

Abstract
Purpose
The chemopreventive effects of selenium have been extensively examined but its role in cancer development or as a chemotherapeutic agent have only recently been explored. Because Selenium Binding Protein 1 (SELENBP1, SBP1, hSP56) has been shown to bind selenium covalently and selenium deficiency has been associated with esophageal adenocarcinoma (EAC), we examined its role in EAC development and its potential effect on chemosensitivity in the presence of selenium.
Experimental Design
SELENBP1 expression level and copy number variation were determined by oligonucleotide microarrays, real-time RT-PCR, tissue microarrays, immunoblotting and SNP arrays. Bisulfite sequencing and sequence analysis of RT-PCR-amplified products explored epigenetic and post-transcriptional regulation of SELENBP1 expression, respectively. WST-1 cell proliferation assays, senescence-associated β-galactosidase staining, immunoblotting, and flow cytometry were performed to evaluate the biological significance of SELENBP1 overexpression in selenium-supplemented EAC cells.
Results
SELENBP1 expression decreased significantly in Barrett's esophagus to adenocarcinoma progression. Both epigenetic and post-transcriptional mechanisms appeared to modulate SELENBP1 expression. Stable overexpression of SELENBP1 in methylseleninic acid-supplemented Flo-1 cells resulted in enhanced apoptosis, increased cellular senescence, and enhanced cisplatin cytotoxicity. Although inorganic sodium selenite similarly enhanced cisplatin cytotoxicity, these 2 forms of selenium elicited different cellular responses.
Conclusions
SELENBP1 expression may be an important predictor of response to chemoprevention or chemosensitization with certain forms of selenium in esophageal tissues.
Introduction
Esophageal adenocarcinoma (EAC) is increasing in incidence in Western countries and remains a highly lethal malignancy. Despite advances in endoscopic surveillance programs, surgical therapy and multimodality treatment, the prognosis of patients with EAC remains poor, with an overall 5-year survival of 5-12% (1). Serum selenium deficiency, in particular, has been associated with esophageal adenocarcinoma (EAC) as well as its precursor lesion, dysplastic Barrett's esophagus (2). Organic selenium compounds such as methylseleninic acid appear to enhance response to chemotherapeutic agents such as cisplatin and paclitaxel possibly by down-regulation of antiapoptotic signals (3, 4).
Selenium binding protein 1 (Chr:1q21; SBP1, SELENBP1, hSP56) has been shown to bind selenium covalently (5, 6) and is expressed in a variety of tissues and cell lines (7, 8). Its expression has been shown to be reduced markedly in multiple tumor types as compared to their corresponding normal tissues and its reduction has been associated with poor outcome in lung cancer (9), ovarian cancer (10), colorectal cancer (11, 12) and pleural mesothelioma (13). Previous studies have reported a correlation between decreased expression of a chromosome 1q21 gene cluster and resistance to preoperative esophageal adenocarcinoma chemoradiotherapy (14).
Higher levels of SELENBP1 expression in non-growing versus rapidly growing cells have been reported in both mouse and human cells (8, 15). While higher SELENBP1 expression has been shown to be associated with normal colonic epithelial differentiation (12), lower expression was associated with poor tumor differentiation in lung adenocarcinomas (9). Although clear discrepancies were observed between the levels of mRNA and protein expression, mouse selenium-binding proteins were found to be associated with the aging process in senescence-accelerated mouse models (16).
Based on such findings, we evaluated the role of SELENBP1 in the tumorigenesis of esophageal adenocarcinoma as well as the impact of differential SELENBP1 expression and selenium supplementation on cell viability and chemosensitivity. We observed that SELENBP1 expression was decreased in primary esophageal adenocarcinoma tumor samples. Decreased expression appeared to be regulated both by methylation of the SELENBP1 promoter and by alternative splicing of SELENBP1 mRNA. When SELENBP1 expression was reconstituted in vitro, tumor cells responded to selenium treatment with increased apoptosis, increased cellular senescence and increased sensitivity to cisplatin.
Methods and Materials
Patients and Tissues
Written patient consent and approval of the Institutional Review Board were obtained to collect specimens from patients undergoing esophagectomy at the University of Michigan Medical Center (Ann Arbor MI). Specimens were transported to the laboratory in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad CA) on ice. A portion of each sample was frozen in ornithine carbamoyl-transferase (OCT) compound (Miles Inc., Elkhart IN) for cryostat sectioning. The remainder was frozen in liquid nitrogen and stored at -80°C.
Cell Lines and Treatment
Flo-1 was derived from a patient with esophageal adenocarcinoma and has been described previously (17). OE33 was derived from an esophageal adenocarcinoma and is maintained by The European Collection of Cell Cultures (Sigma-Aldrich, Corp.; St Louis MO). Het-1A (derived from SV40 large T antigen-transfected esophageal epithelial cells) (18), SW480 (established from a primary colon adenocarcinoma), and H460 (derived from the pleural fluid of a patient with large cell lung carcinoma) are all maintained by the American Type Culture Collection (Manassas VA). Cell lines were grown in DMEM or RPMI supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Norcross GA) and 1% penicillin/streptomycin/fungizone (Invitrogen) at 37°C in 5% carbon dioxide/95% air.
Cells were seeded at either 4000 cells per well in a 96-well plate format, 50,000 cells per well in a 12-well format, or 325,000 cells per well in 60 mm plates for 24 hours (T0 time point). All experiments performed included a 72 hour treatment with either 2.5 μM methylseleninic acid (MSA) or 10 μM sodium selenite (NaS) and/or a 12-24 hour treatment with 20 μg/mL cisplatin (cis-Diammineplatinum(II) dichloride; CDDP). The demethylating agent 5-aza-2-deoxycytidine (5-Aza) and the histone deacetylase (HDAC) inhibitors trichostatin A (TSA) and valproic acid (VPA) were used at 5 μM, 300 nM, and 5 mM, respectively.
RNA Extraction and Oligonucleotide Microarray
Total RNA was isolated from esophageal adenocarcinomas (37 cases) and Barrett's esophageal samples (9 metaplasia, 15 low-grade and 7 high-grade dysplasia) as previously described (19) using Trizol (Invitrogen) followed by RNeasy column purification (Qiagen, Valencia CA) per the manufacturers' instructions. cRNA was generated and hybridized to GeneChip HG-U133A oligonucleotide microarrays (Affymetrix, Santa Clara CA). Image analysis was performed by the University of Michigan DNA Microarray Core Facility. A filtering algorithm was used to select genes with either increased or decreased expression in adenocarcinomas or dysplastic Barrett's mucosa when compared to Barrett's metaplasia samples. To normalize the microarray data, a summary statistic was calculated using the robust multichip average (RMA) method (20) as implemented in the Affymetrix library of Bioconductor1 which provides background adjustment, quantile normalization, and summarization. Expression values for each sample were then compared to the mean expression value for the seven Barrett's metaplasia samples. Fold reduction of 50% was considered significant (21).
Quantitative Reverse Transcription Polymerase Chain Reaction (RT-PCR) and DNA Sequencing
Total RNA from treated cells was isolated and column purified using the RNeasy Mini Kit (Qiagen) per the manufacturer's instructions. RNA was eluted from the spin column using RNase-free dH2O and reverse-transcribed with 5 U/μL SuperScript II reverse transcriptase (Invitrogen). Real time PCR amplification using 20 ng total RNA, Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen), and 0.2 μM both forward and reverse primers (Supplemental Table 1) was performed on the Corbett Rotor-Gene 6000 (Qiagen). Significant differences of relative quantification were determined using the 2(-ΔΔC(T)) method (22) with normalization to GAPDH or β-actin. PCR amplification of SELENBP1 was performed using 20 ng total RNA or genomic DNA, 0.025 U/μL GoTaq DNA polymerase (Promega Corp., Madison WI), 0.2mM dNTPs, and 0.36 μM both forward and reverse primers (Supplemental Table 1). Agarose gel (1%)-purified PCR products were submitted to the University of Michigan DNA Sequencing Core. Sequencing results were analyzed with FinchTV software and NCBI BLAST software. Agarose gel image densitometry of full-length and alternative splice products was performed using ImageJ software2.
