse β-actin (1:30,000; Abcam).
Immunohistochemistry.
Paraffin-embedded brain sections (10 μm) were deparaffinized, alcohol dehydrated, and immunostained with monoclonal or polyclonal antibodies raised against microtubule associated protein 2 (MAP2; 1:200) and synaptophysin (1:200), anti-GFAP (1:500) (Sigma) and CBP (1:200; Santa Cruz Biotechnology). For fluorescence immunostaining on MAP2 and synaptophysin, the brain sections were incubated with either Alexa Fluor 488 or 594 anti-mouse secondary antibodies (Invitrogen) and then analyzed with a 510 confocal laser scanning microscope (Zeiss) and a BX50 microscope (Olympus). For MAP2 and synaptophysin staining, fields from the cortex and the hippocampal CA1 area were collected from three sections per mouse. The iris and gain levels were adjusted to obtain images with a pixel intensity within a linear range. The images were transferred and analyzed using ImageJ (NIH). The mean pixel intensity was calculated for each image. For immunostaining on CBP, brain sections were incubated with biotinylated secondary antibodies and developed by using the peroxidase avidin–biotin reagent and the Vectastain Elite ABC kit (Vector Laboratories).
Nissl staining and stereological neuron counting.
Mice were killed by CO2 and perfused transcardially with PBS. The brain was fixed in 10% neutral buffered formalin for 2 h. Fixed brain was dehydrated and embedded in paraffin. The left hemibrain was sectioned sagittally using a sliding vibratome (Leica Microsystems). Serial sections (10 μm) were baked at 58°C for 1 h. Sections were deparaffinized in histoclear and rehydrated. Sections were rinsed in PBS for 5 min and soaked in 0.5% cresyl violet for 12 min. Sections were rehydrated and then dehydrated. After the sections were cleared in histoclear, they were coverslipped with Fisher SP Permount (Fisher Scientific).
Cortical neuron counts were performed in six 10 μm cresyl violet-stained sagittal sections spaced 400 μm apart using optical dissector technique (Irizarry et al., 1997). The number of neurons in neocortex was estimated using ∼12 optical dissectors. Each optical dissector was a 50 × 50 μm sampling box. Using a 100× oil-immersion lens, neurons with a visible nucleolus were counted if they were not present in the initial plane of focus, but came into focus as the optical plane moved through the tissue. The estimation of total neurons was calculated by multiplying the volume density of the neurons in the layers by the volume of the layers. Neuron counts were reported for a single hemisphere. The coefficient of error from the counting technique was <0.10.
The Morris water maze.
The water maze was a circular pool, 160 cm in diameter. During the hidden platform training, the platform (10 cm in diameter) was kept submerged under water and maintained in the same position. The mice were given training six trials per day (two trials per block, ∼2 h interblock interval) for 5 d. During the 60 s training period, if the mice were unable to locate the hidden platform, they were guided to the platform by hand and allowed to remain on it for 30 s. The swimming of the mice was monitored using an automated tracking system (HVS Image). Following training d 5, the mice were subjected to a 60 s probe trial in which the platform was removed and the mice were allowed to search for it. In the visible platform test, the platform was raised above water and marked by a black and white golf ball.
Contextual fear conditioning.
The mice were placed into the conditioning chamber for 3 min before the onset of an unconditioned stimulus (footshock; 1 s, 1 mA) to allow them to explore the testing chamber and develop a representation of the context. After the footshock, the mice were left in the chamber for additional 2 min and then returned to their home cages. For short-term memory test, the mice were tested 30 or 60 min after a single footshock in the same conditioning chamber. For long-term memory test, the mice were tested 24 h after a single footshock. For remote memory test, the mice were tested 4 weeks after a single footshock. Freezing responses were recorded and scored using the FreezeFrame automated system (Coulbourn Instruments).
Novel-object recognition.
The novel-object recognition task was conducted in an open-field chamber (42 cm × 42 cm). Mice were habituated in the empty chamber for 2 d (15 min per day) before testing. The task consisted of a sample phase and a choice phase. In the sample phase (Ennaceur and Delacour, 1988; Chen et al., 2000), mice were allowed to explore two identical objects placed into the chamber at fixed locations for 10 min. Active exploration, i.e., sniffing and biting of the objects, was scored. Several different sets of identical objects were used for different delays. The objects measured ∼10–15 cm in height and were washed in ethanol (75%) after each trial. After the sample phase, the mice and the objects were removed from the chamber and the mice were returned to the home cage during the memory delays (1 or 24 h). In the choice phase, a third copy of the earlier object was placed into the chamber at one location and a new object at another. The mice were placed in the chamber and allowed to explore for 10 min. Memory of the familiar object was associated with increased exploration of the new object and a preference index (PI) was calculated using the formula PI = 100 × (new object inspection time/total inspection time).
Open-field test.
VersaMax system from AccuScan Instruments was used to study locomotor functions of the mice. Two 42 × 42 cm Plexiglas chambers were set in a quiet laboratory room. The open-field chamber was divided by 16 horizontal infrared beams from left to right and 16 horizontal infrared beams from front to back on the floor. The area 7 cm from the walls was defined as marginal area. The area 10 cm away from the wall is defined as the central area. The mice were allowed to get habituated in the experimental room for 30 min before the test started. During the test, the mouse was placed in the center of the open-field chamber and allowed to ambulate in it for 15 min. After each testing trial, the chambers were thoroughly cleaned to remove any odors left by animals tested in a previous trial. The total distance traveled and the time spent in the marginal area by the animals during the 15 min testing period was tracked by the VersaMax system.
Rotarod test.
The rotarod test was conducted in the same experimental room for the open-field test. The mice were placed in a neutral position on a stationary 6-cm-diameter accelerating rotarod (Economex; Columbus Instruments), which was equipped with individual timers and allowed for four mice to be tested at a time. Mice were first trained to stay on the rod at a constant rotation speed of 5 rpm. Mice unable to stay on the rod for 2 min were placed back on the rod until they did not fall from the rod for 2 min (pretraining). After successful pretraining, mice were placed back on the rod at a rotation speed of 5 rpm. The rotating speed was then increased at a rate of 0.2 rpm/s. The latency to fall was measured. All mice were tested for three trials in total on the same experimental day.
Prepulse inhibition.
The mice were placed in startle chambers where acoustic stimuli could be produced by a high-frequency speaker. The startle-eliciting stimulus was a 50 ms noise pulse of 110 dB intensity. The prepulse was a 50 ms of 80 dB intensity, presented 100 ms before the eliciting stimulus. The standard test procedure consisted of a total of 50 trials presented in counterbalanced order. Half of the trials were presented with the eliciting stimulus alone and the other half with the prepulse preceding the eliciting stimulus. The stimulus-alone trials served as the baseline startle for the prepulse trials. The prepulse inhibition (PPI) was obtained as the mean difference in startle amplitude between stimulus-alone and prepulse trials and expressed as the percentage of the mean stimulus-alone startle level. Intertrial interval was 30 s.
Trichostatin A treatment.
Trichostatin A (TSA), purchased from Biomol, was solubilized in dimethylsulfoxide (DMSO). CBP cKO mice (TSA, n = 7; DMSO, n = 9) and control mice (DMSO, n = 9) were treated with intraperitoneal injections of either TSA [2 μg/g body weight (BW), a dose that was identical to what was previously used in Korzus et al. (2004)] or DMSO once a day for 3 consecutive days. The contextual fear conditioning test started on d 3. After the behavioral test was completed on d 4, the mice were killed and brain lysates were prepared for analysis of acetylated histone levels (∼26 h after the footshock). A subgroup of CBP cKO mice (n = 3) were treated with intraperitoneal injections of TSA (2 μg/g BW; 3 injections) and were killed ∼2 h after the last injection. The experimenter (G. Chen) was blind to the genotype of mice and to the drug status, which were coded by a third party (H. Zhao).
