

Genetic diseases are illnesses caused by abnormalities in genes or chromosomes, including hemophilia, Huntington’s disease, and cancer. They can be caused by both genetic and environmental predispositions. Small interfering RNA (siRNA) is a powerful tool to inhibit gene function because it can be easily applied to any therapeutic target, providing revolutionary potency and selectivity of improved targeted therapeutics.[1] However, poor intracellular uptake, instability, and non-specific immune stimulation are obstacles associated with current methods of siRNA oligonucleotide delivery. Therefore, developing a stable, highly effective siRNA delivery system with optimal cellular integration and low toxicity is of utmost importance. In the present study, we successfully use a nanocomplex assembly of polyethylenimine (PEI) and mesoporous silica nanoparticles (MSNs) to protect and deliver siRNA into human cells, and effectively shutdown both exogenous and endogenous gene signals.
Among various delivery systems, nanoparticle-mediated gene therapy has attracted much research interest for its potential application in biomedicine. Because nanoparticles have been shown to be effective DNA vectors, it is only logical that they be applied to siRNA therapeutics. siRNA molecules cannot penetrate into the cell efficiently, necessitating the use of a carrier system for its delivery. Viral and nonviral vectors have been investigated for gene silencing.[2–5] Viral vectors enable stable inhibition of gene expression;[2,3] however, numerous studies have shown that viral vectors are associated with concerns of systemic toxicity and immunogenicity: even when locally injected into the tumor site, viral dissemination still occurs.[5–7]
Nonviral vectors are generally considered safer than viral vectors. Utilizing mesoporous silica nanoparticles (MSNs) as a delivery vehicle for siRNA represents a relatively new form of gene therapy.[8–10] MSNs have many advantages for intra-cellular delivery, such as large surface area,[11] tunable pore sizes,[12] easy modification of surface,[13] and encapsulation of guest molecules such as drugs,[14] proteins,[15] and biogenic molecules.[16–18]
The surface of MSNs can be chemically modified with various functional groups, such as cancer-specific antibodies or ligands, for targeting purposes.[19–26] Further, the size of MSNs used is approximately 100 nm, which is the appropriate size for accumulation in tumors, based on the enhanced permeability and retention (EPR) effect of tumors.[27,28] Moreover, the synthesis of controllable nanomachines based on MSNs provides an effective external or cellular stimuli-controlled delivery system.[9,22,24,25,29] Previous studies have shown that MSNs are nontoxic and biocompatible,[30,31] and sufficiently excreted by renal routine;[32] MSNs are therefore a promising candidate for delivery applications.[32,33] Following internalization into the cell by endocytosis, MSNs escape the endolysosomal compartment and slowly release the encapsulated payload in the cytoplasm.[34] These observations suggest that MSNs could be suitable for siRNA-mediated gene-silencing applications as well.
Our MSNs were formulated using a base-catalyzed sol–gel hot aqueous solution with modification of published procedures.[21] In a typical synthesis, 100 mg cetyltrimethylammonium bromide (CTAB, Aldrich, 95%) was dissolved in a solution of 48 mL water and 350 μL sodium hydroxide (2 M) and heating to 80 °C. 0.5 mL of tetraethylorthosilicate (Aldrich, 98%) was then added into the aqueous solution containing CTAB surfactants. After 15 min, 127 μL of 3-(trihydroxysilyl)propyl methylphosphonate (Aldrich, 42%) was added into the mixture, and the solution was stirred for another 2 h. The resulting particles were centrifuged and washed with methanol. The CTAB surfactants were then removed from the pores by refluxing the MSNs in acidic methanol, the success of which was confirmed by Fourier transform infrared spectroscopy (FTIR). Electron microscopy and X-ray diffraction (XRD) analyses showed that the particle shape and hexagonal arrays of the pores in the MSNs remained intact after the surfactant-removal process (Figure 1B). The nanoparticles were roughly spherical in shape and smaller than 130 nm in diameter. An average pore diameter of around 2.5 nm was observed by using transmission electron microscopy (TEM) and an interplanar spacing of d (100) ≈4 nm was calculated from the XRD pattern.
Figure 1.

A) Conceptual diagram of siRNA-mediated EGFP gene silencing utilizing mesoporous silica nanoparticles as delivery vehicles. Polycation adjuncts (i.e., PEI) allow binding between nucleic acids and MSNs. B) Scanning electron microscopy images of the MSNs. C) Quantitative binding assay between PEI-surface-modified MSNs and EGFP siRNA. siRNA and PEI-MSNs were mixed and rotated overnight at 4 °C, and then spun down at 5000 rpm for 1 min to pellet the MSNs. The supernatant containing unbound siRNA is quantified and subtracted from the starting amount of siRNA to determine the percentage of siRNA bound to PEI-MSNs. The majority of siRNA remains attached to PEI-MSNs. D) Binding between nanoparticles and siRNA is mediated by an electrostatic interaction between positively charged PEI on the surface of the MSNs and negatively charged siRNA reagents. E) Schematic image of part of the surface of PEI-MSNs.