Immunohistochemistry and Tissue Microarray (TMA)
Tissue microarrays were constructed, as previously described (19, 23), with formalin-fixed, paraffin-embedded tissues from 73 patients including 64 tumor, 8 lymph node metastases, 8 dysplastic Barrett's mucosa, and 11 nondysplastic Barrett's metaplasia samples. Multiple samples from representative areas of esophageal adenocarcinoma, metaplasia, or dysplasia were included for 33 patients. Normal esophagus was also included from 3 patients who had undergone esophagectomy for benign indications.
Immunohistochemical staining was performed on the DAKO Autostainer (DAKO, Carpinteria CA) using DAKO LSAB+ and diaminobenzadine (DAB) as the chromagen. Dewaxed and rehydrated sections of the TMA at 4-mm thickness were labeled with SELENBP1 antibody (mouse monoclonal antibody, clone 4D4, 1:200 dilution; MBL International, Woburn MA). We performed microwave citric acid epitope retrieval for 20 minutes. Slides were lightly counterstained with hematoxylin. Each sample was scored independently by two readers using a scale of 0 (no staining), 1+ (<10% staining), 2+ (10-50%) staining, or 3+ (50% or greater).
Protein Extraction and Immunoblot Analysis
Total cellular protein was extracted in lysis buffer (150 mM NaCl; 20 mM Tris, pH 7.5; 1 mM EDTA; 1 mM EGTA; 2.5 mM Na4P2O7; 1 mM glycerol 2-phosphate disodium salt hydrate; 1 mM Na3VO4; 1% Triton X-100) supplemented with Protease Inhibitor Cocktail (20 mL/1 mL lysis buffer; Sigma-Aldrich Corp.). Twenty to 40 μg total cell lysate were boiled in 6x sample buffer, resolved on 8-16% Tris-glycine gels (Invitrogen), and transferred to Immobilon-P membranes (Millipore). Mouse monoclonal SELENBP1 (MBL) and rabbit polyclonal PARP (Cell Signaling, Beverly MA) antibodies were used at 1:2000 dilution. Mouse monoclonal β-actin antibody (Abcam, Cambridge MA) was used at 1:10000 dilution as a loading control. Both HRP-conjugated mouse (Southern Biotechnology Associates, Birmingham, AL) and rabbit (Vector Laboratories, Burlingame, CA) secondary antibodies were used at 1:10000-1:25000 dilutions. Antigen-antibody complexes were detected using the SuperSignal West Pico Chemiluminescent substrate (Pierce Biotechnology, Inc; Rockford IL).
Construction of SELENBP1 Stable Cell Line
A SELENBP1 mammalian expression construct was created and stably tranfected into Flo-1 cells. The SELENBP1 clone, MGC-9270 (American Type Culture Collection), was PCR-amplified using primers containing EcoRI and BamHI restriction sites for directional cloning into the pEGFP-C2 vector containing enhanced green fluorescent protein (EGFP) (Clontech, Mountain View, CA). The pEGFP-SELENBP1 or empty vector construct was transfected into Flo-1 cells using FuGENE 6 transfection reagent (Roche, Indianapolis IN). Selected clones were maintained in growth media containing 200 μg/ml Geneticin.
Proliferation Assay
The cell proliferation reagent WST-1 (Roche) was used for spectrophotometric quantification of cell proliferation, viability, and chemosensitivity in accordance with the manufacturer's directions. Relative proliferation rates were calculated as a percentage of the initial T0 reading within each cell line. Chemosensitivity analysis of a 3-day treatment with either MSA or sodium selenite and/or 12-24 hour treatment with CDDP was similarly performed using WST-1 reagent. Treatments were initiated following T0 absorbance readings. Viability was calculated as a percentage of mock-treated cells at 72 hours within each cell line.
Wound Healing Assay
Cells were densely plated and allowed to grow to 100% confluence. Following serum-starvation for 24 hours, cell monolayers were wounded with sterile p200 tips. Digital images of predetermined locations in each well were taken at both 4× and 10× optical magnification on an Olympus CK2 inverted microscope using a SPOT Idea 1.3MP camera and analyzed with SPOT Basic software (Diagnostic Instruments, Sterling Heights, MI) immediately after wounding and, subsequently, every 24 hours until the wounds were closed. The percentage of initial wound width was determined by averaging measurements per well at each time point.
Single Nuclear Polymorphism (SNP) Analysis
SNP arrays were performed at the Genetic Analysis Platform of the Broad Institute of MIT and Harvard in accordance with the manufacturer's instructions as previously described (24, 25). Briefly, 73 EAC DNAs were processed for Affymetrix genome-wide 250K Sty I SNP arrays. The data were annotated and visualized using Integrative Genomics Viewer (IGV) software3.
Bisulfite Sequencing of Genomic DNA
Genomic DNA was isolated using the DNeasy kit (Qiagen) and 1ug of each sample DNA was treated with bisulfite using the EZ DNA Methylation-Gold Kit (Zymo Research Corp., Orange, CA) according to the manufacturers' instructions. The 5′ untranslated region of SELENBP1 was analyzed using the Transcriptional Regulatory Element Database4 and promoter prediction software from the Berkeley Drosophila Genome Project5. One of the highest-scored sequences (0.98) spanned -39 to +11. A 186 bp fragment (-95 to +90), which covered 158 bp upstream and 25 bp downstream (inside the 1st intron) of the ATG start codon of SELENBP1, was PCR-amplified using bisulfite-treated DNA and primers designed by MethPrimer software6 (Supplemental Table 1). PCR products were purified by electrophoresis on 1% agarose gels, ligated into pCR2.1pTOPO-TA vectors (Invitrogen), and introduced into TOP10 One Shot (Invitrogen) competent bacteria. Insert-containing plasmid DNA was extracted using the Quick Plasmid Miniprep kit (Invitrogen) and eluted in 30 μl buffer. A minimum of ten clones selected at random was sequenced by the University of Michigan DNA Sequencing Core.
Flow Cytometry
Following treatment, DNA from whole cell nuclei was harvested and stained using modified Krishan buffer (0.1% citric acid trisodium salt dihydrate, 0.02 mg/mL RNase A, 0.3% NP-40, 0.05 mg/mL propidium iodide) at the indicated time points. Apoptosis and cell cycle analyses were performed by the University of Michigan Flow Cytometry Core and measured on the FACSCalibur Cytometer (BD Biosciences) using CellQuest and Modfit Light softwares, respectively.
Senescence Assay
Senescence-associated β-galactosidase activity was determined with the Senescence Detection Kit (BioVison Research Products, Mountain View, CA) per the manufacturer's instructions. Digital images were taken at 20x optical magnification and stained cells were counted and expressed as a percentage of total cell number in three independent fields per well per treatment group to obtain an average value for β-galactosidase staining activity.
Experimental Statistical Analysis
Two-way analysis of variance (ANOVA) and Student's t-test were performed using SPSS for Windows 15 (SPSS, Inc., Chicago IL) and Microsoft Excel 2002 (Microsoft Corp., Redmond WA).
Results
Patient demographics
Clinical and pathologic features for the patients whose tissue samples were used for this study are provided in Supplemental Table 2.
Down-regulation of SELENBP1 expression was observed during the progression from metaplastic Barrett's esophagus to esophageal adenocarcinoma
Affymetrix HG-U133A oligonucleotide microarray indicated diminished SELENBP1 expression in esophageal adenocarcinoma relative to both metaplastic and dysplastic Barrett's esophagus (Figure 1A). SELENBP1 expression in esophageal adenocarcinoma decreased by 25% in samples of high-grade dysplasia (p=0.022) and by nearly 50% in esophageal adenocarcinoma (p=0.005) compared with metaplastic Barrett's esophagus (Figure 1B). The array results were validated using quantitative RT-PCR (Figure 1C). Reduced SELENBP1 expression in esophageal adenocarcinoma was reaffirmed using a second set of patient tissues in oligonucleotide microarrays (Figure 1D). Consistent with many primary esophageal adenocarcinomas, Flo-1 cells demonstrated minimal expression of SELENBP1 (Figure 1C). SELENBP1 expression was decreased significantly in adenocarcinomas compared with metaplastic or dysplastic Barrett's esophagus using immunohistochemistry (IHC) of TMAs (Figure 2A-C). Minimal-to-low SELENBP1 expression was observed more frequently in adenocarcinoma (p<0.001, two-way ANOVA) and dysplastic Barrett's esophagus (p<0.05, two-way ANOVA), compared with SELENBP1 expression in samples of Barrett's metaplasia (Supplemental Table 3). Of note, two samples on the oligonucleotide microarray with increased SELENBP1 expression (36T and 39T) had coordinately increased SELENBP1 protein expression by IHC in TMA cores.