Microarray analysis.
Total RNA was purified from the hippocampus of CBP cKO and control mice using the TRI reagent (Sigma). RNA concentration was determined by a spectrophotometer (ND-1000; NanoDrop). RNA integrity was determined by 260/280 = 1.85–2.01 using the ND-1000. Isolated RNAs were kept at −80°C until needed for analysis. Gene expression profiling was performed with the GeneChip Mouse Genome 430 2.0 array (Affymetrix). RNAs (5 μg) were used for each array hybridization. Preparation of biotinylated cRNA, GeneChip hybridization, staining, and scanning of the arrays were performed according to recommended Affymetrix protocols by the Harvard Medical School, Partners Healthcare Center for Personalized Genetic Medicine (Cambridge, MA; http://www.hpcgg.org/Microarrays/overview.jsp).
Data analysis.
Statistical analyses were performed using ANOVA to measure effects on genotype, trial, day, and age, or any interaction effects. Unless otherwise specified, two-tailed Student's t test was conducted for the pairwise comparisons in the behavioral and biochemical results. A p < 0.05 is considered significant. All the data were reported as mean ± SE throughout.
Results
Generation of postnatal forebrain-specific CBP cKO mice
To generate postnatal forebrain-specific CBP cKO mice, we crossed floxed CBP (fCBP/fCBP) mice (Kang-Decker et al., 2004) with Cre Tg mice, in which Cre-recombinase is expressed under the control of the α-calcium-calmodulin-dependent kinase II (αCaMKII) promoter in excitatory neurons of the postnatal forebrain (Yu et al., 2001). To examine levels of CBP mRNAs in the cortex of CBP cKO mice, we first conducted Northern analysis of total RNA derived from the cortex of mutant and control mice aged 2 months. We observed a dramatic reduction of CBP transcripts in the cerebral cortex of CBP cKO mice (p < 0.05) (Fig. 1A). In contrast, we did not find any significant change in CBP mRNA levels in the cerebellum (p > 0.3) (Fig. 1A), suggesting that CBP is selectively inactivated in the cerebral cortex. We then used an antibody raised against the N terminus of CBP for immunoblotting and found markedly decreased protein levels of CBP in cortical lysates, which were prepared from both the neocortex and the hippocampus (control, 100% ± 14.7%; cKO, 52.8 ± 10.1%; p < 0.05) (Fig. 1B). The residue amount of CBP in cortical lysates is likely due to normal expression of CBP in interneurons and/or glia, in which Cre is not expressed (Yu et al., 2001; Saura et al., 2004; Tabuchi et al., 2009). To examine whether inactivation of CBP has any effects on p300, a homolog of CBP (Goodman and Smolik, 2000), we performed Western analysis of p300 and found normal levels of p300 in the cerebral cortex of CBP cKO mice (control, 100% ± 7.5%; cKO, 97.9 ± 7.7%; p > 0.2) (Fig. 1C), suggesting that there is no compensatory upregulation of p300 in the absence of CBP.
Figure 1.

Molecular characterization of forebrain-specific CBP cKO mice. A, Northern analysis of CBP transcripts. There is a dramatic reduction on CBP mRNA levels in the cerebral cortex of CBP cKO mice (n = 3 per group, p < 0.05). There is no significant change in CBP mRNA levels in the cerebellum (p > 0.3). GAPDH serves as the internal control (C). B, Immunoblotting of CBP using cortical lysates of CBP cKO and control mice at 2 months of age. CBP protein levels are significantly reduced in CBP cKO mice (n = 5 per group, p < 0.05). Valosin-containing protein (VCP) serves as loading controls. C, Immunoblotting of p300 using cortical lysates of CBP cKO and control mice at 2 months of age. Protein levels of p300 are not significantly affected (n = 5 per group, p > 0.2). Valosin-containing protein serves as loading controls. D, Immunostaining of CBP using brain sections of control and CBP cKO mice at 2 months of age (2× magnification). A cortical inset and a hippocampal inset are enlarged in E and F. E, Immunostaining of CBP in the neocortex at 60× magnification. F, Immunostaining of CBP in the hippocampal CA1 area at 60× magnification. Scale bar, 100 μm.
To determine the spatial inactivation pattern of CBP in the brain of CBP cKO mice, we performed immunohistochemical analysis at 2 months of age. Under a low magnification (2×), it was already clearly visible that pyramidal neurons in the hippocampus and the neocortex of control mice show strong CBP immunoreactivity, but this immunoreactivity is lacking in pyramidal neurons of the CBP cKO cortex (Fig. 1D). Under a higher magnification (60×), abundant CBP-positive staining was clearly seen in cell bodies of pyramidal neurons in the neocortex of control mice, whereas little staining was detected in those of CBP cKO mice (Fig. 1E). Similarly, in the hippocampus, only cell bodies of pyramidal neurons of control mice showed intense CBP immunoreactivity (Fig. 1F). In contrast, there was no detectable change in CBP-positive staining in the cerebellum of CBP cKO mice (data not shown). Collectively, these data indicate that the CBP gene was successfully inactivated in excitatory neurons of the cortex in CBP cKO mice.
No age-related cortical neurodegeneration in CBP cKO mice
Previous studies showed that CBP+/− mice display growth retardation (Oike et al., 1999). To determine whether conditional inactivation of CBP in the postnatal forebrain affects general physical growth, we examined body weight and did not find any significant changes in CBP cKO mice at 2–8 months, indicating that restriction of CBP inactivation to the postnatal forebrain does not cause growth retardation. To determine whether conditional loss of CBP in the cerebral cortex affects neuronal morphology, we conducted histological analysis at 2 months of age. Nissl staining revealed no abnormal morphology in the cerebral cortex of CBP cKO mice at this age (Fig. 2A). We used an unbiased stereological neuron counting method to determine cortex volume and total cortical neuron number in 2- to 3-months-old CBP cKO mice and found no significant changes in cortex volume (n = 3–6 per group, p > 0.2) (Fig. 2B) and total cortical neuron number (n = 3–6 per group, p > 0.2) (Fig. 2E) in CBP cKO mice.
Figure 2.
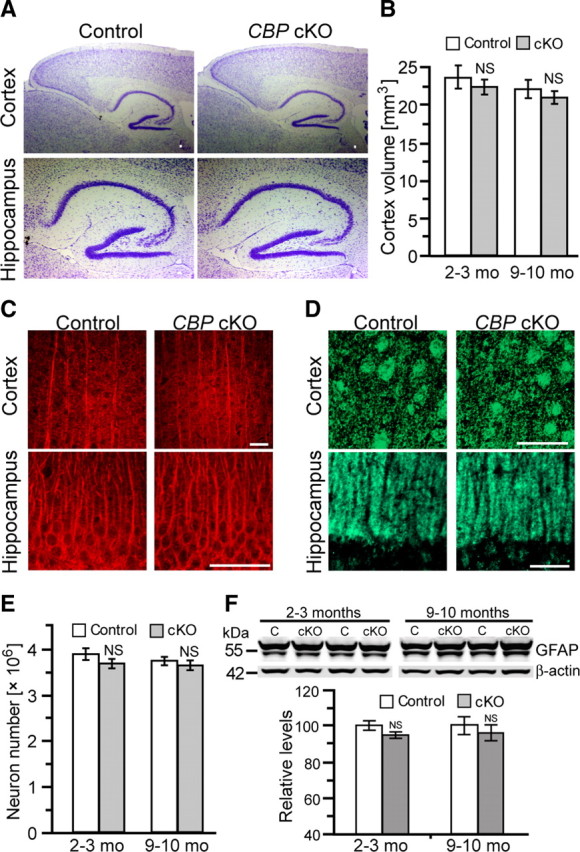
Normal brain morphology and no age-related neurodegeneration in CBP cKO mice. A, Nissl staining from paraffin-embedded brain sections of control and CBP cKO mice. Representative images for the cortex and the hippocampus are shown for control and CBP cKO mice. Scale bar, 50 μm. B, Cortical volumes for control and CBP cKO mice. Stereological neuron counting was used to measure the cortex volume at 2–3 and 9–10 months of age. There was no significant difference on the cortex volume between CBP cKO and control mice at 2–3 or 9–10 months of age (p > 0.1). NS, Not significant. C, MAP2 staining for the neocortex and the hippocampus from control and CBP cKO mice. There was no significant difference between control and CBP cKO mice. Scale bars, 50 μm. D, Synaptophysin staining for the neocortex and the hippocampus from control and CBP cKO mice. There was no significant difference between control and CBP cKO mice. Scale bars, 50 μm. E, Total cortical neuron number for control and CBP cKO mice. There was no significant difference between control and CBP cKO mice either at 2–3 or 9–10 months (p > 0.5). F, Western analysis of GFAP in CBP cKO and control (C) mice at 2–3 and 9–10 months of age. Top, Immunoblotting of GFAP and β-actin. Protein levels of GFAP were normalized to β-actin. There was no significant difference between the two genotypes at either age (p > 0.1).
