

Abstract
Annexin A1 (ANXA1), a mediator of the anti-inflammatory action of glucocorticoids, is important in cancer development and progression, whereas NF-κB regulates multiple cellular phenomena, some of them associated with inflammation and cancer. We demonstrated that glucocorticoids and the chemopreventive modified NSAIDs, such as nitric oxide donating aspirin (NO-ASA) and phospho-aspirin, induced ANXA1 in cultured human colon and pancreatic cancer cells. ANXA1 associated with NF-κB and suppressed its transcriptional activity by preventing NF-κB binding to DNA. The induction of ANXA1 by glucocorticoids was proportional to their anti-inflammatory potency, as was the suppression of NF-κB activity, which was accompanied by enhanced apoptosis and inhibition of cell growth mediated by changes in NF-κB-dependent cell signaling. The proposed novel mechanism was operational in the intestinal mucosa of mice treated with dexamethasone or NO-ASA. ANXA1-based oligopeptides displayed the same effects as ANXA1 on NF-κB. One such tripeptide (Gln-Ala-Trp) administered to nude mice inhibited the growth of SW480 human colon cancer xenografts by 58% compared to control; p<0.01). Our findings reveal that ANXA1 is an inducible endogenous inhibitor of NF-κB in human cancer cells and mice, provide a novel molecular mechanism for the action of anti-inflammatory agents, and suggest the possibility of mechanism-driven drug development.
Keywords: Annexin A1, NF-κB, cancer, inflammation, anti-inflammatory drugs
INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAIDs) have emerged as important agents for the prevention of several human cancers (1). A derivative of aspirin, nitric oxide-donating aspirin (NO-ASA), consisting of aspirin and a NO-donating moiety covalently attached to it (Fig. 3A), prevents various cancers in preclinical models and displays anti-inflammatory properties (2). Inhibition of NF-κB by NO-ASA appears important for its chemopreventive effect; NO-ASA inhibits NF-κB in various cancer cell lines and animal models of cancer (3).
Fig. 3. ANXA1 mediates the apoptosis and cell death induced by anti-inflammatory agents.

A: NO-ASA inhibited the growth of HT-29 human colon cells (top). The chemical structure of NO-ASA is shown. NO-ASA enhanced HT29 cell apoptosis in a concentration-dependent manner (middle). This effect was associated with the suppressed expression of several NF-κB dependent antiapoptotic genes (bottom), determined by immunoblotting over a 6 h observation period; BxPC-3 cells were treated with NO-ASA 30 µM. *, p<0.04 and **, p<0.001 compared to control. B: The expression of ANXA1 was knocked down in HT-29 cells using ANXA1 specific siRNA as in Methods, but not in cells transfected with control siRNA. The cells were treated for 3 h with NO-ASA 30 µM or dexamethasone 4 µM, and apoptosis was determined using an ELISA assay. *, p<0.001 compared to control. C: BxPC-3 cells were transfected with an ANXA1 cDNA or with empty vector, while controls were not transfected. The expression status of ANXA1 was evaluated by immunoblotting (middle panel) confirming its overexpression by the ANXA1 cDNA construct. Cell death was induced only in the cells overexpressing ANXA1 as shown in photomicrograph #3 in the upper panel (the numbers in each panel correspond to those inside each column in the bottom panel). Cell growth was determined by the MTT assay, as in Methods. In Fig. 3, all values are mean±SD (*p<0.01; n=3).
NF-κB plays a role in autoimmune responses, cell proliferation and apoptosis and represents a plausible link between inflammation and carcinogenesis (4, 5). NF-κB is sequestered inactive in the cytoplasm bound to IκB proteins, an interaction that regulates its activity. Multiple stimuli activate NF-κB signaling, which consists of translocation of NF-κB to the nucleus where it binds to κB binding sites in the enhancer or promoter regions of target genes, regulating their transcription (6, 7). The NF-κB signal transduction pathway is dysregulated in various human cancers (8, 9). In most such cases, NF-κB is constitutively active and resides in the nucleus, whereas in others the enhanced NF-κB activity is due to changes in the IKK pathway. The sustained NF-κB activation not only protects cancer cells from apoptotic cell death, but may also enhance their proliferation.