Figure 1.
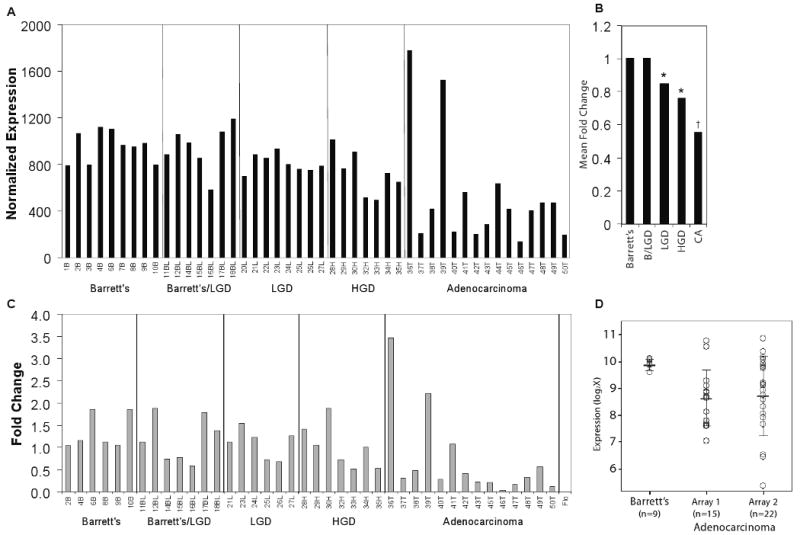
SELENBP1 expression was decreased in the progression from Barrett's metaplasia to adenocarcinoma. A, Normalized relative SELENBP1 (probe set: 214433_s_at) expression as determined by HG-U133A (Affymetrix) oligonucleotide microarray analysis of primary tissue samples from patients with Barrett's metaplasia (B, n=9), Barrett's metaplasia with indeterminate or low-grade dysplasia (BL, n=7), low-grade dysplasia (L, n=8), high-grade dysplasia (H, n=7) and esophageal adenocarcinoma (T, n=15). B, Mean fold-change for each group compared to Barrett's metaplasia (*p<0.05 and †p<0.005 vs. Barrett's metaplasia). C, Quantitative real-time PCR analysis of SELENBP1 expression in a panel of primary tissue samples from the oligonucleotide microarray analysis compared to normal intestinal tissue. Relative quantitative differences were determined using the 2(-ΔΔC(T)) method. D, Comparison of SELENBP1 expression in samples of non-dysplastic Barrett's metaplasia and esophageal adenocarcinoma from 2 independent HG-133A oligonucleotide microarray analyses.
Figure 2.
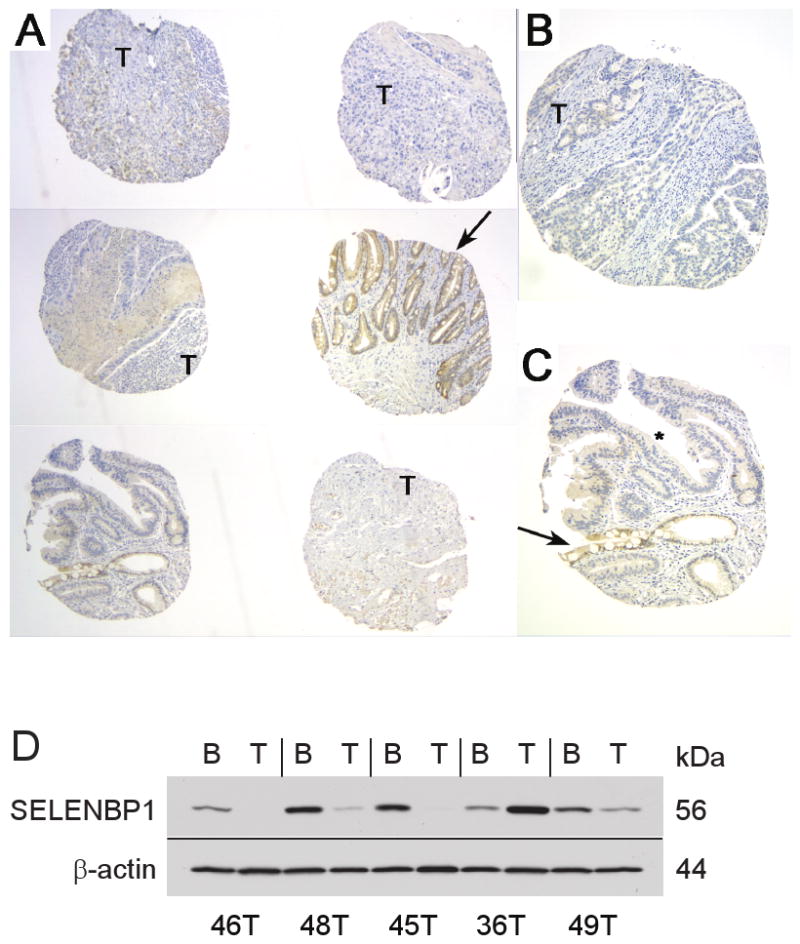
SELENBP1 expression was decreased in samples of high-grade dysplasia (*) and adenocarcinoma (T), when compared with areas of non-dysplastic Barrett's esophagus (arrows) as indicated by IHC analysis using tissue microarray (TMA) of esophageal surgical specimens from 73 patients. Representative sections from TMA for SELENBP1 expression at A, 4×, B and C, 10× magnification. D, Western immunoblotting demonstrated specific binding to SELENBP1 in patient specimens of Barrett's metaplasia (B) and esophageal adenocarcinoma (T). β-actin was used as a loading control.
Immunoblot analysis of paired esophageal adenocarcinoma and Barrett's metaplasia protein extracts indicated expression of a single SELENBP1 protein band (Figure 2D) with reduced expression in tumor samples relative to Barrett's metaplasia in four of the five cases analyzed. In the fifth case (36T), SELENBP1 expression was higher in the tumor compared to an adjacent non-carcinoma section, corresponding to the gene expression findings observed by both oligonucleotide microarray and quantitative RT-PCR.
Decreased DNA copy number changes indicating loss of heterozygosity (LOH) were not detected by SNP array analysis
Since frequent LOH has been recently reported in regions near the SELENBP1 gene (14, 26), we analyzed SNP arrays of 73 esophageal adenocarcinoma samples to determine if the down-regulated SELENBP1 expression was due to decreased DNA copy number at 1q21-q22. No samples were found to have DNA copy number changes less than log2 ratio of -0.737, representing 1 copy if ∼30% of the sample DNA contained contaminating normal esophagus. Nine patient samples exhibited allelic gain (log2 ratio > 0.7) or amplification (log2 ratio > 0.848) at the SELENBP1 locus (Supplemental Figure 1). Eight tumors were examined by both SNP array and oligonucleotide microarray analyses (36T, 40T, 41T, 43T, 44T, 45T, 46T, 50T). The differential expression of SELENBP1 in these samples could not be explained by changes in DNA copy number, suggesting transcriptional or post-transcriptional modulation.
DNA methylation and chromatin remodeling of SELENBP1 may down-regulate gene expression
Sequence analysis of SELENBP1 revealed several CpG sites near the predicted promoter region of this gene, suggesting that DNA methylation may be another mechanism for suppressed SELENBP1 expression in EAC. Bisulfite sequencing of genomic DNA prepared from the EAC cell line Flo-1 and the immortalized esophageal squamous cell line Het-1A indicated that methylation occurred in CpG sites at -95 to +90 in the 5′ untranslated region of SELENBP1. In contrast, very little if any methylation was detected in the colon cancer cell line SW480 (Figure 3A). In addition, treatment of Flo-1 cells with the demethylating agent 5-aza and/or the HDAC inhibitors TSA or VPA increased SELENBP1 expression as determined by quantitative RT-PCR analysis (Figure 3B). Combination treatments that resulted in the greatest increase in SELENBP1 mRNA expression levels were not able to produce detectable SELENBP1 protein (Figure 3C). Despite this, treatment of Flo-1 cells with demethylating and acetylase-enhancing agents increased sensitivity to apoptosis with cisplatin as determined by PARP cleavage (Figure 3D). These results indicate that SELENBP1 expression may be regulated at the epigenetic level but that the level of inducible expression was still too low to detect by protein immunoblotting.