To examine whether loss of CBP function affects neuronal and synaptic morphology, we used markers for dendrites and presynaptic terminals and performed immunohistochemical analysis. Using an antibody against MAP2, a dendritic marker, we examined dendritic structure in the neocortex and the hippocampus. CBP cKO mice showed no changes on MAP2 staining in the neocortex or the hippocampus (Fig. 2C). Quantitative analysis showed no difference in optical density (p > 0.1) in the neocortex or the hippocampal CA1 area between the two genotypic groups. We then used an antibody against synaptophysin, a presynaptic marker, and performed fluorescence immunostaining. We did not find any significant changes on synaptophysin immunostaining in the neocortex or the hippocampus (p > 0.2) (Fig. 2D). Quantitative analysis further indicated no significant difference in optical density (p > 0.2) in the neocortex or hippocampal CA1 area between the two genotypic groups. These results indicate normal brain cytoarchitecture, neuronal number, and morphology in CBP cKO mice at this age.
To investigate whether loss of CBP function in the forebrain affects neuron survival in older mice, we performed stereological analysis on Nissl-stained brain sections at 9–10 months. We found no significant changes in the cortex volume (n = 6 per group) (Fig. 2B) or the total cortical neuron number (Fig. 2E) in CBP cKO mice, indicating no significant age-related cortical neuronal loss. We further examined signs of gliosis, which is often accompanied by neuronal loss (Beglopoulos et al., 2004; Saura et al., 2004; Tabuchi et al., 2009). We performed Western analysis on levels of GFAP, a marker for reactive astrocytes. Our results showed no significant changes on GFAP levels in CBP cKO mice at either 2–3 (control, 100 ± 2.5%; cKO, 95.0 ± 2.1%; p > 0.15) (Fig. 2F), 9–10 (control, 100 ± 5.1%; cKO, 96.1 ± 4.3%; p > 0.5), or 15 (control, 100 ± 4.8%; cKO, 93.9 ± 5.7%; p > 0.5) months of age. We also performed immunohistochemical analysis on GFAP using brain sections of CBP cKO mice at 2, 10, and 15 months of age (n = 3–6 per group). There were no significant increases in GFAP immunoreactivity in the cortex of CBP cKO mice at any of these ages (data not shown), confirming the absence of gliosis in CBP cKO brains. In contrast, striking cortical atrophy, astrocytosis, and neuronal loss were found in the cerebral cortex of conditional KO mice lacking presenilins (Beglopoulos et al., 2004; Saura et al., 2004) or nicastrin (Tabuchi et al., 2009) at 6 months of age. These results indicate that conditional inactivation of CBP in the adult cerebral cortex does not cause age-related neurodegeneration, suggesting that CBP is not a key mediator for neuronal survival promoted by presenilin or its γ-secretase activity.
Impaired spatial learning and memory in CBP cKO mice
To examine whether a complete loss of CBP in excitatory neurons affects spatial learning and memory, we tested two different cohorts of CBP cKO mice using the Morris water maze task, one at 2–3 and the other at 8–10 months. The mice were trained to learn the location of a hidden platform in the water maze for 5 consecutive days. First, we performed an ANOVA to evaluate overall effects for all mice. The ANOVA revealed no significant age effect (F < 1) or age × genotype interaction effect (F < 1). Therefore, we pooled the data for the two different ages together for each genotype (Figs. 3A, acquisition curve). The ANOVA showed a highly significant within-subject effect on training days (F = 53.2, df 4/180, p < 0.0001), suggesting effective learning during training. Interestingly, the overall ANOVA also revealed a highly significant main genotype effect (F = 15.0, df1/45, p < 0.0005), suggesting impaired spatial learning in CBP cKO mice.
Figure 3.

Impaired spatial learning and memory in CBP cKO mice. A, Escape latency for CBP cKO and control mice (acquisition curves). The animals were trained using a hidden platform task in the Morris water maze for 5 consecutive days. CBP cKO mice show significantly longer latency in finding the hidden platform than control animals. B, Quadrant occupancy of the water maze during a probe trial in CBP cKO and control mice. A probe trial was conducted 2 h after the training. The animals were allowed to spend 60 s searching for the location of the hidden platform. CBP cKO mice spent significantly less time in the target quadrant than control animals. AL, Adjacent left quadrant; T, target quadrant; OP, opposite quadrant; AR, adjacent right quadrant. C, Platform crossings in different quadrants of the water maze during a probe trial in CBP cKO and control mice. CBP cKO mice show significantly fewer number of crossings for the location of the platform in the target quadrant than control animals (*p < 0.05, ***p < 0.005).
After training, the mice were subjected to a probe trial, in which the hidden platform was removed from the water maze. To compare the time spent in the target quadrant and the other three quadrants (Fig. 3B), we conducted an overall ANOVA for all mice. We found a significant quadrant effect (F = 14.5, df3/135, p < 0.001), suggesting a searching preference for the target quadrant after the training. We then compared the time spent in the target quadrant between the control and cKO mice. We found a significant genotype effect (F = 4.2, df1/47, p < 0.05), suggesting impaired spatial memory in CBP cKO mice. We further compared the number of platform crossings during the probe trial between the control and cKO mice. Again, we found a significant genotype effect (F = 4.1, df1/47, p < 0.05) (Fig. 3C), confirming impaired spatial memory in CBP cKO mice.
Impaired short- and long-term associative and object-recognition memory in CBP cKO mice
Previous studies using partial loss-of-function mutants suggest that CBP is not required for short-term memory formation (Oike et al., 1999; Bourtchouladze et al., 2003; Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005). However, it was unknown whether a complete loss of CBP in the cerebral cortex affects short-term memory. Since memory is acquired during several days of training, it is difficult to use the water maze protocol to differentiate short- and long-term memory. To distinguish the role of CBP in short- and long-term memory formation, we used contextual fear conditioning and novel-object-recognition tasks, both of which allow assessment of these two types of memory.
In contextual fear conditioning (CFC), a well established behavioral task known to be dependent on intact amygdala and hippocampus (Phillips and LeDoux, 1992), robust hippocampus-dependent associative memory can be acquired in a single trial; thus, both short- and long-term memory can be assessed. To examine long-term fear memory, we trained a total of 18 adult naive mice in the conditioning chamber at a 24 h retention interval. Control and cKO mice exhibited no difference in responses to the shock, as all the mice showed comparable levels of vocalization and running immediately after the shock. CBP cKO mice did not show different freezing levels during the training session (p > 0.1) (Fig. 4A). However, cKO mice displayed significantly less freezing than littermate controls during the testing session (control, 59.3 ± 12.2%; cKO, 24.0 ± 7.4%; p < 0.025) (Fig. 4A), suggesting impaired long-term memory. We further tested whether loss of CBP affects remote memory by examining a different cohort of cKO mice. One single footshock was applied to a total of 16 naive mice and remote memory was examined 4 weeks later. Although CBP cKO mice did not differ from the controls in freezing responses during the training session (p > 0.15) (Fig. 4B), they exhibited markedly less freezing during the testing session compared with control mice (control, 51.9 ± 8.2%; cKO, 4.8 ± 1.9%; p < 0.0001) (Fig. 4B).