It is necessary for efficient cellular uptake of the particles that the MSNs do not aggregate in the buffer solution. By modifying only the surfaces of the fluorescent MSNs (FMSNs) with trihydroxysilylpropyl methylphosphonate (THMP) after particle formation, we reduced the aggregation and increased the stability of the particles in aqueous solution.[14]
In order to bind negatively charged nucleic acids and improve uptake, silica materials are typically modified with positively charged organic adjuncts, such as poly-l-lysine (PLL) or polyethyleneimine (PEI).[35–37] Consequently, we incorporated the cationic polymer, PEI (Figure 2A), to effect siRNA encapsulation in MSNs (PEI-MSNs; Figure 2B). Additionally, PEI is reported to promote nanoparticles’ endosomal escape due to its “proton sponge effect”,[36,38,39] although in our previous study, the MSNs were observed to be able to release loaded cargo out of endolysosomes into cytosol.[14] As discussed below, PEI-MSNs are biocompatible, possess high affinity for their particular payload, sequester their payload, and allow delivery of their contents. PEI/siRNA complexes alone have been used for non-viral gene delivery but their cytotoxicity has been an obstacle to in vivo applications (Supporting Information, Figure S1B).[40] Compared to other nonviral transfection vehicles, such as Lipofectamine 2000 and PEI/siRNA complexes, PEI-MSNs showed much less cytotoxicity to cells (Supporting Information, Figures S1 and S4),[9] suggesting that PEI-MSNs are a less toxic alternative for siRNA delivery. Moreover, having the porous structure would provide the possibility of both binding of siRNA on the nanoparticles surface and loading of small molecules, such as chemotherapeutic drugs, within the pores, providing dual delivery of drugs and nucleic acids.[8,41]
Figure 2.
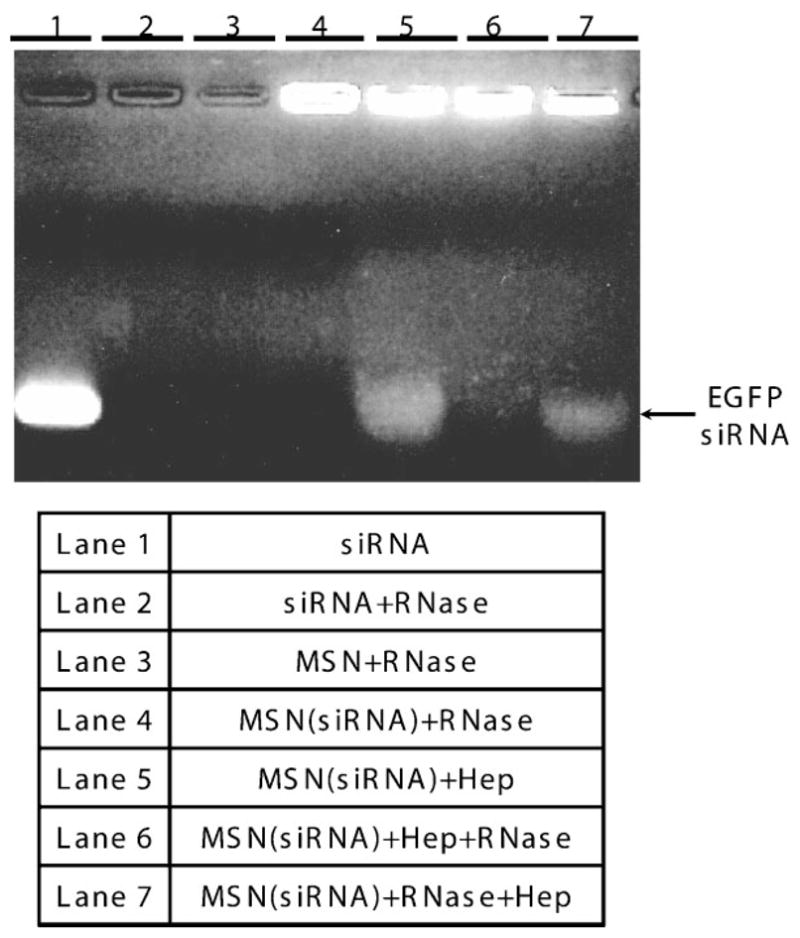
PEI-MSNs can bind and protect siRNA from cleavage by RNase-A. PEI-MSNs loaded with siRNA are treated with RNase A (Lane 4), or with heparin (Lane 5), or with RNase A followed by heparin (Lane 7), or heparin followed by RNase A (Lane 6), as described in the Experimental Section. The samples were run on a gel and visualized. Lane 1 shows gel migration of siRNA. In Lane 2, siRNA after RNase A treatment was analyzed by gel electrophoresis. siRNA bound to PEI-MSNs was retained in the gel wells and showed no sign of degradation (Lane 4). PEI-MSNs-bound siRNA was intact following incubation with RNase A and subsequent dissociation by heparin(Lane 7). siRNA dissociated from PEI-MSNs by heparinis shown in lane 5. On the other hand, siRNA that dissociated from PEI-MSNs by heparin was degraded when exposed to RNase A again (Lane 6).