Figure 3.
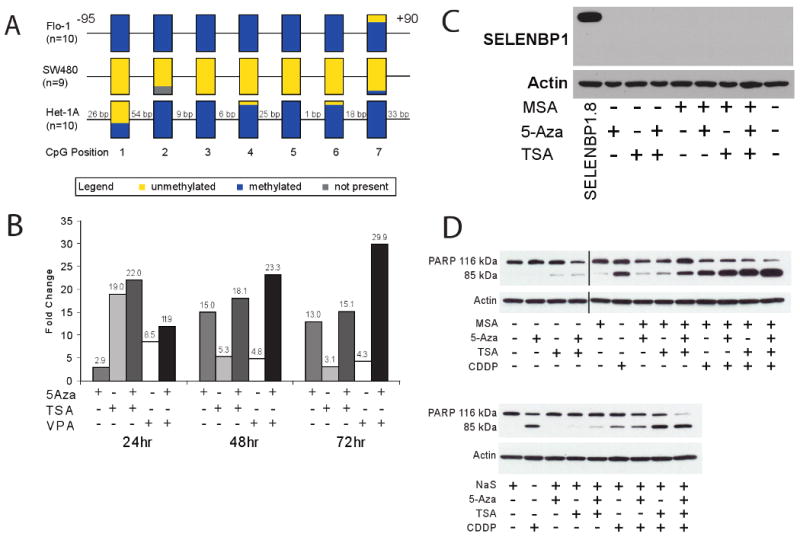
A, Bisulfite sequencing of Flo-1 (EAC), SW480 (colon adenocarcinoma) and Het-1A (immortalized esophageal squamous epithelium) suggested that hypermethylation of the 5′ upstream promoter region of SELENBP1 occurs in esophageal tissues. B, Treatment of Flo-1 cells with 5 μM 5-aza-2-deoxycytidine (5-Aza), 300 nM trichostatin A (TSA) and 5 mM valproic acid (VPA) induced SELENBP1 expression compared to vehicle-treated controls, as determined by qRT-PCR. C, Treatment of Flo-1 cells with 2.5 μM methylseleninic acid (MSA), 5 μM 5-Aza, and/or 300 nM TSA did not lead to detectable levels of SELENBP1 protein. Stably-transfected SELENBP1.8 cells were used as a positive control. β-actin was used as a loading control. D, Treatment of Flo-1 cells with combinations of 2.5 μM MSA, 5 μM 5-Aza, 300 nM TSA, 10 μM sodium selenite (NaS) and/or 20 μg/mL cisplatin (CDDP) induced apoptosis, as determined by PARP cleavage.
Alternative splicing of SELENBP1 mRNA may diminish gene expression
To determine whether the loss of SELENBP1 expression was due to internal exon deletion or mutation in the coding region of the gene, RT-PCR-amplified cDNA products from patient tumor samples and cell lines were sequenced and compared to the NCBI Genbank database for SELENBP1 using the BLAST program. Five overlapping primer pairs were designed to cover the entire coding sequence (Figure 4A). PCR amplification of 2 regions denoted as the L and mid2 fragments (Figure 4B) indicated several variants (Figure 4C) including an exon 4 deletion (L, exon 4-), deletions of both exons 3 and 4 (L, exon 3-/4-), and an exon 7 deletion (mid2, exon 7-), suggesting the possibility of alternative splicing of the SELENBP1 transcript. While deletion of exon 4 was in-frame, deletion of exons 3 and 4 or of exon 7 introduced early termination codons that might translate truncated protein products.
Figure 4.
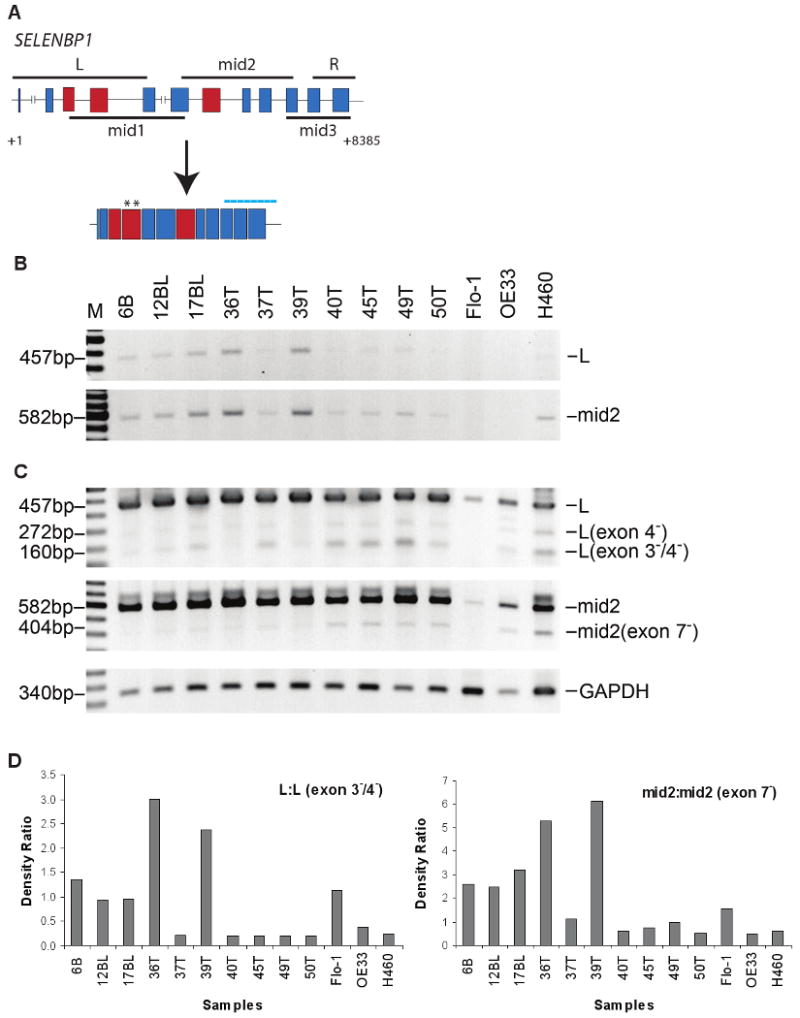
PCR amplification and sequence analysis of the coding region of SELENBP1 suggested that full length mRNA transcripts were alternatively spliced in patient tissue samples and cell lines. A, Schematic diagram of the SELENBP1 gene. Boxes and intervening lines represent exons and introns, respectively. Solid black lines both above and below the diagram represent locations of various overlapping PCR products that were generated using the designated primer set. Darker shaded boxes represent exons that were deleted by alternative splicing. Asterisks represent the location of one set of SELENBP1 qRT-PCR primer pairs. Dotted line represents the location of the Affymetrix oligonucleotide array probe sets for SELENBP1. B, Agarose gel electrophoreses of PCR-amplified full-length L (21 cycles) and mid2 (25 cycles) products in patient tissues confirmed SELENBP1 expression levels detected by microarray and qRT-PCR analyses. C, Extended PCR amplification (35 cycles) and sequencing analyses revealed truncated products for both L (exon 4- and/or exons 3-/4-) and mid2 (exon 7-) primer pairs predominantly in patient tissues with lower levels of full-length products. Flo-1 (EAC), OE33 (EAC), and H460 (lung carcinoma) cell lines were included for comparison. B: non-dysplastic Barrett's esophageal tissue; BL: Barrett's with low-grade dysplasia; T: esophageal adenocarcinoma. PCR amplification of GAPDH (22 cycles) was used as a loading control. D, Integrated densities of electrophoresed PCR products were determined by Image J software and plotted as the density ratio of full length to alternative-splice product.