Figure 4.

Impaired long-term memory in CBP cKO mice. A, Percentage of time spent freezing for CBP cKO and control mice. The animals received one single footshock at 1 mA intensity for 1 s during the training session. There is no significant difference in the percentage of time freezing between the two genotypic groups (p > 0.1). The mice were returned to the conditioning chamber 24 h after the footshock. CBP cKO mice showed significantly less time freezing than the control during the testing session. *p < 0.05. B, Percentage of time spent freezing for CBP cKO and control mice. There is no significant difference in percentage of time freezing between the two genotypic groups during the training session (p > 0.1). CBP cKO mice displayed significantly less time freezing than the control during the testing session. ****p < 0.0001. C, PI for CBP cKO and control mice at a 24 h retention delay using a novel-object-recognition task. The animals spent equal amounts of time exploring objects during the sample phase. However, CBP cKO mice spent significantly less time inspecting the novel objects than the control did during the choice phase at 24 h posttraining. ****p < 0.001. PI = (time spent on novel object/total inspection time for objects) × 100. NS, Not significant.
To compare the performance of the mice at the two long-term retention intervals in the CFC, we performed an overall ANOVA. The overall ANOVA revealed not only a highly significant genotype effect (F = 23.3, df 1/30, p < 0.0001) but also a significant interval effect (F = 4.3, df 1/30, p < 0.05). Separate ANOVAs for each genotype showed a significant interval effect only in the cKO groups (F = 5.2, df1/16, p < 0.05), suggesting an interval-dependent decline of memory (data not shown for the control groups). The ability of CBP cKO mice to retain memory declines with time, as CBP cKO mice exhibit basal levels of freezing responses after 4 weeks.
We also performed a novel-object-recognition task (Ennaceur and Delacour, 1988). CBP cKO mice and littermate controls were allowed to explore objects for 10 min during the sample and choice phases. Twenty-four hours after the training (sample phase), we found that CBP cKO mice showed significantly reduced level of PI to the novel object during the choice phase (control, 76.7 ± 2.3%; cKO, 58.9 ± 3.4%; p < 0.001) (Fig. 4C). There was no significant difference during the sample phase (control, 49.9 ± 1.3%; cKO, 51.1 ± 0.9%) (Fig. 4C). These data further suggest impairment of long-term recognition memory in CBP cKO mice.
To determine whether CBP is required for short-term memory, we tested CBP cKO and control mice in both the CFC and novel-object-recognition tasks. We used two different retention intervals (30 and 60 min) in the CFC. First, we applied a single footshock and examined freezing responses 30 min after the shock. A total of 16 naive adult mice were tested. Interestingly, we found that CBP cKO mice spent significantly less time freezing than control mice during the testing session (control, 57.9 ± 7.3%; cKO, 23.1 ± 6.0%; p < 0.005) (Fig. 5A). Second, we applied a single footshock to another cohort of naive mice and measured freezing responses 60 min later. Again, the cKO group froze significantly less than the control (control, 54.3 ± 5.8%; cKO, 30.7 ± 4.1%; p < 0.005) (Fig. 5B). Together, these results demonstrate that short-term associative memory is impaired in CBP cKO mice.
Figure 5.

Impaired short-term memory in CBP cKO mice. A, Percentage of time spent freezing at 30 min delay. The animals received one single footshock at 1 mA intensity for 1 s during the training session. There was no significant difference in the percentage of time freezing between the two genotypic groups (p > 0.1). CBP cKO mice exhibit significantly less time freezing than the control during the testing session 30 min after the footshock. ***p < 0.005. B, Percentage of time spent freezing after a 60 min delay. There was no significant difference in the percentage of time freezing between the two genotypic groups during the training session (p > 0.1). CBP cKO mice showed significantly less time freezing than the control mice did during the testing session. ***p < 0.005. C, Preference index for CBP cKO and control mice at a 1 h retention delay using object-recognition task. The animals spent an equal amount of time exploring objects during the sample phase. However, CBP cKO mice spent significantly less time inspecting the novel objects than the control mice did during the choice phase at 1 h posttraining. ***p < 0.005.
In the object-recognition task, we further performed a 1 h retention delay experiment. We found that CBP cKO mice exhibited significantly reduced levels of preference to the novel object during the testing phase (control, 73.3 ± 1.2%; cKO, 61.9 ± 2.8%; p < 0.005) (Fig. 5C), but showed unchanged preference during the training phase (control, 49.9 ± 1.0%; cKO, 49.8 ± 0.7%; not significant) (Fig. 5C). These results suggest that short-term memory for object recognition is also impaired in CBP cKO mice. These results together provided the first experimental support for an essential role of CBP in short-term memory formation.
Unimpaired sensorimotor functions in CBP cKO mice
As abnormal sensorimotor functions, including hyperactivity, high level of anxiety, and impaired vision acuity, may significantly affect performance in the contextual fear conditioning (Ikegami et al., 2000) and the water maze (Chen et al., 2007), we further tested whether memory deficits in CBP cKO mice could be secondary to sensorimotor or locomotor abnormalities. Therefore, we examined locomotion, anxiety level, sensorimotor functions, and motor coordination in these mice.
To examine locomotor ability, we conducted an open-field task using mice 2–3 and 8–10 months old. We examined total distance traveled and thigmotaxis (time spent in the marginal area proximal to the walls) during the 15 min testing period. An overall ANOVA revealed no significant effects on genotype (F = 2.2, df 3/49, p > 0.1) or genotype × age interaction (F = 2.3, df 1/49, p > 0.1). Both genotypic groups traveled equivalent distance in the open field (control, 14.8 ± 1.2 m; cKO, 13.4 ± 1.5 m; p > 0.5), suggesting unimpaired locomotor ability. We also analyzed thigmotaxis and the overall ANOVA showed no significant effects on genotype (F < 1) and genotype × age interaction (F < 1) (control, 72.9 ± 3.5%; cKO, 73.2 ± 4.5%; p > 0.5), suggesting unimpaired level of anxiety in the open field in CBP cKO mice.
To examine sensorimotor functions in the water maze, we first analyzed thigmotaxis (time spent in the area 10 cm within the walls) during the hidden platform training (Fig. 6A). We observed a highly significant within-subject effect on training days (F = 53.5, p < 0.001), confirming improvement of learning across days. However, we did not find any significant effects on genotype (F < 1) or genotype × age interaction (F < 1), suggesting that CBP cKO mice showed normal anxiety level in the water maze. Second, we conducted a visible cue task in the water maze to assess sensorimotor functions and basic learning. After the hidden platform training was complete, a subset of the animals was trained for a total of four trials in the cue task. Although we observed a highly significant within-subject effect on trials (F = 6.0, df 3/87, p < 0.001) (Fig. 6B), we did not find any significant effects on genotype (F = 2.3, df 1/29, p > 0.1), age (F = 2.3, df 1/29, p > 0.1), or genotype × age interaction (F < 1), suggesting normal vision acuity and unimpaired basic leaning ability in CBP cKO mice.
Figure 6.