The ability of PEI-MSNs to complex with siRNA was confirmed by UV absorption measurements and gel assays. PEI-MSNs were incubated with enhanced green fluorescent protein (EGFP) siRNA overnight at 4 °C to allow loading. They were then washed with phosphate buffered saline (PBS) solution to remove any unbound siRNA. Double-stranded siRNA has 11 base pairs per turn, with a diameter of 2.6 nm.[42] Since our nanoparticles have an average pore diameter of 2.5 nm, it is plausible for siRNA to enter MSN pores if they adopt the necessary orientation. However, because PEI coats both the external and pore nanoparticle surfaces, it is more likely that the majority of bound siRNA attaches to the external surface since no particular siRNA orientation is required. The gel assay showed negligible release of siRNA when bound to PEI-MSNs during electrophoresis (Figure 2, Lane 4). To confirm the binding between PEI-MSNs and siRNA, absorption measurements determined that 175 μg of PEI-MSNs could sequester approximately 0.35 nmol of siRNA (approximately 2 pmol μg−1 siRNA/MSN; Figure 1C).
Agarose gel electrophoresis analysis also established that PEI-MSNs could protect siRNA from enzymatic cleavage. It has been previously reported that carbosilane dendrimer nanoparticles exhibit a protective effect on nucleic acids.[43] We sought to determine if the same was true for MSNs following the published protocol.[43] Similar to our UV absorption experiment, PEI-MSNs were loaded with EGFP siRNA by incubating them overnight at 4 °C. After washing with PBS to remove unbound siRNA, RNase A was added to the complex in solution, incubated at 37 °C for 1 h, and heparin was used to liberate siRNA from the PEI-modified nanoparticles immediately before running on the gel.
Naked siRNA was completely degraded by RNase A treatment prior to gel electrophoresis (Figure 2, Lane 2), while siRNA retained in the gel wells due to their union with MSNs showed no signs of RNase degradation whatsoever (Figure 2, Lane 4). The small amount of siRNA that was not bound to the MSNs in the sample was presumably degraded due to RNase A (Figure 2, Lane 4). Finally, the bound siRNA was found to be still intact following exposure to RNase A treatment and subsequent dissociation from MSNs by heparin (Figure 2, Lane 7). The size of the released siRNA was the same as that released from PEI-MSNs (Lane 5). As a control, we also released bound siRNA with heparin then exposed the dissociated complex to RNase to confirm that free siRNA could still be degraded (Figure 2, Lane 6). Thus, we have shown that PEI-modified MSNs are able to efficiently complex with siRNAs and protect their siRNA payload from enzymatic degradation, an important finding if PEI-MSNs are to be utilized in in vivo therapies.
Next, we wanted to determine the intracellular fate of PEI-MSNs as siRNA carriers. In particular, it was of great importance to provide evidence of activity of the siRNA inside cells. This capability was tested in PANC-1 cells stably expressing EGFP (PANC-1/EGFP) through Lipofectamine 2000-mediated transfection. To test the specificity of knockdown, two different types of siRNA were used for nanocomplex formation: siRNA against EGFP and a scrambled siRNA sequence as a control. A homogenous suspension of the EGFP siRNA-loaded PEI-MSNs in fetal bovine serum (FBS)-free media was added to PANC-1/EGFP cells to determine if the nanoparticles were able to transport siRNA into cancer cells and silence EGFP expression. The EGFP transgene was chosen for our initial in vitro experiments because EGFP’s fluorescence properties facilitated analysis of silencing efficiency (Figure 1A). As EGFP emits strong green fluorescence (509 nm) under UV excitation (395 nm), we used fluorescence microscopy to monitor the distribution of EGFP-expressing cells following treatment (Figure 3A). As seen in Figure 3Ag and h, siRNA-loaded PEI-MSNs caused dramatic decrease in the number of fluorescent cells (compare Figure 3Ad and h). As a comparison, a suspension of the same concentration of EGFP siRNA in FBS-free media was transfected into the cells by lipofectamine 2000 (Figure 3Ae, f). It appears that the use of siRNA-PEI-MSNs provides comparable or even better efficacy of delivering siRNA in silencing EGFP compared with Lipofectamine (Figure 3A, B, the down-regulation by siRNA-PEI-MSNs in PANC-1 cells is 61.7% (Supporting Information, Figure S1B)). Control formulations with scrambled siRNA introduced through lipofectamine or PEI-MSNs revealed that gene silencing was highly specific (Supporting Information, Figure S3). These results suggested that siRNA-loaded PEI-MSNs are capable of sequestering and protecting siRNA, and delivering it intracellularly to affect transgene shutdown.
Figure 3.