Although truncated PCR products were detected in all tumor specimens, tumors with higher levels of exon 3-/4- and exon 7--deleted PCR products (Figure 4C) had lower levels of full length PCR products (Figure 4B). These tumors also had reduced expression by both oligonucleotide microarray and quantitative RT-PCR analyses. Conversely, the 2 tumors that expressed the highest levels of SELENBP1 (36T and 39T) had lower levels of both exon 3-/4- and exon 7--deleted PCR products but higher levels of their respective full length products (Figure 4D). Although at seemingly reduced levels, alternatively spliced fragments were also detected in normal esophageal and gastric tissues (data not shown), suggesting that SELENBP1 is transcriptionally unstable. Interestingly, direct comparison of tumor sample 45T with its matching normal gastric tissue showed a marked increase in the levels of both exon 3-/4-- and exon 7--deleted PCR products in tumor cDNA (data not shown). The correlative presence or absence of the alternatively spliced L (exon 3-/4-) and mid2 (exon 7-) fragments in tumors remains unexplained. The oligonucleotide microarray probes for SELENBP1 that were used in this study were located 3′ of the mid2 fragment (Figure 4A) and their hybridization should not have been affected by these spliced regions. It is possible that alternative splicing affects the stability of the resulting transcript. Further investigation of this gene silencing mechanism is warranted.
The intronic regions surrounding exons 3, 4 and 7 of SELENBP1 were sequenced to examine potential modifications near splice junctions. Although a base pair insertion in intron 3 and several polymorphic sites were identified (Supplemental Table 4), representing both cassette alternative exon and intron retention splicing (27), no biological correlation to the different levels of alternatively spliced fragments in the tumors was found.
Overexpression of SELENBP1 potentiated the anti-proliferative effect of MSA in vitro and increased chemosensitivity to cisplatin
As noted earlier, a subset of tumor samples (36T and 39T) had higher expression of SELENBP1. To examine the biological significance of SELENBP1 expression in esophageal adenocarcinoma, Flo-1 cells were stably-transfected with either an eGFP-SELENBP1 or a corresponding empty vector plasmid, and levels of SELENBP1 expression were determined by immunoblot analysis. Clone SELENBP1.8 expressed moderate levels while SELENBP1.1 expressed high levels of eGFP-SELENBP1 compared to empty vector and untransfected Flo-1 cells (Figure 5A). Cellular proliferation and migration did not change appreciably with varying levels of SELENBP1 overexpression compared to empty vector controls as determined by WST assay (Supplemental Figure 2) and by wound healing assay (data not shown), respectively. Since it has been shown that SELENBP1 covalently binds selenium and that a selenium-replete form of SELENBP1 was required for protein-protein interactions (6), we evaluated whether selenium supplementation could alter the cellular response to SELENBP1 overexpression in esophageal adenocarcinoma. Overexpression of SELENBP1 appeared to potentiate an anti-proliferative response to selenium supplementation with methylseleninic acid (MSA) but not with sodium selenite (NaS), particularly in the higher-expressing clone, SELENBP1.1 (Figure 5C).
Figure 5.
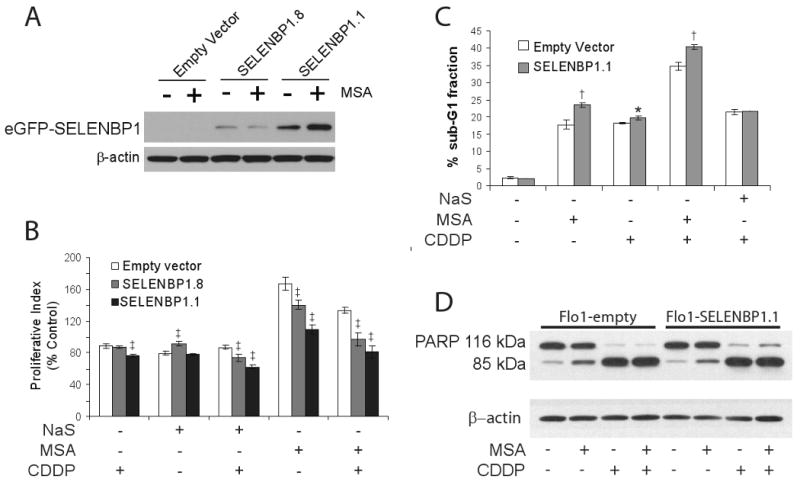
Flo-1 cells were stably-transfected with an eGFP-tagged empty vector or SELENBP1-containing plasmid. The transfection efficiency was estimated to be 60% to 70%. A, Immunoblot analysis of SELENBP1 protein expression in geneticin-selected clones in the presence or absence of 2.5 μM MSA. β-actin served as a loading control. B, Overexpression of SELENBP1 potentiated the anti-proliferative effect of MSA (2.5μM) and NaS (10 μM) particularly during treatment with CDDP (10 μg/mL) as determined by WST-1 assays. Cells were treated with either MSA or NaS for 72 hrs, and CDDP for 24 hrs. WST-1 data endpoints represent four independent wells. C, Small but statistically significant increases in apoptosis were observed by flow cytometry in SELENBP1-expressing cells treated with 2.5 μM MSA, 20 μg/mL CDDP or both agents, but were not discernible by D, immunoblot analysis of PARP cleavage. β-actin was used as a loading control. Each flow cytometry data point represents 3 independently-treated wells. All data were confirmed in repeated experiments. *, p<0.05; †, p<0.01; ‡, p<0.005; Student's t-test vs. empty vector control.
To determine if the decrease in cell viability detected after MSA treatment of SELENBP1-overexpressing cells could enhance the cytotoxicity of CDDP, we examined the sub-G1 fraction of treated cells by flow cytometry. Although SELENBP1 overexpression itself did not have an effect, treatment of Flo-1 cells with MSA, CDDP, and the combination of MSA and CDDP resulted in slight but statistically significant increases in apoptosis in SELENBP1.1 cells when compared with empty vector-transfected controls (Figure 5D). The proportion of apoptotic cells increased with combination treatment, but there was no apparent synergy between MSA and CDDP. The increase in apoptosis following NaS/CDDP treatment appeared to be SELENBP1-independent.
Both MSA and CDDP treatments culminated in PARP cleavage, but this measure of apoptosis was not sensitive enough to detect differences between SELENBP1- and control- transfected Flo-1 cells (Figure 5E). WST-1 analysis revealed that overexpression of SELENBP1 enhanced cisplatin-mediated cytotoxicity (Figure 5C). Additionally, overexpression of SELENBP1 increased cancer cell CDDP sensitivity when treated with either MSA or NaS in comparison to the empty vector-transfected control.
SELENBP1 overexpression potentiated selenium-induced senescence
Expression of selenium-binding proteins has been shown to be associated with accelerated senescence in mice (16). To determine if SELENBP1 overexpression activated cellular senescence in human cells, we stained stably-transfected Flo-1 cells for senescence-associated β-galactosidase (SA-β-Gal). Whereas baseline senescence was not consistently different between empty vector-transfected and SELENBP1-transfected Flo-1 cells, SELENBP1-transfected cells had increased levels of senescence following treatment with MSA but not NaS (Figure 6A and B). These data suggest that MSA and SELENBP1 work together to activate this growth inhibitory pathway.
Figure 6.