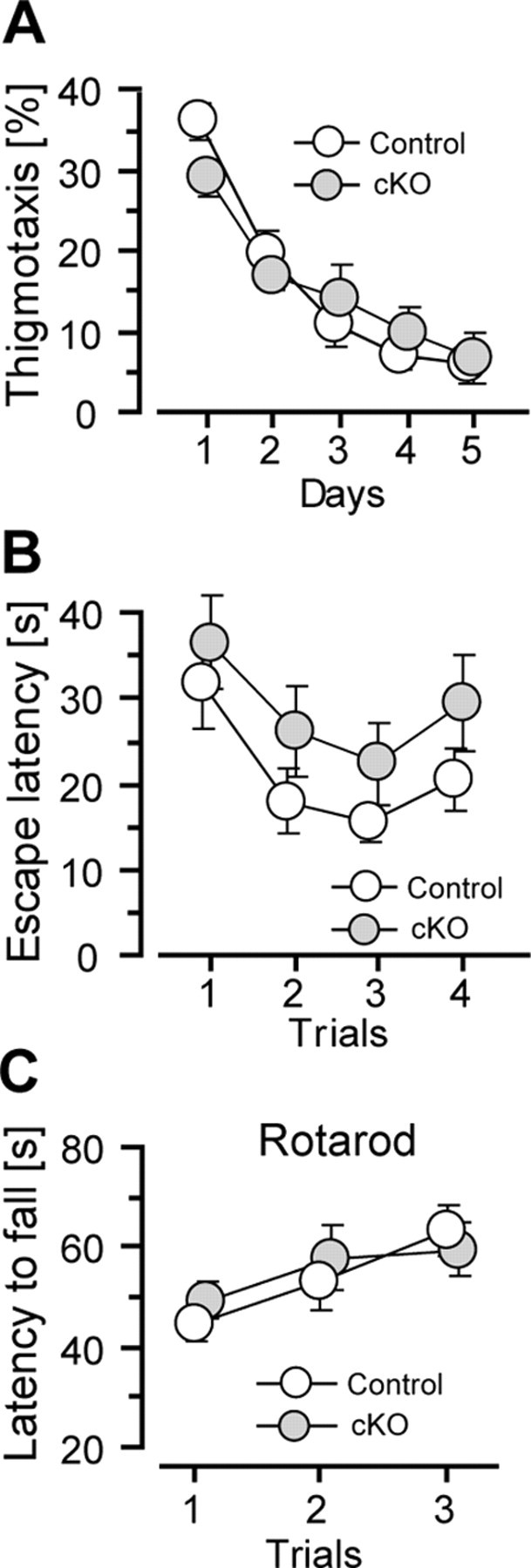
Unimpaired locomotor and sensorimotor functions in CBP cKO mice. A, Thigmotaxis (percentage time spent in the area 10 cm within the walls) during the hidden platform training. The mice were tested at 2–3 and 8–10 months of age. There were no significant differences in genotype (control, n = 24; CBP cKO, n = 25; F < 1), age (F = 3.6, p > 0.05), or genotype × age interaction (F < 1) between CBP cKO and control mice. The data for two ages were pooled together for each genotype. B, Performance on a cued task in the water maze. A subset of CBP cKO and control mice were examined for four trials using a visible platform. There were no significant differences in genotype (control, n = 16; CBP cKO, n = 17; F = 1.3, p > 0.2), age (F = 2.4, p > 0.1), or genotype × age interaction (F < 1) between CBP cKO and control mice. C, Rotarod performance in CBP cKO and control mice. The ANOVA showed a significant effect in training trials (F = 8.8, p < 0.001), suggesting effective motor learning across trials. However, there was no significant difference between the two genotypic groups (control, n = 25; CBP cKO, n = 28) on latency to fall from the rotarod (F < 1).
To examine whether a complete loss of CBP function in excitatory neurons affects motor coordination, we conducted a rotarod task. We found that CBP cKO mice stayed on the rotating rod for as long as control mice did (F < 1) (Fig. 6C), suggesting unimpaired motor coordination. We then performed a startle reflex task to measure PPI using CBP cKO mice at 2–3 and 8–10 months of age. We did not observe any significant effects of PPI on genotype (F = 2.4, p > 0.1; data not shown) or genotype × age interaction (F < 1). In summary, these data suggest that CBP cKO mice display normal locomotor functions in the open field and unimpaired sensorimotor functions in the water maze, rotarod, and startle reflex.
Effects of a HDAC inhibitor, TSA, on memory impairment in CBP cKO mice
Recent studies have shown that use of HDAC inhibitors was able to reverse the long-term memory impairment in CBP+/− mice and CBP Tg mice expressing truncated CBP lacking the HAT domain (Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005), suggesting that the memory impairment is caused by the reduced HAT activity in these mice. To test whether the short- and long-term memory impairment observed in CBP cKO mice is also due to reduced HAT activity, we treated CBP cKO mice with a HDAC inhibitor, TSA (Korzus et al., 2004). TSA was daily administered into CBP cKO mice for 3 d and the mice were subjected to the contextual fear conditioning test. On the third day, a footshock was given to the mice ∼1 h after the last TSA injection. Short- and long-term memory was assessed 1 and 24 h after the footshock (Fig. 7A). No significant differences in freezing responses were observed between the TSA- and vehicle (veh)-treated groups either 1 h (cKO-veh, 29.7 ± 7.2%; cKO-TSA, 19.8 ± 5.7%; not significant) (Fig. 7B) or 24 h (cKO-veh, 29.8 ± 4.4%; cKO-TSA, 36.5 ± 7.8%; not significant) (Fig. 7B) after the training session. Compared with control mice, CBP cKO mice, including TSA- and vehicle-treated, showed significantly reduced percentages of time spent in freezing 1 (control, 52.1 ± 5.1%; cKO, 25.4 ± 7.2%; p < 0.005) (Fig. 7C) and 24 h (control: 65.5 ± 4.8%; cKO, 32.7 ± 6.3%; p < 0.00001) (Fig. 7C) following the training session. These results indicate that TSA treatment did not improve short- or long-term memory impairment in CBP cKO mice.
Figure 7.

Effects of TSA treatment on memory and histone acetylation in CBP cKO mice. A, Experimental design. The mice were treated with either TSA or DMSO for 3 d. On d 3, a footshock was given to the mice during training session of the CFC task. The mice were tested 1 (to assess short-term memory) and 24 h (to assess long-term memory) after the shock. B, Percentage of time spent freezing for TSA- and vehicle-treated CBP cKO mice. There were no significant differences in the percentage time of freezing between the two treatment groups during the training session, 1 and 24 h after the shock (p > 0.5). C, Percentage of time freezing for CBP cKO and control mice. The data for the two CBP cKO groups were pooled together. There were significant differences in the percentage of time freezing between the control and cKO groups 1 and 24 h testing sessions. ***p < 0.005. D, Western analysis for protein levels of acetylated histones AcH3 and AcH2B. Top, Immunoblotting of AcH3 and AcH2B. Protein levels were normalized to β-actin. Significant reductions in levels of AcH3 (control, 100 ± 12.6%; cKO-veh, 56.3 ± 8.9%; p < 0.025; n = 8 per group) and AcH2B (control, 100 ± 8.2%; cKO-veh, 53.0 ± 4.3%; p < 0.005) are seen in vehicle-treated CBP cKO mice. By contrast, treatment of TSA significantly enhances levels of AcH3 (cKO-TSA, 220.9 ± 12.9%; p < 0.0001) and AcH2B (cKO-TSA, 153.7 ± 21.7%; p < 0.0001) in TSA-treated CBP cKO mice. *p < 0.05; ***p < 0.005; ******p < 0.0001.
To determine the effect of TSA treatment on histone acetylation levels, we measured levels of acetylated histones and total histones using cortical lysates obtained from vehicle- and TSA-treated mice. Western analysis showed that relative levels of acetylated histone H3 (AcH3) and acetylated histone H2B (AcH2B) to total histones in the cortex of CBP cKO mice are significantly lower compared with control mice (p < 0.05) (Fig. 7D). Following the TSA treatment, relative levels of AcH3 and AcH2B to total histones were much higher (p < 0.0001) (Fig. 7D). No significant differences of total histones H3 and H2B were found among the groups. These results demonstrate that TSA treatment fully rescued and dramatically improved the reduction of histone acetylation in CBP cKO mice.