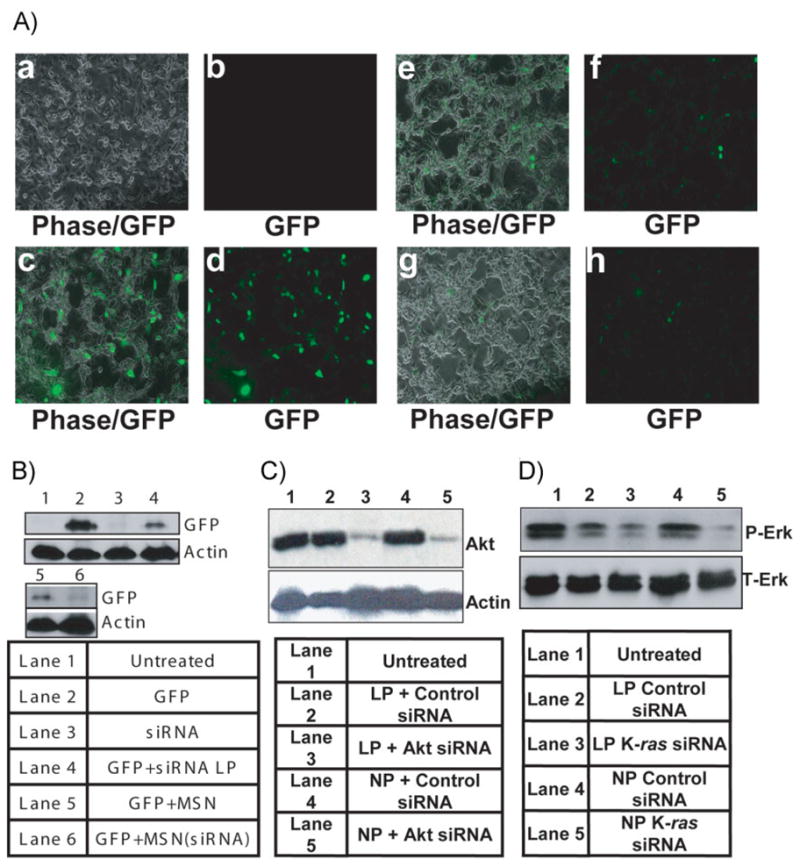
A) Visualization of PEI-MSN-mediated EGFP silencing with fluorescence microscopy. a,b) Untreated PANC-1 cells as a negative control. c,d) EGFP-expressing PANC-1 Cells. e,f) EGFP-expressing PANC-1 cells transfected with EGFP siRNA by lipofectamine as a positive control. g,h) EGFP-expressing PANC-1 cells treated with EGFP siRNA-loaded PEI-MSNs. Left: phase images; right: fluorescent images. B) Western-blot analysis of PANC-1cells transfected with GFP and subsequently knocked down with eGFP-specific siRNA delivered by the Lipofectamine 2000 or PEI-MSNs loaded with eGFP siRNA. C) Western-blot analysis of PANC-1cells treated with siRNA against Akt delivered by the chemical transfection reagent, Lipofectamine 2000 (Lane 3) or siRNA-loaded PEI-MSNs (Lane 5). Lane 1 shows Akt in untreated cells. Lane 2 shows Akt in cells treated with scrambled siRNA delivered by Lipofectamine. Lane 4 shows Akt in cells treated with PEI-MSNs loaded with scrambled siRNA. D) Western-blot analysis of p-Erk in cells treated with siRNA against K-ras delivered by Lipofectamine (Lane 3) or with PEI-MSNs loaded with siRNA against K-ras (Lane 5). Lanes 2 and 4 show p-Erk in cells treated with scrambled siRNA delivered by Lipofectamine and PEI-MSNs, respectively.
Having proven that PEI-MSNs can deliver siRNA to silence transgene expression, the next logical step was to silence an endogenous gene. Accumulating genetic and cancer biology studies indicate a prominent role for Akt in cancer cell growth and survival, and have culminated in the aggressive development of PI3K/Akt pathway inhibitors as cancer therapies.[44] Therefore, we decided to test firstly the effect of delivering siRNA against Akt by PEI-MSNs to human cancer cells. A homogenous suspension of the Akt siRNA-loaded PEI-MSNs in FBS-free media was added to PANC-1 cells and incubated as described in the Experimental Section. Western-blot analysis indicated that upon incubation with siRNA-loaded nanoparticles, Akt expression was attenuated (Figure 3C, compare Lanes 1, 4, and 5). As a positive control, Akt siRNA was also introduced through Lipofectamine 2000 transfection into PANC-1 cells, and Akt expression was reduced to a similar degree as with nanoparticles (Lane 3). Thus, siRNA-loaded PEI-MSNs are capable of delivering siRNA into cancer cells to effect gene silencing of endogenous targets. To further investigate the shutdown of endogenous gene by PEI-MSN delivery of siRNA, we next tested K-ras signaling with a siRNA against K-ras, which is another critical target for cancer therapeutics, because activated mutations of K-ras are found in various cancers, including pancreatic cancers.[45] As shown in Figure 3D, expression of p-Erk, a direct downstream molecule of K-ras, was significantly reduced by PEI-MSN-mediated siRNA (compare Lanes 1, 4, and 5). The total Erk level, on the other hand, was unchanged.