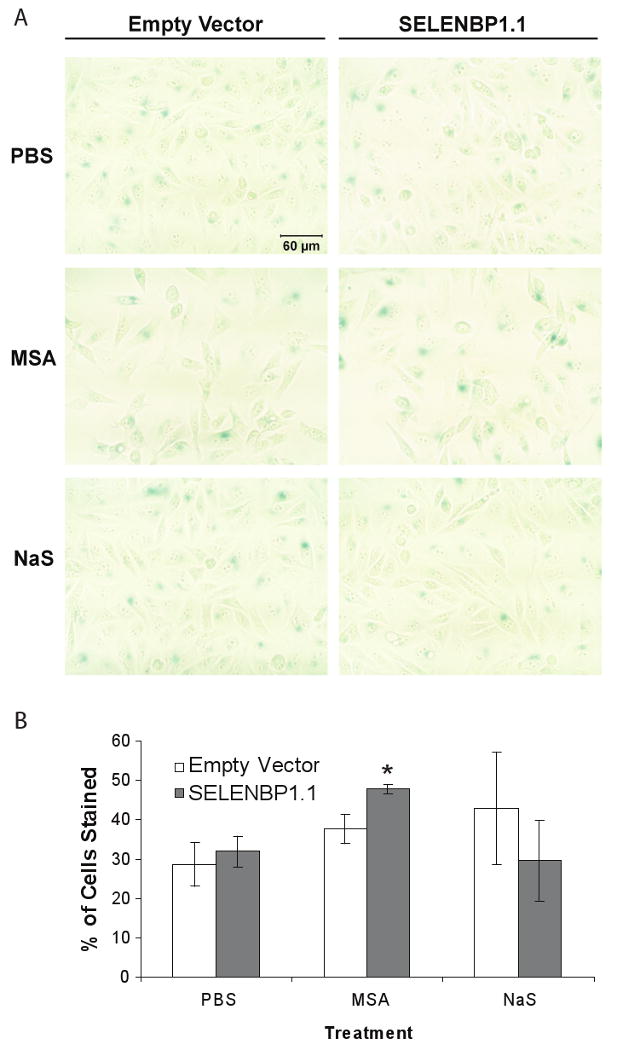
SELENBP1 enhanced MSA-induced cell senescence in Flo-1 cells. Stably-transfected Flo-1 cells were treated with 2.5 μM MSA, 10 μM NaS, or vehicle for 3 days followed by fixation and staining for senescence-associated β-galactosidase expression. A, Representative digital images of treatment groups (20× magnification) B, The number of blue-stained cells versus total cell count per image in 3 non-overlapping areas per well were recorded for each treatment group. Although variability was high, these observations were confirmed in triplicate experiments. *, p<0.05; Student's t-test vs. empty vector control.
Discussion
In this study, we have demonstrated that expression of Selenium Binding Protein 1 (SELENBP1) is decreased in the progression from non-dysplastic Barrett's esophagus to Barrett's esophagus with high-grade dysplasia and esophageal adenocarcinoma at both mRNA and protein levels. Down-regulation of SELENBP1 does not appear to be related to LOH at the 1q21 SELENBP1 locus as demonstrated by SNP array analysis, suggesting that regulation is more likely a transcriptional or post-transcriptional event.
Regulation of SELENBP1 may be affected by epigenetic mechanisms. Gene hypermethylation in the “epidermal differentiation complex,” which is located within 700 Kb of the SELENBP1 locus, has been previously reported (28, 29). Bisulfite sequencing of the EAC cell line Flo-1, which demonstrated very low endogenous levels of SELENBP1, revealed the presence of methylated CpG sites in the 5′ upstream sequence of SELENBP1, suggesting that diminished SELENBP1 expression might be due, in part, to its promoter methylation. In support of this, significantly increased SELENBP1 mRNA expression was observed after 5-Aza-induced demethylation and TSA- or VPA-induced histone acetylation. Increased levels of corresponding protein products were not detected, suggesting that this degree of SELENBP1 re-expression was not sufficient to be detected by Western blotting.
Alternative splicing of primary transcripts is a common mechanism in higher eukaryotic organisms to increase cell type- and developmental stage-specific protein diversity (27). Unbalanced alternative splicing can lead to the overexpression of antagonistic splice variants of genes involved in differentiation, apoptosis, invasion, and metastasis, often correlating with poor prognosis in cancers (30). We observed an inverse correlation between the levels of 2 alternatively spliced PCR products (exon 3-/4- or exon 7-) and their respective full-length sequences in esophageal adenocarcinoma tissues. The expression levels of the full-length PCR products correlated with the SELENBP1 expression levels determined by oligonucleotide microarray and quantitative RT-PCR. Resulting cDNA sequences from these 2 alternatively spliced variants predicted altered amino acid sequences and truncated protein products. Similar N-terminally truncated variants have been reported in the closely related mouse homolog Selenbp2, suggesting that this particular gene is susceptible to alternative splicing (31). These truncated variants may serve to reduce directly the number of full-length transcripts that would be translated into functional protein, or they may serve as dominant negative inhibitors of wild-type SELENBP1. The full significance and functional relevance of these splice variants in EAC tumorigenesis is yet to be determined. Based on these data, strategies to inhibit specific splicing events might serve as another mechanism for SELENBP1 re-expression in esophageal adenocarcinoma.
Population-based studies suggest a correlation between higher serum selenium concentration and lower incidence for a variety of epithelial malignancies (32) as well as pre-malignant diseases such as Barrett's esophagus with high-grade dysplasia (2). Comparison of the cancer chemoprotective effects of different forms of selenium in vitro has revealed marked differences between methylselenol precursors, such as MSA, and those compounds metabolized predominantly to hydrogen selenide, such as NaS (33). Although both MSA and NaS have been shown to inhibit tumor invasion (34, 35), induce apoptosis (36), and cause cell cycle arrest (33, 37), methylselenol precursors have been reported to be more effective tumor inhibitors than selenite or selenomethionine (38). Whereas MSA treatment induced cell retraction and detachment followed by p53-independent caspase-mediated apoptosis, NaS treatment resulted in cytoplasmic vacuole formation, ROS production leading to single strand DNA breaks, and caspase-independent cell death (36). Additionally, MSA but not NaS increased the potency of SN38 (topoisomerase I inhibitor), etoposide (topoisomerase II inhibitor), and the microtubule inhibitor, paclitaxel, in prostate cancer cells (39).
We observed many of these selenium compound-specific differences following treatment of Flo-1 esophageal adenocarcinoma cells. MSA alone caused cell detachment and subsequent PARP cleavage, while NaS-treated cells showed characteristic vacuole formation with no apparent PARP cleavage. Although reconstitution of SELENBP1 alone did not significantly alter cellular proliferation in Flo-1 cells, addition of MSA to these cells resulted in significant inhibition as compared to non-SELENBP1-expressing cells. NaS treatment did not have this differential effect. Although non-toxic doses of both selenium compounds enhanced cisplatin cytotoxicity to some extent, the effects were more pronounced with MSA treatment and were further enhanced by SELENBP1 overexpression. Also, MSA but not NaS treatment of SELENBP1-transfected Flo-1 cells enhanced senescence as assayed by β-galactosidase staining. These differences emphasize the importance of the chemical form of selenium in cancer chemoprevention and chemotherapy strategies.
The relationship between selenium levels and SELENBP1 expression in cancer progression is still unclear. Although levels of selenite did not affect the levels of selenium-binding proteins in rodent cells (40), the level of expression of a 58-kDa selenoprotein correlated with the level of selenite supplementation and with inhibition of DNA synthesis in mouse mammary epithelial cells (41). Huang et al. have also shown that SELENBP1 expression is increased with methylselenocysteine treatment in human ovarian cancer cells (10). It has been reported that selenium compounds promote DNA and histone hypomethylation (42) and may act as histone deacetylase inhibitors in human prostate cancer cells (43, 44), possibly potentiating the re-expression of epigentically silenced genes such as SELENBP1. Selenium may also exert cell growth inhibition by modifying the function of pre-existing proteins. Supporting this is a recent report that selenium-replete SELENBP1 is required for the interaction between the von Hippel-Lindau protein-interacting deubiquinating enzyme, VDU1, and SELENBP1 (6). Many of the tumor suppressive mechanisms analyzed in our studies were either enhanced by or were observed only in the presence of MSA. We have also observed increased SELENBP1 expression following MSA treatment. Our findings suggest that selenium affects both SELENBP1 activity and expression in esophageal adenocarcinoma.
Recently, SELENBP1 was shown to contain hypoxia response elements (HREs) in its promoter region and was found to be a target gene for the transcription factor, Hypoxia-Inducible Factor, 1α (HIF-1α) (45). SELENBP1 mRNA and protein expression was increased over 10-fold in the keratinocytes of transgenic mice expressing constitutive HIF-1α compared with non-transgenic littermates. These transgenic mice demonstrated increased resistance in the transformation from papilloma to carcinoma (45). Because HIF-1α appears to be redox sensitive and the metabolic reduction of some forms of selenium resulted in ROS production (45-47), it is plausible that selenium may also indirectly modify SELENBP1 expression through HIF-1α.