Decreased expression levels of calcium-calmodulin-dependent kinases and glutamate receptors in CBP cKO mice
To further explore how CBP is involved in the regulation of memory formation, we conducted gene-chip microarray analysis using mRNAs isolated from the hippocampus of CBP cKO and control mice. Consistent with the reported roles of CBP in tumor suppression, cell differentiation, and cell cycle regulation (Goodman and Smolik, 2000), we found that expression of many transcription factors for genes involved in these processes is reduced in the absence of CBP, suggesting that CBP regulates expression of these transcription factors (Table 1).
Table 1.
Downregulated gene expression on transcription factors in CBP cKO mice
| Classification | Gene description | Relative level | GenBank access number |
|---|---|---|---|
| Tumor suppressor | Cyclin D binding myb-like transcription factor 1 | 10.6% | BB248138 |
| Oct2.4 transcription factor | 22.1% | X57939.1 | |
| Transcription factor AP-2, gamma (Tcfap2c) | 32.2% | NM_009335.1 | |
| Transcription factor 7, T-cell specific (Tcf7) | 39.4% | NM_009331.1 | |
| Transcription factor P45 NF-E2 (Nfe2) exons 1 through 3 | 43.7% | L09600.1 | |
| Thyroid transcription factor 1 (Titf1) | 55.3% | NM_009385.1 | |
| Transcription factor Oct-1 isoform 7 (Pou2f1) | 55.3% | AF095460.1 | |
| Transcription factor AP-2, alpha (Tcfap2a) | 63.3% | AI507504 | |
| NK-3 transcription factor, locus 1 (Nkx3-1) | 66.6% | NM_010921.1 | |
| Oct2.3 transcription factor | 66.9% | X57938.1 | |
| Cell differentiation | E74-like factor 4 (ets domain transcription factor) | 27.3% | BF578163 |
| Homeobox transcription factor NKX2-3 (Nkx2-3) | 33.8% | AF202036.1 | |
| GATA binding protein 5 (Gata5) | 35.9% | NM_008093.1 | |
| HMG-box transcription factor TCF4B (Tcf4) | 36.4% | AF107298.1 | |
| NK2 transcription factor related, locus 3 | 42.2% | Y11117.1 | |
| Transcription factor Genesis (Foxd3) | 58.9% | U41047.1 | |
| Transcription factor AP-2delta | 63.3% | AF421891.1 | |
| Trans-acting transcription factor 6 | 66.7% | BF464465 | |
| NK2 transcription factor related, locus 5 (Nkx2-5) | 68.3% | NM_008700.1 | |
| Transcription factor AP-2 beta | 72.2% | AV334599 | |
| Nuclear transcription factor-Y alpha | 73.5% | D78642.1 | |
| Myelin transcription factor 1 (Myt1) | 74.0% | NM_008665.1 | |
| POU domain, class 3, transcription factor 4 (Pou3f4) | 75.4% | NM_008901.1 | |
| GATA binding protein 4 (Gata4) | 77.5% | NM_008092.1 | |
| Met-mesencephalon-olfactory transcription factor 1 (Ebf2) | 77.7% | U71189.1 | |
| Transcription factor NF 1 mRNA (Nfic) | 77.7% | AF358459.1 | |
| Insulin expression | ISL1 transcription factor, LIMhomeodomain, (Islet-1) | 39.5% | BQ176915 |
| LIM homeobox transcription factor 1 alpha (Lmx1a) | 54.7% | NM_033652.1 | |
| ISL1 transcription factor, LIMhomeodomain (Isl1) | 58.7% | NM_021459.1 | |
| Others | pituitary specific transcription factor 1 (Pit1) | 30.6% | NM_008849.1 |
| Transcription factor kA1 (A7) | 35.0% | D29919.1 | |
| Transcription factor NFAT1-D | 38.2% | AF289078.1 | |
| Helix-loop-helix transcription factor (Hxt) | 43.8% | U43714.1 | |
| Transcription factor GATA-4 | 48.7% | AB075549.1 | |
| Prostate specific ets transcription factor (Pse-pending) | 50.2% | NM_013891.1 | |
| Spalt transcription factor | 55.8% | X97581.1 | |
| Basic transcription factor 3 | 57.4% | AW556975 | |
| POU domain, class 3, transcription factor 2 (Pou3f2) | 58.6% | NM_008899.1 | |
| Pre B-cell leukemia transcription factor 2 (Pbx2) | 63.5% | NM_017463.1 | |
| Homeodomain transcription factor (Nkx6-1) | 65.7% | AF357883.1 | |
| Activating transcription factor 4 | 66.9% | AV026735 | |
| General transcription factor II A, 1-like factor (Gtf2a1lf) | 68.9% | NM_023630.1 | |
| Transcription factor Sox6 | 79.0% | AJ010605.1 | |
| Forkheadwinged-helix transcription factor 2 (Foxp2) | 79.2% | AY079003.1 |
Interestingly, expression of a number of calcium-calmodulin-dependent kinases (CaMKs) and excitatory glutamate receptors, which are well known to play critical roles in learning and memory (Mayford et al., 1996; Tsien et al., 1996; Mayford and Kandel, 1999; Martin et al., 2000), is also reduced in the hippocampus of CBP cKO mice. Specifically, levels of mRNAs for many CaMKs, including CaMK1γ, CaMKIIβ, CaMKIIγ, and CaMKIIδ (Table 2), NMDA receptors, and AMPA receptors (Table 3), are reduced. Levels of postsynaptic density protein (PSD), such as PSD93 and PSD95, and glutamate receptor interacting proteins, including Grid1, Grik5, Grip1, and Grid2ip, are also reduced (Table 3). To validate these findings, we performed immunoblotting to compare protein levels. We found significantly reduced protein levels for CaMK1γ (control, 100% ± 5.2%; cKO, 71.7 ± 4.9%; p < 0.005) (Fig. 8), CaMKIIβ (control, 100% ± 1.6%; cKO, 77.6 ± 2.0%; p < 0.00001), CaMKIIγ (control, 100% ± 4.6%; cKO, 75.1 ± 2.9%; p < 0.001), and CaMKIIδ (control, 100% ± 8.3%; cKO, 74.3 ± 1.7%; p < 0.005). We also found a significant reduction on CaMKKα levels (control, 100% ± 3.9%, cKO; 66.2 ± 3.7%; p < 0.0001) (Fig. 8) and a small decrease on CaMKKβ levels (control, 100% ± 2.9%; cKO, 91.0 ± 2.9%; p < 0.05) in CBP cKO mice.
Table 2.
Downregulated gene expression on Calm, CaMK, and CaMKK
| Classification | Gene description | Relative level | GenBank access number |
|---|---|---|---|
| Calm | Calmodulin 4 (Calm4) | 54.7% | NM_020036.1 |
| CaMK | Calcium/calmodulin-dependent protein kinase I gamma (CaMK1γ) | 55.1% | AF428262.1 |
| Calcium/calmodulin-dependent protein kinase II beta (CaMKIIβ) | 92.5% | NM_007595.1 | |
| Calcium/calmodulin-dependent protein kinase II delta (CaMKIIδ) | 74.3% | BM239047 | |
| Calcium/calmodulin-dependent protein kinase II gamma (CaMKIIγ) | 84.2% | BC025597.1 | |
| CaMKK | Calcium/calmodulin-dependent protein kinase kinase alpha (CaMKKα) | 62.5% | NM_018883.1 |
| Calcium/calmodulin-dependent protein kinase kinase beta (CaMKKβ) | 63.5% | BC023103.1 |
Table 3.