While studying the mechanism by which NO-ASA suppresses NF-κB activation, we noted that NO-ASA induces the expression of annexin A1 (ANXA1), a 37 kDa protein originally identified as a mediator of the anti-inflammatory effect of gulcocorticoids (10). ANXA1 has diverse functions including the regulation of cell division, apoptosis and cell growth. Although there is no proof that ANXA1 is a disease-causing gene, it is clear that altering its expression or the localization of the protein it encodes can contribute to the pathogenesis of inflammatory diseases and cancer (11, 12). The induction of endogenous ANXA1 is part of the mechanism of action of gulcocorticoids such as dexamethasone (13, 14). Dexamethasone-induced apoptosis and anti-inflammatory responses are associated with the inhibition of NF-κB. Here, we show that ANXA1 is required for the inhibition of NF-κB activity by anti-inflammatory drugs; propose a novel mechanism of NF-κB inhibition; and demonstrate that an ANXA1-based peptide suppresses the growth of colon cancer xenografts in nude mice.
MATERIALS AND METHODS
Reagents
NO-ASA was synthesized by us; all others were from Sigma-Aldrich. The N-terminal peptides of ANXA1, Ac2-26 and Ac2-12, were from Phoenix Pharmaceuticals. ANXA1 short peptides were synthesized by GenScript Corp. mAbs were from Cell Signaling or Santa Cruz Biochemical.
Cell lines
All were from ATCC (Manassas, VA) and were grown according to their specifications.
MTT assay
We employed a kit from Sigma and followed the recommended protocol.
Apoptosis
We used an ELISA method (Roche) and followed the manufacturer’s protocol.
Cell fractionation
Cell fractions were obtained as described (15). Briefly, cells treated with the test drug were harvested, washed and re-suspended in buffer A (10 mM HEPES, pH 7.9; 10 mM KCl; 1.5 mM MgCI2; 0.5 mM DTT; 0.5 mM PMSF) with a protease inhibitor cocktail (Sigma) and incubated on ice for 15 min. Cell lysates were spun at 5,000 rpm three times. The supernatants were the cytoplasmic extracts. Nuclear pellets were washed with buffer A and resuspended in buffer C (20 mM HEPES, pH 7.9; 450 mM NaCl; 1.5 mM MgCl2; 0.2 mM EDTA; 0.5 mM DTT; 0.5 mM PMSF; 25% glycerol) with the protease inhibitor cocktail and incubated on ice for 30 min. Nuclear extracts were cleared by centrifugation.
Immunoblotting
It was performed following standard protocols. β-actin was used as the loading control.
NF-κB activity
NF-κB activity was measured using a kit from Panomics (Redwood City, CA) and following the manufacturer’s protocol. Nuclear extracts were incubated in a plate coated with NF-κB probe. The primary antibody against NF-κB was incubated for 1 h, followed by HRP-conjugated secondary antibody for 1 h.
siRNA or cDNA clone transfections
siRNA (Santa Cruz) was transfected according to product protocol and after 24 h the transfected cells were treated with NO-ASA for 3 h. Whole cell lysates were used for protein determination and NF-κB activity measurement. cDNA clones (OreGen) were transfected into BxPC-3 cells using Lipofectamine according to product protocol (Invitrogen) and incubated for 24 h. Whole cell lysates were used to determine the protein level and NF-κB activity. Control vector: PCMV6XL5.
Electrophoretic mobility shift assay (EMSA)
EMSAs were carried out according to the manufacturer’s protocol (Panomics). p65 double stranded oligonucleotide probe: 5′-CATCGGAAATTTCCGGAAATTT CCGGAAATTTCCGGC-3′.
Co-immunoprecipitation
Cell extracts were incubated overnight with agarose-conjugated anti-p65 antibody (Santa Cruz). The precipitate was washed, dissolved in 2× Laemmli buffer, boiled, and separated by SDS-PAGE and detected by immunoblotting.