We have found that SELENBP1 is down-regulated in the progression from Barrett's metaplasia to esophageal adenocarcinoma. Although decreased expression of SELENBP1 appeared to be related to epigenetic modification, we also observed increased alternative splice products with predicted premature truncation in a significant number of tumors with diminished SELENBP1 expression. In vitro overexpression of SELENBP1 enhanced cisplatin-mediated cell death, particularly when cells were also treated with MSA. In conclusion, diminished SELENBP1 expression in esophageal adenocarcinoma appeared to blunt the cellular response to some forms of selenium supplementation, and loss of SELENBP1 might be a marker for limited response to ongoing attempts to utilize selenium as a chemopreventive or chemosensitization agent. Alternatively, the presence of full-length SELENBP1 in cancer tissues might predict better response to chemotherapy treatment and would support selenium-mediated secondary chemoprevention strategies.
Supplementary Material
Supplemental Figure 1. Decreased SELENBP1 DNA copy number changes were not detected by SNP array analysis. Genomic DNA from 73 EAC tissues were processed and hybridized to Affymetrix genome-wide 250k Sty I SNP arrays. Data were annotated and the region of chromosome 1 encompassing SELENBP1 (1q21-q22) was visualized using Integrative Genomics Viewer (IGV) software. Blue and red regions represent decreased and increased copy numbers, respectively, as compared to the normal diploid state. Log2 copy numbers of -0.737, 0.70, and 0.848 were used as thresholds for deletion, gain, and amplification, respectively.
Supplemental Table 1. SELENBP1 primer sequences for real-time PCR, methlylation-specific PCR, and regular PCR of both cDNA and genomic DNA
Supplemental Figure 2. Overexpression of SELENBP1 itself did not inhibit cellular proliferation when compared to empty vector controls as determined by WST-1 assays at incubation periods of 24, 48 and 72 hours. WST-1 data endpoints represent four independent wells.
Supplemental Table 2. Subject characteristics and demographics
Supplemental Table 3. SELENBP1 expression in tissue microarray
Supplemental Table 4. Sequence alterations detected in PCR-amplified SELENBP1 cDNA fragments and in PCR-amplified SELENBP1 genomic DNA in and around the splice junctions of exons 3, 4, and 7
Acknowledgments
We would like to thank Ms. Laura Lynch and Mr. Amrit Misra, for their technical assistance in the experiments presented in this manuscript.
Grant support: By the NIH through 5R01CA071606 (DGB), 5P30CA046592 (TJG), 5K08CA134931(AJB), 5K08CA127212 (ACC) and the Thoracic Surgery Foundation for Research and Education (ACC)
Footnotes
version 1.3, www.bioconductor.org
Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997-2008
Statement of Translational Relevance: The overall long-term survival for patients with esophageal adenocarcinoma remains poor. Despite improved understanding regarding the progression from Barrett's esophagus to dysplasia and then adenocarcinoma, patients present commonly in advanced stage. Population-based studies have found that lower serum concentration of selenium is associated with greater incidence of esophageal cancer, including adenocarcinoma, suggesting a possible basis for selenium chemoprevention. Selenium binding protein 1, which binds selenium covalently, is reduced in the progression from dysplastic Barrett's esophagus to adenocarcinoma, but its function has not been well-characterized. The present study shows that SELENBP1 expression is regulated at both the epigenetic and post-transcriptional levels. Gene overexpression increased in vitro esophageal adenocarcinoma cell death in response to selenium supplementation and cisplatin. These studies suggest that differential expression of SELENBP1 may impact cellular response to selenium supplementation both in the chemoprevention and in the treatment of esophageal cancer.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer Statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Rudolph RE, Vaughan TL, Kristal AR, et al. Serum selenium levels in relation to markers of neoplastic progression among persons with Barrett's esophagus. J Natl Cancer Inst. 2003;95:750–7. doi: 10.1093/jnci/95.10.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer JL, Mihelc EM, Pollok KE, Smith ML. Chemotherapeutic selectivity conferred by selenium: a role for p53-dependent DNA repair. Mol Cancer Ther. 2007;6:355–61. doi: 10.1158/1535-7163.MCT-06-0472. [DOI] [PubMed] [Google Scholar]
- 4.Hu H, Li Gx, Wang L, Watts J, Combs GF, Jr, Lu J. Methylseleninic acid enhances taxane drug efficacy against human prostate cancer and down-regulates antiapoptotic proteins Bcl-XL and survivin. Clin Cancer Res. 2008;14:1150–8. doi: 10.1158/1078-0432.CCR-07-4037. [DOI] [PubMed] [Google Scholar]
- 5.Behne D, Kyriakopoulos A. Mammalian selenium-containing proteins. Annu Rev Nutr. 2001;21:453–73. doi: 10.1146/annurev.nutr.21.1.453. [DOI] [PubMed] [Google Scholar]
- 6.Jeong JY, Wang Y, Sytkowski AJ. Human selenium binding protein-1 (hSP56) interacts with VDU1 in a selenium-dependent manner. Biochem Biophys Res Comm. 2009;379:583–8. doi: 10.1016/j.bbrc.2008.12.110. [DOI] [PubMed] [Google Scholar]
- 7.Chang PW, Tsui SK, Liew C, Lee CC, Waye MM, Fung KP. Isolation, characterization, and chromosomal mapping of a novel cDNA clone encoding human selenium binding protein. J Cell Biochem. 1997;64:217–24. doi: 10.1002/(sici)1097-4644(199702)64:2<217::aid-jcb5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 8.Yang M, Sytkowski AJ. Differential expression and androgen regulation of the human selenium-binding protein gene hSP56 in prostate cancer cells. Cancer Res. 1998;58:3150–3. [PubMed] [Google Scholar]
- 9.Chen G, Wang H, Miller CT, et al. Reduced selenium-binding protein 1 expression is associated with poor outcome in lung adenocarcinomas. J Pathol. 2004;202:321–9. doi: 10.1002/path.1524. [DOI] [PubMed] [Google Scholar]
- 10.Huang KC, Park DC, Ng SK, et al. Selenium binding protein 1 in ovarian cancer. Int J Cancer. 2006;118:2433–40. doi: 10.1002/ijc.21671. [DOI] [PubMed] [Google Scholar]
- 11.Kim H, Kang HJ, You KT, et al. Suppression of human selenium-binding protein 1 is a late event in colorectal carcinogenesis and is associated with poor survival. Proteomics. 2006;6:3466–76. doi: 10.1002/pmic.200500629. [DOI] [PubMed] [Google Scholar]
- 12.Li T, Yang W, Li M, et al. Expression of selenium-binding protein 1 characterizes intestinal cell maturation and predicts survival for patients with colorectal cancer. Mol Nutr Food Res. 2008;52:1289–99. doi: 10.1002/mnfr.200700331. [DOI] [PubMed] [Google Scholar]
- 13.Pass HI, Liu Z, Wali A, et al. Gene expression profiles predict survival and progression of pleural mesothelioma. Clin Cancer Res. 2004;10:849–59. doi: 10.1158/1078-0432.ccr-0607-3. [DOI] [PubMed] [Google Scholar]
- 14.Luthra MG, Ajani JA, Izzo J, et al. Decreased expression of gene cluster at chromosome 1q21 defines molecular subgroups of chemoradiotherapy response in esophageal cancers. Clin Cancer Res. 2007;13:912–9. doi: 10.1158/1078-0432.CCR-06-1577. [DOI] [PubMed] [Google Scholar]
- 15.Morrison DG, Oborn CJ, Medina D. Selenite distribution in log and confluent growth phase murine mammary epithelial cells. Cancer Lett. 1988;43:227–36. doi: 10.1016/0304-3835(88)90175-9. [DOI] [PubMed] [Google Scholar]
- 16.Cho YM, Bae SH, Choi BK, et al. Differential expression of the liver proteome in senescence accelerated mice. Proteomics. 2003;3:1883–94. doi: 10.1002/pmic.200300562. [DOI] [PubMed] [Google Scholar]