Downregulated gene expression on glutamate receptors and interacting proteins
| Classification | Gene description | Relative level | GenBank access number |
|---|---|---|---|
| NMDA receptors | Glutamate receptor, ionotropic, NMDA1 (Grin1) | 78.5% | NM_008169 |
| Glutamate receptor, ionotropic, NMDA2A (Grin2a) | 79.9% | NM_008170 | |
| Glutamate receptor, ionotropic, NMDA2C (Grin2c) | 40.7% | NM_010350 | |
| Glutamate receptor, ionotropic, NMDA2D (Grin2d) | 87.9% | NM_008172 | |
| AMPA receptors | Glutamate receptor, ionotropic, NMDA3B (Grin3b) | 77.2% | BB325206 |
| Glutamate receptor, ionotropic, AMPA2 (Gria2) | 77.3% | AK014389 | |
| Glutamate receptor, ionotropic, AMPA3 (Gria3) | 83.8% | NM_016886 | |
| Glutamate receptor, metabotropic, type 1 (Grm1) | 88.8% | AF320126 | |
| Postsynaptic density | PSD-93 | 85.0% | NM_011807 |
| PSD-95 | 83.2% | BC014807 | |
| Interacting proteins | Glutamate receptor, ionotropic, delta 1 (Grid1) | 85.0% | NM_008166 |
| Glutamate receptor, ionotropic, kainate 5 (Grik5) | 54.8% | NM_008168 | |
| Glutamate receptor interacting protein 1 (Grip1) | 70.6% | NM_130891 | |
| Glutamate receptor Grid2 interacting protein 1 (Grid2ip) | 49.4% | NM_133355 |
Figure 8.

Reduced protein levels of CaM kinases proteins in CBP cKO mice. Cortical lysates were used to conduct immunoblotting to determine protein levels. Protein levels were normalized to β-actin. There are significant reductions on protein levels for CaMK1γ, CaMKIIβ, CaMKIIγ, CaMKIIδ, CaMKKα, and CaMKKβ in CBP cKO mice (*p < 0.05; ***p < 0.005; ****p < 0.001; control, n = 5–9; cKO, n = 8–9).
We further detected significantly reduced protein levels on NMDA1 (GluN1) receptor (control, 100% ± 15.4%; cKO, 66.5 ± 6.3%; p < 0.05) (Fig. 9), NMDA2A (GluN2A) receptor (control, 100% ± 10.3%; cKO, 73.6 ± 5.6%; p < 0.01), AMPA2 (GluA2) receptor (control, 100% ± 4.9%; cKO, 77.0 ± 3.9%; p < 0.01), and AMPA3 (GluA3) receptor (control, 100% ± 4.0%; cKO, 79.6 ± 3.7%; p < 0.005). We found that PSD95 (control, 100% ± 4.1%; cKO, 70.5 ± 3.3%; p < 0.001) (Fig. 9) and SynGAP (control, 100% ± 6.5%; cKO, 57.2 ± 4.6%; p < 0.00001) protein levels were also significantly reduced. However, we did not find significant changes on many presynaptic proteins, such as SNAP25, Snapsin, or Rab3A in CBP cKO mice (data not shown).
Figure 9.

Reduced protein levels of glutamate receptors and postsynaptic density proteins in CBP cKO mice. There were significant reductions in protein levels for NMDAR1 (GluN1), NMDAR2A (GluN2A), AMPAR2 (GluA2), AMPAR3 (GluA3), PSD95, and SynGAP in CBP cKO mice (*p < 0.05; **p < 0.01; ****p < 0.001; control, n = 5–9; cKO, n = 8–9).
Discussion
CBP is a transcription coactivator (Goodman and Smolik, 2000). CBP is implicated in cell death in neurodegenerative diseases (Ferrante et al., 2003; Taylor et al., 2003; Bates et al., 2006; Rouaux et al., 2007; Pallos et al., 2008) and in mental retardation in Rubinstein-Taybi syndrome (Petrif et al., 1995; Oike et al., 1999). However, it remains unknown whether CBP is required for protection of mature neurons and formation of different types of memory. Because germline deletion of the CBP gene causes embryonic lethality (Tanaka et al., 1997; Yao et al., 1998; Oike et al., 1999), we used the Cre-loxP system to generate viable CBP cKO mice. Unlike partial loss-of-function CBP+/− (Oike et al., 1999; Alarcón et al., 2004) and dominant-negative CBP transgenic mice (Korzus et al., 2004; Wood et al., 2005), CBP inactivation is complete and restricted to excitatory neurons of the adult cerebral cortex in CBP cKO mice (Fig. 1). We found that loss of CBP function in the adult cortex does not cause age-dependent neurodegeneration up to 15 months of age (Fig. 2 for 2–10 months; data not shown for 15 months). However, CBP cKO mice exhibit robust impairment in short- and long-term memory in multiple hippocampus-dependent memory paradigms (Figs. 3–5). Restoration of the reduced level of histone acetylation by HDAC inhibitors failed to rescue impaired short- and long-term memory (Fig. 7). Our further microarray followed by Western analysis showed that expression of CaMK isoforms and glutamate receptor subunits was decreased (Figs. 8, 9; Tables 2, 3). Collectively, our study demonstrated that CBP is required for both short- and long-term memory, and that CBP likely promotes memory formation through its transcriptional activation of genes important for learning and memory.
CBP is not required for neuronal survival in the adult cerebral cortex
We previously found that conditional inactivation of presenilins in the adult cerebral cortex causes memory impairment and aged-related neurodegeneration (Beglopoulos et al., 2004; Saura et al., 2004). One consistent molecular change we observed in the cerebral cortex of presenilin double cKO (PS cDKO) mice at multiple ages was the reduced expression of CBP and several CREB target genes, including c-fos, BDNF, NF-1, and egr-1 (Saura et al., 2004). The presence of the putative RBP-Jκ binding site in the CBP promoter suggested a possibility that presenilin may regulate transcription of CBP and CREB target genes through γ-secretase-dependent production of the Notch intracellular domain, which relieves transcriptional suppression by RBP-Jκ (Saura et al., 2004). We therefore tested whether CBP is required for neuronal survival and memory formation using a complete loss-of-function conditional CBP mutant mouse.
Interestingly, we found no significant loss of cortical neuron in CBP cKO mice by 9–15 months of age (Fig. 2 for 2–10 months; data not shown for 15 months). In contrast, PS cDKO mice exhibit progressive loss of cortical neurons and cortical volume (e.g., ∼9% and ∼18–24% reduction of cortical neurons at 4 and 6–9 months, respectively) (Saura et al., 2004; Wines-Samuelson et al., 2010). Furthermore, we did not observe any significant increases in GFAP levels in CBP cKO mice by the age of 15 months, indicating the lack of neurodegeneration-related astrogliosis. In contrast, PS cDKO mice display an approximately tenfold increase in GFAP levels at the age of 6 months (Beglopoulos et al., 2004; Saura et al., 2004). Together, these data suggest that CBP alone is not required for neuronal survival in the aging cerebral cortex. However, we cannot rule out a possibility that the normal expression of the CBP functional homolog, p300, in the cortex of CBP cKO mice may be sufficient to protect cortical neurons from age-related neurodegeneration (Fig. 1). Thus, CBP is unlikely to serve as a key downstream target of PS in mediation of neuronal protection. Consistent with this notion, our most recent studies have indicated that, PS may regulate the CREB pathway indirectly (Watanabe et al., 2009) and exert its function on neuronal protection through a γ-secretase-dependent mechanism (Tabuchi et al., 2009).