Confocal Microscopy
Cells exposed to NO-ASA for 2 h were fixed, permeabilized, blocked and incubated with mouse monoclonal IgG2b anti-ANXA1 (Santa Cruz) and rabbit monoclonal IgG anti-p65 (Cell Signaling, Beverly, MA) at room temperature for 1 h. After washes, cells were incubated with Alexa555 conjugated anti-mouse IgG (Molecular Probes) and Alexa488 conjugated anti-rabbit IgG for 1 h at room temperature. Images were acquired with a Zeiss LSM 510 META NLO Two-Photon Laser Scanning Image Confocal Microscope. Co-localization scores were generated by the Co-localization Macro Program.
Mice
Min and C57BL/6J mice (Jackson Laboratory) were treated with NO-ASA 100 mg/kg or dexamethasone 10 mg/kg/day for 7 days once a day intraperitoneally, when they were euthanized and small intestinal mucosa was harvested by scraping. We evaluated the induction of ANXA1 and the interaction between ANXA1 and NF-κB p65 in mucosal whole cell lysates. We determined NF-κB activity in the nuclear fraction. We established xenografts in female 6-week-old BALB/c nude mice (Jackson Laboratory) by injecting subcutaneously 2×106 SW480 cells in 100 µl of PBS. Tumor volume (V) was determined using the formula V= L×W(L+W/2)×0.56 (L=length; W=width) (16).
Statistical analyses
We used the two-tailed unpaired Student t-test. Differences were considered significant at p<0.05.
RESULTS
NO-ASA and glucocorticoids induce ANXA1 in human cancer cells
Initially, we assessed the effect of NO-ASA, conventional ASA, other NSAIDs and various glucocorticoids on the expression of ANXA1 in human pancreatic and colon cancer cell lines. As shown in Fig. 1, NO-ASA induced the expression ANXA1 in a concentration- and time-dependent manner. In the cytoplasm, the induction of ANXA1 was rapid and its levels were maximal at 3 h remaining relatively stable for at least 8 h. In the nucleus, ANXA1 became detectable at 2 h, peaked sharply at 3 h and declined rapidly thereafter, likely indicating a time-dependent transport process. A similar effect was observed in HT-29 human colon cancer cells. Dexamethasone, the synthetic corticosteroid with the highest anti-inflammatory potency (17) also induced ANXA1 in BxPC-3 cells, but only in the nuclei; its cytoplasmic levels did not show any significant change. The same effect was observed in HT-29 cells (data not shown). We also evaluated phosphoaspirin (structurally similar to NO-ASA bearing in the place of the NO-donating moiety diethyl phosphate), which has strong anticancer properties (16, 18). Phosphoaspirin induced ANXA1 similar to NO-ASA (data not shown). However, conventional ASA up to 5 mM, cortisone up to 100 µM, and six additional conventional NSAIDs, each at 1 mM for 6 h, failed to induce the expression of ANXA1 in BxPC-3 cells (Suppl. Fig. 1).
Fig. 1. NO-ASA and glucocorticoids induced ANXA1 in human cancer cells.

The expression of ANXA1 was determined by immunoblotting in the nuclei (n) and cytoplasm (c); β-actin was the loading control. NO-ASA induced the expression of ANXA1 in BxPC-3 pancreatic (A, top) and HT-29 colon cancer cells (A, bottom). Dexamethasone was studied in BxPC-3 cells (B). The concentrations in the time-response experiments were: NO-ASA=30 µM; dexamethasone=4 µM. The numbers in italics below the immunoblots represent the band intensity compared to that of the corresponding loading control. C: BxPC-3 cells were treated for 6 h with glucocorticoids, each at 4 µM; ANXA1 was evaluated by immunoblotting. The anti-inflammatory potency of these glucocorticoids, shown in parentheses, correlates with the levels of ANXA1, which they induce. ANXA1 levels, determined by densitometry, are expressed in arbitrary units (AU). D: The effect of these glucocorticoids on NF-κB activity of these cells was determined in parallel by ELISA. Values are mean±SEM (n=3). The numbers in the abscissa, reflecting those in G, denote the corresponding glucocorticoids. The association between NF-κB activity and ANXA1 levels as determined in G and I is statistically significant.