- 17.Hughes S, Nambu Y, Soldes O, et al. Fas/APO-1 (CD95) is not translocated to the cell membrane in esophageal adenocarcinoma. Cancer Res. 1997;57:5571–8. [PubMed] [Google Scholar]
- 18.Stoner G, Kaighn M, Reddel R, et al. Establishment and characterization of SV40 T-antigen immortalized human esophageal epithelial cells. Cancer Res. 1991;51:365–71. [PubMed] [Google Scholar]
- 19.Lin J, Raoof DA, Wang Z, et al. Expression and effect of inhibition of the ubiquitin-conjugating enzyme E2C on esophageal adenocarcinoma. Neoplasia. 2006;8:1062–71. doi: 10.1593/neo.05832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostat. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 21.Risinger JI, Maxwell GL, Chandramouli GV, et al. Microarray analysis reveals distinct gene expression profiles among different histologic types of endometrial cancer. Cancer Res. 2003;63:6–11. [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-[Delta][Delta]CT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Kononen J, Bubendorf L, Kallionimeni A, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844–7. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- 24.Weir BA, Woo MS, Getz G, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009 doi: 10.1038/ng.465. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maru DM, Luthra R, Correa AM, et al. Frequent loss of heterozygosity of chromosome 1q in esophageal adenocarcinoma: loss of chromosome 1q21. 3 is associated with shorter overall survival Cancer. 2009;115:1576–85. doi: 10.1002/cncr.24122. [DOI] [PubMed] [Google Scholar]
- 27.He C, Zuo Z, Chen H, et al. Genome-wide detection of testis- and testicular cancer-specific alternative splicing. Carcinogenesis. 2007;28:2484–90. doi: 10.1093/carcin/bgm194. [DOI] [PubMed] [Google Scholar]
- 28.Marenholz I, Zirra M, Fischer DF, Backendorf C, Ziegler A, Mischke D. Identification of human epidermal differentiation complex (EDC)-encoded genes by subtractive hybridization of entire YACs to a gridded keratinocyte cDNA library. Genome Res. 2001;11:341–55. doi: 10.1101/gr.114801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elder JT, Zhao X. Evidence for local control of gene expression in the epidermal differentiation complex. Exp Dermatol. 2002;11:406–12. doi: 10.1034/j.1600-0625.2002.110503.x. [DOI] [PubMed] [Google Scholar]
- 30.Venables JP. Unbalanced alternative splicing and its significance in cancer. Bioessays. 2006;28:378–86. doi: 10.1002/bies.20390. [DOI] [PubMed] [Google Scholar]
- 31.Mattow J, Demuth I, Haeselbarth G, Jungblut PR, Klose J. Selenium-binding protein 2, the major hepatic target for acetaminophen, shows sex differences in protein abundance. Electrophoresis. 2006;27:1683–91. doi: 10.1002/elps.200500703. [DOI] [PubMed] [Google Scholar]
- 32.Blot WJ, Li JY, Taylor PR, et al. Nutrition intervention trials in Linxian, China: Supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Natl Cancer Inst. 1993;85:1483–91. doi: 10.1093/jnci/85.18.1483. [DOI] [PubMed] [Google Scholar]
- 33.Lu J, Jiang C, Kaeck M, et al. Dissociation of the genotoxic and growth inhibitory effects of selenium. Biochem Pharmacol. 1995;50:213–9. doi: 10.1016/0006-2952(95)00119-k. [DOI] [PubMed] [Google Scholar]
- 34.Park JM, Kim A, Oh JH, Chung AS. Methylseleninic acid inhibits PMA-stimulated pro-MMP-2 activation mediated by MT1-MMP expression and further tumor invasion through suppression of NF-kappaB activation. Carcinogenesis. 2007;28:837–47. doi: 10.1093/carcin/bgl203. [DOI] [PubMed] [Google Scholar]
- 35.Yoon SO, Kim MM, Chung AS. Inhibitory effect of selenite on invasion of HT1080 tumor cells. J Biol Chem. 2001;276:20085–92. doi: 10.1074/jbc.M101143200. [DOI] [PubMed] [Google Scholar]
- 36.Jiang C, Wang Z, Ganther H, Lu J. Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 2001;61:3062–70. [PubMed] [Google Scholar]
- 37.Zhu Z, Jiang W, Ganther HE, Thompson HJ. Mechanisms of cell cycle arrest by methylseleninic acid. Cancer Res. 2002;62:156–64. [PubMed] [Google Scholar]
- 38.Ip C, Hayes C, Budnick RM, Ganther HE. Chemical form of selenium, critical metabolites, and cancer prevention. Cancer Res. 1991;51:595–600. [PubMed] [Google Scholar]
- 39.Hu H, Jiang C, Ip C, Rustum YM, Lu J. Methylseleninic acid potentiates apoptosis induced by chemotherapeutic drugs in androgen-independent prostate cancer cells. Clin Cancer Res. 2005;11:2379–88. doi: 10.1158/1078-0432.CCR-04-2084. [DOI] [PubMed] [Google Scholar]
- 40.Bansal M, Mukhopadhyay T, Scott J, Cook R, Mukhopadhyay R, Medina D. DNA sequencing of a mouse liver protein that binds selenium: implications for selenium's mechanism of action in cancer prevention. Carcinogenesis. 1990;11:2071–3. doi: 10.1093/carcin/11.11.2071. [DOI] [PubMed] [Google Scholar]
- 41.Morrison DG, Dishart MK, Medina D. Intracellular 58-kd selenoprotein levels correlate with inhibition of DNA synthesis in mammary epithelial cells. Carcinogenesis. 1988;9:1801–10. doi: 10.1093/carcin/9.10.1801. [DOI] [PubMed] [Google Scholar]
- 42.Davis CD, Uthus EO. Dietary folate and selenium affect dimethylhydrazine-induced aberrant crypt formation, global DNA methylation and one-carbon metabolism in rats. J Nutr. 2003;133:2907–14. doi: 10.1093/jn/133.9.2907. [DOI] [PubMed] [Google Scholar]
- 43.Lee JI, Nian H, Cooper AJ, et al. Alpha-keto acid metabolites of naturally occurring organoselenium compounds as inhibitors of histone deacetylase in human prostate cancer cells. Cancer Prev Res (Phila Pa) 2009;2:683–93. doi: 10.1158/1940-6207.CAPR-09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiang N, Zhao R, Song G, Zhong W. Selenite reactivates silenced genes by modifying DNA methylation and histones in prostate cancer cells. Carcinogenesis. 2008;29:2175–81. doi: 10.1093/carcin/bgn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scortegagna M, Martin RJ, Kladney RD, Neumann RG, Arbeit JM. Hypoxia-Inducible Factor-1{alpha} suppresses squamous carcinogenic progression and epithelial-mesenchymal transition. Cancer Res. 2009;69:2638–46. doi: 10.1158/0008-5472.CAN-08-3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 47.Li GX, Hu H, Jiang C, Schuster T, Lu J. Differential involvement of reactive oxygen species in apoptosis induced by two classes of selenium compounds in human prostate cancer cells. Int J Cancer. 2007;120:2034–43. doi: 10.1002/ijc.22480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Decreased SELENBP1 DNA copy number changes were not detected by SNP array analysis. Genomic DNA from 73 EAC tissues were processed and hybridized to Affymetrix genome-wide 250k Sty I SNP arrays. Data were annotated and the region of chromosome 1 encompassing SELENBP1 (1q21-q22) was visualized using Integrative Genomics Viewer (IGV) software. Blue and red regions represent decreased and increased copy numbers, respectively, as compared to the normal diploid state. Log2 copy numbers of -0.737, 0.70, and 0.848 were used as thresholds for deletion, gain, and amplification, respectively.
Supplemental Table 1. SELENBP1 primer sequences for real-time PCR, methlylation-specific PCR, and regular PCR of both cDNA and genomic DNA
Supplemental Figure 2. Overexpression of SELENBP1 itself did not inhibit cellular proliferation when compared to empty vector controls as determined by WST-1 assays at incubation periods of 24, 48 and 72 hours. WST-1 data endpoints represent four independent wells.
Supplemental Table 2. Subject characteristics and demographics
Supplemental Table 3. SELENBP1 expression in tissue microarray
Supplemental Table 4. Sequence alterations detected in PCR-amplified SELENBP1 cDNA fragments and in PCR-amplified SELENBP1 genomic DNA in and around the splice junctions of exons 3, 4, and 7