CBP is required for both short- and long-term memory
Our behavioral analysis demonstrated that CBP cKO mice exhibit impairment in three types of hippocampus-dependent long-term memory, spatial learning and memory, fear-associative memory, and object-recognition memory (Figs. 3, 4). Consistent with previous observations from partial loss-of-function mutants, CBP+/−, and dominant-negative transgenic mice (Oike et al., 1999; Bourtchouladze et al., 2003; Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005), long-term memory impairment is more severe in CBP cKO mice. For example, CBP+/− mice exhibited normal spatial learning and memory in the water maze task (Alarcón et al., 2004). Our analysis of CBP cKO mice further showed impairment of short-term associative and object-recognition memory in the absence of CBP (Fig. 5), in contrast to normal short-term associative memory in CBP+/− and dominant-negative transgenic mice, in which CBP is only partially inactivated (Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005). Thus, our analysis of a CBP complete loss-of-function mutant revealed a previously unappreciated role of CBP in short-term memory. Together, these findings indicate a critical role of CBP in both short- and long-term memory.
Mechanisms by which CBP controls memory formation
The role of histone acetylation in memory consolidation has been widely studied (Alarcón et al., 2004; Korzus et al., 2004; Levenson et al., 2004; Wood et al., 2006; Vecsey et al., 2007). To explore molecular mechanisms involved in CBP-dependent short- and long-term memory, we first assessed histone acetylation levels in CBP cKO mice. We compared levels of histone acetylation between CBP cKO (this study) and other CBP partial loss-of-function mutant mice (Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005). The reduction in levels of acetylated histone H2B (∼47%) and acetylated histone H3 (∼45%) in the cortex of CBP cKO mice (Fig. 7D) is more dramatic compared with CBP+/− mice, in which levels of only acetylated histone H2B (∼29%) are reduced (Alarcón et al., 2004, their Fig. 6). Since histone acetylation was not reported in the original reports (Korzus et al., 2004; Wood et al., 2005), it remains unclear whether histone acetylation is affected in the cortex of CBP dominant-negative transgenic mice.
To determine whether decreased HAT activities of CBP account for short- and long-term memory deficits in the cKO mice, we conducted a rescue experiment. Administration of HDAC inhibitors fully rescued acetylated histone levels in CBP cKO mice, but failed to improve short- or long-term memory (Fig. 7). Although reduced HAT activity might indeed not underlie the memory deficits observed in CBP cKO mice, it remained possible that HDAC inhibitors did not rescue histone acetylation at promoters of specific genes involved in memory formation in these mice. Prior studies showed that HDAC inhibitors improved memory in either CBP+/− (Alarcón et al., 2004) or dominant-negative transgenic mice (Korzus et al., 2004), raising the possibility that remaining CBP's HAT activity in these partial loss-of-function mutant mice may be required for the rescue of the memory deficits by HDAC inhibitors. Interestingly, CBP has recently been reported to bind to neuronal activity-regulated enhancers and recruit RNA polymerase II to these sites for transcribing enhancer RNAs (Kim et al., 2010). Our molecular analysis showed reduced expression levels of multiple CaMK isoforms and glutamate receptor subunits in CBP cKO mice (Figs. 8, 9; Tables 2, 3). The importance of CaMKs, NMDA receptors, AMPA receptors, and PSDs in learning and memory has been established by a large number of prior studies (Migaud et al., 1998; Zamanillo et al., 1999; Malinow and Malenka, 2002; Reisel et al., 2002; Lee et al., 2003; Shimshek et al., 2006). Thus, decreased expression of these proteins important for learning and memory may contribute to the short- and long-term memory deficits in CBP cKO mice.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS41783 (J.S.). We thank Wen Cheng for breeding the mice and Youren Tong, Huailong Zhao, and Lan Wang for coding the mice. We thank all members of the Shen lab for helpful discussions.
The authors declare that they have no competing financial interests.
References
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Bates EA, Victor M, Jones AK, Shi Y, Hart AC. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J Neurosci. 2006;26:2830–2838. doi: 10.1523/JNEUROSCI.3344-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglopoulos V, Sun X, Saura CA, Lemere CA, Kim RD, Shen J. Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem. 2004;279:46907–46914. doi: 10.1074/jbc.M409544200. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci U S A. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Chen G, Chen KS, Kobayashi D, Barbour R, Motter R, Games D, Martin SJ, Morris RG. Active beta-amyloid immunization restores spatial learning in PDAPP mice displaying very low levels of beta-amyloid. J Neurosci. 2007;27:2654–2662. doi: 10.1523/JNEUROSCI.3710-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1. Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini ME. Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol. 2002;3:673–684. doi: 10.1038/nrm910. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Ikegami S, Harada A, Hirokawa N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett. 2000;279:129–132. doi: 10.1016/s0304-3940(99)00964-7. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related Abeta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Kang-Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA, Taylor WR, Brindle PK, van Deursen JM. Loss of CBP causes T cell lymphomagenesis in synergy with p27Kip1 insufficiency. Cancer Cell. 2004;5:177–189. doi: 10.1016/s1535-6108(04)00022-4. [DOI] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AL, Rebel VI, Bronson RT, Ch'ng LE, Sieff CA, Livingston DM, Yao TP. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272–277. [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Mayford M, Kandel ER. Genetic approaches to memory storage. Trends Genet. 1999;15:463–470. doi: 10.1016/s0168-9525(99)01846-6. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Kandel E. CaMKII function in the nervous system explored from a genetic perspective. Cold Spring Harb Symp Quant Biol. 1996;61:219–224. [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, Yasue H, Nabeshima T, Araki K, Yamamura K. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet. 1999;8:387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrif F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, Breuning MH. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Reisel D, Bannerman DM, Schmitt WB, Deacon RM, Flint J, Borchardt T, Seeburg PH, Rawlins JN. Spatial memory dissociations in mice lacking GluR1. Nat Neurosci. 2002;5:868–873. doi: 10.1038/nn910. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–6549. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouaux C, Panteleeva I, René F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci. 2007;27:5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Saura CA, Chen G, Malkani S, Choi SY, Takahashi RH, Zhang D, Gouras GK, Kirkwood A, Morris RG, Shen J. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimshek DR, Jensen V, Celikel T, Geng Y, Schupp B, Bus T, Mack V, Marx V, Hvalby Ø, Seeburg PH, Sprengel R. Forebrain-specific glutamate receptor B deletion impairs spatial memory but not hippocampal field long-term potentiation. J Neurosci. 2006;26:8428–8440. doi: 10.1523/JNEUROSCI.5410-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Simpson EM, Takahashi JS, Lipp HP, Nakanishi S, Wehner JM, Giese KP, Tully T, Abel T, Chapman PF, Fox K, Grant S, Itohara S, Lathe R, Mayford M, McNamara JO, Morris RJ, Picciotto M, Roder J, Shin HS. Mutant mice and neuroscience: recommendations concerning genetic background. Neuron. 1997;19:755–759. doi: 10.1016/s0896-6273(00)80958-7. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Chen G, Südhof TC, Shen J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J Neurosci. 2009;29:7290–7301. doi: 10.1523/JNEUROSCI.1320-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Maekawa T, Masuya H, Shiroishi T, Ishii S. Abnormal skeletal patterning in embryos lacking a single Cbp allele: a partial similarity with Rubinstein-Taybi syndrome. Proc Natl Acad Sci U S A. 1997;94:10215–10220. doi: 10.1073/pnas.94.19.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Taye AA, Campbell C, Kazemi-Esfarjani P, Fischbeck KH, Min KT. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev. 2003;17:1463–1468. doi: 10.1101/gad.1087503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB: CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Smith MJ, Heilig E, Beglopoulos V, Kelleher RJ, 3rd, Shen J. Indirect regulation of presenilins in CREB-mediated transcription. J Biol Chem. 2009;284:13705–13713. doi: 10.1074/jbc.M809168200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wines-Samuelson M, Schulte EC, Smith MJ, Aoki C, Liu X, Kelleher RJ, 3rd, Shen J. Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS One. 2010;5:e10195. doi: 10.1371/journal.pone.0010195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem. 2006;13:609–617. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- Yu H, Saura CA, Choi SY, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, Younkin S, Kandel ER, Kirkwood A, Shen J. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Köster HJ, Borchardt T, Worley P, Lübke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]