We investigated the effect on ANXA1 expression of seven glucocorticoids, representing a broad range of anti-inflammatory potencies (17). They included (anti-inflammatory potency in parentheses): cortisol (1), cortisone (0.8), prednisone (4), triamincinolone (5), fludrocortisone (10), betamethasone (25) and dexamethasone (25). BxPC-3 cells were treated for 6 h with these compounds, each at 4 µM applied individually. The induction of ANXA1 was proportional to their relative anti-inflammatory potency, with dexamethasone having the greatest effect (Fig. 1C,D). Indeed, the anti-inflammatory potency of these glucocorticoids and the corresponding cellular levels of ANXA1 were significantly correlated (R2=0.91; p<0.001).
ANXA1 is required for the inhibition of NF-κB by anti-inflammatory agents
We investigated whether ANXA1 mediates the inhibition of NF-κB by NO-ASA and dexamethasone. BxPC-3 and HT-29 cells were treated with NO-ASA or dexamethasone for 3 h, and NF-κB activity was determined using an ELISA assay. Both NO-ASA and dexamethasone inhibited the activity of NF-κB in both cell lines (Fig. 2). As shown in Fig. 1, all of the glucocorticoids that we studied inhibited NF-κB activity in a manner that paralleled their anti-inflammatory potency. Of interest, the NF-κB activity was inversely correlated to the levels of ANXA1 that was induced by them (R2=0.93; p<0.001), further suggesting that ANXA1 mediates the NF-κB inhibitory effect of these compounds.
Fig. 2. ANXA1 mediates the inhibition of NF-κB activity by NO-ASA in human cancer cells.
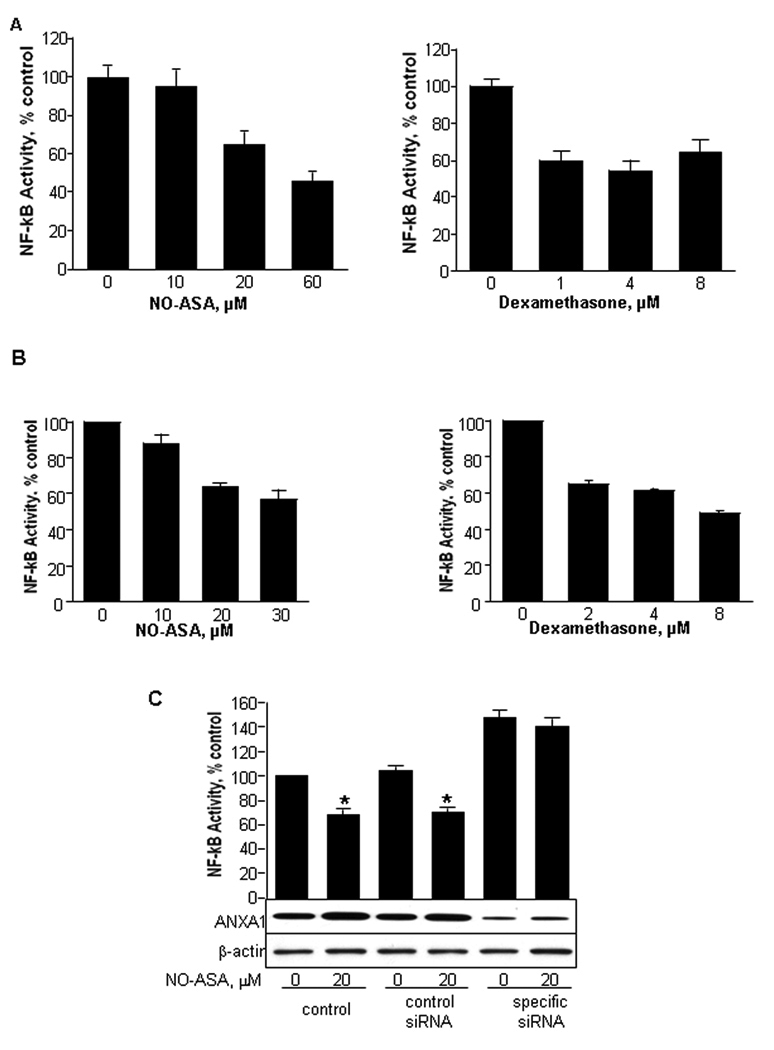
HT-29 human colon (A) or BxPC-3 pancreatic cancer cells (B) were treated for 3 h with NO-ASA or dexamethasone. NF-κB activity was determined by ELISA. C: The expression of ANXA1 in BxPC-3 cells was knocked down by siRNA. NO-ASA reduced NF-κB activity in non-transfected cells and those transfected with nonspecific siRNA. When the expression of ANXA1 was reduced by siRNA, NO-ASA had no effect on NF-κB activity. Compared to the two controls, NF-κB activity was enhanced when ANXA1 expression was suppressed. Values are mean±SD (*, p<0.05; n=3).