In summary, MSNs possess several attractive characteristics for use in the delivery of siRNA. These nanoparticles have large surface areas that can be chemically modified with various functional groups to aid in the attachment of nucleic acid-based reagents. Further, the size and shape of the particles can be customized to maximize cellular uptake by controlling the ratio of the reaction mixture, such as sphere-shaped, short-rod, and long-rod-shaped particles.[46] Unlike nonporous silica nanoparticles, MSNs possess greater surface areas to maximize their binding capacity. Moreover, mesoporous silica nanoparticles can also tolerate many organic solvents. Silica-based materials have already been shown to be effective drug-delivery vehicles, gene-transfection agents,[47] and carriers of molecules.[48]
We have successfully shown nanocomplex assembly between PEI-MSNs and siRNA. Introduction of PEI to MSNs significantly improved the encapsulation of siRNA and its release, compared to siRNA/PEI complex (Supporting information, Figure S1B), while at the same time conferring protection from enzymatic degradation. The latter finding is of notable significance because it supports the possibility of utilizing PEI-MSNs in in vivo silencing applications. Our technique offers a unique opportunity to fabricate well-defined and homogenously distributed nanocarriers for siRNA delivery.
Intracellular siRNA release is the proposed mechanism of gene silencing observed with PEI-MSNs. The exact mechanism and timing surrounding the release of nucleic acids from MSNs remains to be determined; however, gene silencing has been detected, indicating that a dissociation event has occurred. Previous work in our laboratory has shown that MSNs localize to the lysosome of mammalian cells upon cellular uptake before releasing their contents.[14,49] This parallels the suggested theory that PEI confers increased gene delivery to nanoparticles due to its “proton sponge effect”, allowing endosomal escape.[36,38,39] This provides the possibility of using PEI-coated MSNs for in vivo siRNA delivery for biomedical applications.
Experimental Section
Synthesis of nanoparticles and loading of siRNA
All chemicals for the synthesis of the nanoparticles were purchased from Sigma–Aldrich. The PEI-MSNs were synthesized by first dissolving 100 mg cetyltrimethylammonium bromide (CTAB, Aldrich, 95%) in a solution of 48 mL water and 350 μL sodium hydroxide (2 M) and heating to 80 °C. After the temperature had stabilized, 0.5 mL of tetraethylorthosilicate (Aldrich, 98%) was added into the aqueous solution containing CTAB surfactants. After 15 min, 127 μL of 3-(trihydroxysilyl)propyl methylphosphonate (Aldrich, 42%) was added into the mixture, and the solution was stirred for another 2 h. The resulting particles were centrifuged and washed with methanol. The surfactants were removed from the pores by dispersing the particles in a solution of 20 mL methanol and 1 mL hydrochloric acid (12.1 M) and refluxing the mixture for 24 h. The materials were then centrifuged and washed with ethanol. 5 mg of particles were then dispersed in a solution of 2.5 mg polyethyleneimine (PEI, Mn 1200, Mw 1300, Aldrich) and 1 mL absolute ethanol, stirred for 30 min, and then washed with ethanol and deionized water.
The synthetic siRNA and negative control sequences targeting the EGFP transgene were purchased from Ambion, CA (Ambion EGFP siRNA, Cat AM4626). The synthetic siRNA sequences for human K-ras (sense, 5-CUAUGGUCCUAGUAGGAAAtt, Antisense, 5-UUUCCUACUAGGACCAUAGgt) were purchased from Ambion Applied biosystems, for human Akt from Cell Signaling (Signal Silence Akt siRNA II, #6510). All siRNA sequences were prepared in DepC-treated water to a final concentration of 50 μM and stored at the appropriate temperature.
To load siRNA onto nanoparticles, PEI-MSNs were rotated with siRNA at a nanoparticle:siRNA ratio of 1:25 by mass in FBS-free media at 4 °C. After 24 h, the mixture was centrifuged and the supernatant was removed. By using a spectrophotometer, the absorption measurements of the original solution and the supernatant were compared to determine the amount of siRNA that was loaded onto the PEI-MSNs. The siRNA-loaded PEI-MSNs were then resuspended in the original volume and vortexed to disperse the loaded nanoparticles.
Cell culture
The human cancer-cell line PANC-1 was maintained in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) supplemented with 10% FBS (Sigma), 2% L-glutamine, 1% penicillin, and 1% streptomycin stock solutions. The medium was changed every three days, and the cells were passaged by trypsinization before confluence.
Gel electrophoresis and RNase protection assays
For protective-effect experiments, siRNA-loaded PEI-MSNs or siRNA alone were incubated with 0.25% RNase (Sigma) for 1 h and then loaded on a 4% agarose gel containing 0.0125% ethidium bromide and run at 100 V for 30 min. To release the bound siRNA, PEI-MSNs loaded with siRNA were treated with 16.6 U μL−1 heparin in DEPC-treated water immediately before gel electrophoresis.[43]
Fluorescence microscopy
PANC-1 cells were seeded in 8-well plates at a seeding density of 5 × 104 cells/well 24 h prior to the experiment. Following attachment, the medium was changed and the cells were transiently transfected with the EGFP transgene (1.3 μg/mL/well) using Lipofectamine 2000 (Invitrogen) to constitutively express high levels of the protein. Following incubation for 4–6 h at 37 °C, the medium was changed to serum-rich medium to allow the cells to recover. After 24 h, the cells were then treated with nanoparticles loaded with EGFP-targeted siRNA (0.1 μg nanoparticles/μL/well), siRNA (dose equivalent to that in the nanoparticle treatment) along with the transfecting agent Lipofectamine 2000, nanoparticles containing scrambled siRNA, or empty nanoparticles. All the treatments were carried out in the respective growth medium without serum. After 24 h incubation with the treatments, cells were washed with PBS and incubated in serum-rich media for 48 h.
The fluorescence of the nanoparticles at an excitation wavelength of 395 nm was used to confirm the occurrence of GFP expression. PANC-1 cells that were transiently transfected with the EGFP transgene were incubated with PEI-MSNs for 48 h and then washed with DMEM medium and PBS to wash off the nanoparticles that did not enter the cells. The cells were then monitored by fluorescence microscopy with an excitation wavelength of 395 nm, and emission wavelength of 509 nm. The green emission was passed through a 530-nm bandpass filter. The emissions were collected by using two photomultiplier tubes. GFP exhibits intense green fluorescence under UV light. This property allows the use of fluorescence to study the distribution of GFP in PANC-1 cells. PANC-1 cells were incubated with siRNA-loaded PEI-MSNs or siRNA suspended in PBS (160 nm) for 48 h, washed three times with PBS, and then examined by fluorescence microscopy under UV light.
Western-blot analysis
PANC-1 cells were seeded in 12-well plates at a seeding density of 1.5 × 105 cells/well 24 h prior to the experiment. Following attachment, the medium was changed and cells were then treated with nanoparticles containing Akt- or K-ras-targeted siRNA (175 μg nanoparticles/mL/well), siRNA (dose equivalent to that in the nanoparticle treatment) along with the transfecting agent Lipofectamine 2000 (Invitrogen), nanoparticles containing scrambled siRNA, or empty nanoparticles. All the treatments were prepared in serum-free DMEM. After 24 h incubation with the treatments, cells were washed with PBS and incubated in serum-rich DMEM to allow for recovery.
Proteins were separated by gel electrophoresis on a polyacrylamide gel containing SDS and then transferred to nitrocellulose membranes. The membranes were blocked with tris-buffered saline (TBS) containing 5% (w/v) skimmed milk. After being washed with TBS containing 0.1% Tween 20 (Sigma), the membranes were incubated overnight at room temperature with the first antibody (antibody for Akt, K-ras or p-Erk, Cell Signaling) diluted in TBS. After washing, the membranes were incubated for 2 h at room temperature with the second antibody (Santa Cruz Biotechnology, CA). Bands were detected with an ECL system (Amersham Pharmacia Biotech K.K., UK).
Statistical analysis
All results are expressed as mean values ± the standard deviation (SD).
Supplementary Material
Footnotes
This work was supported by US NIH grant CA41996 and CA 133697, NSF grant DMR 0346601, and the UC Lead Campus for Nanotoxicology Training and Research, funded by UC TSR&TP and a gift from Nanopacific Holding Inc.
Supporting information for this article is available on http://www.small-journal.org or from the author.
Contributor Information
Christopher Hom, Department of Microbiology, Immunology, and Molecular Genetics, California NanoSystems Institute, JCCC, University of California, Los Angeles, 609 Charles E. Young Drive East, Los Angeles, CA 90095 (USA).
Dr. Jie Lu, Department of Microbiology, Immunology, and Molecular Genetics, California NanoSystems Institute, JCCC, University of California, Los Angeles, 609 Charles E. Young Drive East, Los Angeles, CA 90095 (USA)
Dr. Monty Liong, Department of Chemistry and Biochemistry, California NanoSystems Institute, University of California, Los Angeles, 605 Charles E. Young Drive East, Los Angeles, CA 90095 (USA)
Hanzhi Luo, School of Basic Medical Sciences, Health Science Center, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191 (P.R. China).
Zongxi Li, Department of Chemistry and Biochemistry, California NanoSystems Institute, University of California, Los Angeles, 605 Charles E. Young Drive East, Los Angeles, CA 90095 (USA).
Prof. Jeffrey I. Zink, Email: zink@chem.ucla.edu, Department of Chemistry and Biochemistry, California NanoSystems Institute, University of California, Los Angeles, 605 Charles E. Young Drive East, Los Angeles, CA 90095 (USA)
Prof. Fuyuhiko Tamanoi, Email: fuyut@microbio.ucla.edu, Department of Microbiology, Immunology, and Molecular Genetics, California NanoSystems Institute, JCCC, University of California, Los Angeles, 609 Charles E. Young Drive East, Los Angeles, CA 90095 (USA)
References
- 1.Castanotto D, Rossi JJ. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pichler A, Zelcer N, Prior JL, Kuil AJ, Piwnica-Worms D. Clin Can Res. 2005;11:4487–4494. doi: 10.1158/1078-0432.CCR-05-0038. [DOI] [PubMed] [Google Scholar]
- 3.Xu D, McCarty D, Fernandes A, Fisher M, Samulski RJ, Juliano RL. Mol Ther. 2005;11:523–530. doi: 10.1016/j.ymthe.2004.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merdan T, Kopecek J, Kissel T. Adv Drug Deliv Rev. 2002;54:715–758. doi: 10.1016/s0169-409x(02)00046-7. [DOI] [PubMed] [Google Scholar]
- 5.Schagen FH, Ossevoort M, Toes RE, Hoeben RC. Crit Rev Oncol Hematol. 2004;50:51–70. doi: 10.1016/S1040-8428(03)00172-0. [DOI] [PubMed] [Google Scholar]
- 6.Yu P, Wang X, Fu YX. Gene Ther. 2006;13:1131–1132. doi: 10.1038/sj.gt.3302760. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Li CY, Yuan F. Conf Proc IEEE Eng Med Biol Soc. 2004;5:3524–3526. doi: 10.1109/IEMBS.2004.1403991. [DOI] [PubMed] [Google Scholar]
- 8.Torney F, Trewyn BG, Lin VSY, Wang K. Nat Nanotechnol. 2007;2:295–300. doi: 10.1038/nnano.2007.108. [DOI] [PubMed] [Google Scholar]
- 9.Slowing I, Vivero-Escoto JL, Wu CW, Lin VS. Adv Drug Deliv Rev. 2008;60:1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Park IY, Kim IY, Yoo MK, Choi YJ, Cho MH, Cho CS. Int J Pharm. 2008;359:280–287. doi: 10.1016/j.ijpharm.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 11.Kobler J, Bein T. ACS Nano. 2008;2:2324–2330. doi: 10.1021/nn800505g. [DOI] [PubMed] [Google Scholar]
- 12.Munoz B, Ramila A, Perez-Pariente J, Diaz I, Vallet-Regi M. Chem Mater. 2002;15:500–503. [Google Scholar]
- 13.Stein A, Melde BJ, Schroden RC. Adv Mater. 2000;12:1403–1419. [Google Scholar]
- 14.Lu J, Liong M, Zink JI, Tamanoi F. Small. 2007;3:1341–1346. doi: 10.1002/smll.200700005. [DOI] [PubMed] [Google Scholar]
- 15.Han YJ, Stucky GD, Butler A. J Am Chem Soc. 1999;121:9897–9898. [Google Scholar]
- 16.Kneuer C, Sameti M, Haltner EG, Schiestel T, Schirra H, Schmidt H, Lehr CM. Int J Pharm. 2000;196:257–261. doi: 10.1016/s0378-5173(99)00435-4. [DOI] [PubMed] [Google Scholar]
- 17.Gao F, Botella P, Corma A, Blesa J, Dong L. J Phys Chem B. 2009;113:1796–1804. doi: 10.1021/jp807956r. [DOI] [PubMed] [Google Scholar]
- 18.Jin S, Leach JC, Ye K. Meth Mol Biol. 2009;544:547–557. doi: 10.1007/978-1-59745-483-4_34. [DOI] [PubMed] [Google Scholar]
- 19.Klichko Y, Liong M, Choi E, Angelos S, Nel AE, Stoddart JF, Tamanoi F, Zink JI. J Am Ceram Soc. 2009;92:2–10. doi: 10.1111/j.1551-2916.2008.02722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coti KK, Belowich ME, Liong M, Ambrogio MW, Lau YA, Khatib HA, Zink JI, Khashab NM, Stoddart JF. Nanoscale. 2009;1:16–39. doi: 10.1039/b9nr00162j. [DOI] [PubMed] [Google Scholar]
- 21.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS Nano. 2008;2:889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angelos S, Johansson E, Stoddart JF, Zink JI. Adv Funct Mater. 2007;17:2261–2271. [Google Scholar]
- 23.Patel K, Angelos S, Dichtel WR, Coskun A, Yang YW, Zink JI, Stoddart JF. J Am Chem Soc. 2008;130:2382–2383. doi: 10.1021/ja0772086. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen TD, Leung KC, Liong M, Pentecost CD, Stoddart JF, Zink JI. Org Lett. 2006;8:3363–3366. doi: 10.1021/ol0612509. [DOI] [PubMed] [Google Scholar]
- 25.Lu J, Choi E, Tamanoi F, Zink JI. Small. 2008;4:421–426. doi: 10.1002/smll.200700903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liong M, Angelos S, Choi E, Patel K, Stoddart JF, Zink JI. J Mater Chem. 2009;19:6251–6257. [Google Scholar]
- 27.Matsumura Y, Maeda H. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 28.Muggia FM. Clin Cancer Res. 1999;5:7–8. [PubMed] [Google Scholar]
- 29.Khashab NM, Belowich ME, Trabolsi A, Friedman DC, Valente C, Lau Y, Khatib HA, Zink JI, Stoddart JF. Chem Commun. 2009:5371–5373. doi: 10.1039/b910431c. [DOI] [PubMed] [Google Scholar]
- 30.Barnes CA, Elsaesser A, Arkusz J, Smok A, Palus J, Lesniak A, Salvati A, Hanrahan JP, Jong WH, Dziubaltowska E, Stepnik M, Rydzynski K, McKerr G, Lynch I, Dawson KA, Howard CV. Nano Lett. 2008;8:3069–3074. doi: 10.1021/nl801661w. [DOI] [PubMed] [Google Scholar]
- 31.Slowing I, Wu CW, Vivero-Escoto JL, Lin VS. Small. 2009;5:57–62. doi: 10.1002/smll.200800926. [DOI] [PubMed] [Google Scholar]
- 32.He X, Nie H, Wang K, Tan W, Wu X, Zhang P. Anal Chem. 2008;80:9597–9603. doi: 10.1021/ac801882g. [DOI] [PubMed] [Google Scholar]
- 33.Burns AA, Vider J, Ow H, Herz E, Penate-Medina O, Baumgart M, Larson SM, Wiesner U, Bradbury M. Nano Lett. 2009;9:442–448. doi: 10.1021/nl803405h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slowing I, Trewyn BG, Lin VSY. J Ame Chem Soc. 2006;128:14792–14793. doi: 10.1021/ja0645943. [DOI] [PubMed] [Google Scholar]
- 35.Bivas-Benita M, Romeijn S, Junginger HE, Borchard G. Eur J Pharm Biopharm. 2004;58:1–6. doi: 10.1016/j.ejpb.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Godbey WT, Wu KK, Hirasaki GJ, Mikos AG. Gene Ther. 1999;6:1380–1388. doi: 10.1038/sj.gt.3300976. [DOI] [PubMed] [Google Scholar]
- 38.Yamazaki Y, Nango M, Matsuura M, Hasegawa Y, Hasegawa M, Oku N. Gene Ther. 2000;7:1148–1155. doi: 10.1038/sj.gt.3301217. [DOI] [PubMed] [Google Scholar]
- 39.Kircheis R, Wightman L, Wagner E. Adv Drug Deliv Rev. 2001;53:341–358. doi: 10.1016/s0169-409x(01)00202-2. [DOI] [PubMed] [Google Scholar]
- 40.Jiang G, Park K, Kim J, Kim KS, Hahn SK. Mol Pharma. 2009;6:727–737. doi: 10.1021/mp800176t. [DOI] [PubMed] [Google Scholar]
- 41.Xia T, Kovochich M, Liong M, Meng H, Kabehie S, George S, Zink JI, Nel AE. ACS Nano. 2009;3:3273–3286. doi: 10.1021/nn900918w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Svintradze DV, Mrevlishvili GM. Int J Biol Macromol. 2005;37:283–286. doi: 10.1016/j.ijbiomac.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 43.Weber N, Ortega P, Clemente MI, Shcharbin D, Bryszewska M, de la Mata FJ, Gomez R, Munoz-Fernandez MA. J Controlled Release. 2008;132:55–64. doi: 10.1016/j.jconrel.2008.07.035. [DOI] [PubMed] [Google Scholar]
- 44.Engelman JA. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 45.He AR, Lindenberg AP, Marshall JL. Exp Rev Anticanc Ther. 2008;8:1331–1338. doi: 10.1586/14737140.8.8.1331. [DOI] [PubMed] [Google Scholar]
- 46.Huang X, Teng X, Chen D, Tang F, He J. Biomaterials. 2010;31:438–448. doi: 10.1016/j.biomaterials.2009.09.060. [DOI] [PubMed] [Google Scholar]
- 47.Radu DR, Lai CY, Jeftinija K, Rowe EW, Jeftinija S, Lin VS. J Am Chem Soc. 2004;126:13216–13217. doi: 10.1021/ja046275m. [DOI] [PubMed] [Google Scholar]
- 48.Lai CY, Trewyn BG, Jeftinija DM, Jeftinija K, Xu S, Jeftinija S, Lin VS. J Am Chem Soc. 2003;125:4451–4459. doi: 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]
- 49.Lu J, Liong M, Sherman S, Xia T, Kovochich M, Nel A, Zink J, Tamanoi F. Nano Bio Technology. 2007;3:89–95. doi: 10.1007/s12030-008-9003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.