To directly assess this possibility, we knocked-down the expression of ANXA1 in BxPC-3 cells using an ANXA1-specific siRNA (Fig. 2C). NO-ASA 20 µM had only a marginal effect on NF-κB activity (10% reduction); in control cells, treated with vehicle or nonspecific siRNA, NO-ASA 20 µM suppressed NF-κB activity by 40%. Dexamethasone had a similar effect (data not shown). The siRNA against ANXA1 enhanced NF-κB activity compared to controls (nonspecific siRNA and not transfected cells), suggesting a baseline inhibitory effect on NF-κB activity by ANXA1.
ANXA1 is required for the induction of apoptosis by anti-inflammatory agents in human cancer cells
NO-ASA inhibits the growth of human cancer cell lines, predominantly through enhanced apoptosis (19). As Fig. 3A confirms, NO-ASA inhibited the growth of HT-29 cells (IC50 = 18.8 µM), inducing apoptosis vigorously (up to 4.5 fold over baseline). Similar results were obtained with BxPC-3 cells (IC50=9.8 µM). In both cell lines, NO-ASA inhibited the expression of Bcl-2, an NF-κB dependent antiapoptotic gene, and of the apoptosis-related proteins survivin, c-IAP-1, c-IAP-2 and TRAF-1 in BxPC-3 cells.
To assess the role of ANXA1 in the cell growth inhibitory effect of these compounds, we knocked down its expression by siRNA (Fig. 3B). Knocking down the expression of ANXA1 completely abrogated the apoptosis induced by either NO-ASA or dexamethasone. This finding suggests that ANXA1 is a key player in the proapoptotic effect of these anti-inflammatory agents. This notion is reinforced by the finding that overexpression of ANXA1 by transfecting ANXA1 cDNA into BxPC-3 cells, cell growth was decreased by ~60% compared to controls. A control plasmid showed no such effect (Fig. 3C).
ANXA1 directly binds to the NF-κB p65 subunit
We obtained several lines of evidence indicating that in order to inhibit the activity of NF-κB, ANXA1 associates physically with the NF-κB dimer. First, we used the 96-well plates of an ELISA NF-κB assay in which double-stranded oligomers containing the κB recognition sequence (5’-CATCGGAAATTTCCGGAA ATTTCCGGAAATTTCCGGC-3’ and its complementary strand) were immobilized on the walls of the reaction wells. Nuclear extracts from BxPC-3 cells treated for 3 h with or without NO-ASA 20 µM were reacted with these κB oligomers. NF-κB dimers bound to the κB oligomers were recognized by anti-p65 or anti-p50 antibodies through a color reaction dependent on a secondary antibody. When, instead of the anti-p65 or anti-p50 antibodies, we used an anti-ANXA1 mAb that did not cross-react with either p50 or p65, we obtained a positive reaction (recognition of the protein bound to the κB oligomers); a nonspecific isotypic antibody gave a negative result (Fig. 4A). These findings suggest either that ANXA1 is associated with the NF-κB dimer or that it cross-binds to the κB oligomers.
Fig. 4. ANXA1 directly binds to NF-κB p65 subunit in human cancer cells.

A: Nuclear proteins from BxPC-3 cells treated for 3 h with NO-ASA 20 µM were incubated with κB oligomers immobilized on the wall of the reaction wells of 96-well plates. NF-κB dimers bound to these oligomers were recognized by anti-p65 or anti-p50 antibodies through a color reaction. An anti-ANXA1 mAb (middle column) that did not cross-react with either p50 or p65 generated a positive reaction. Values are mean±SEM (n≥3). *, p<0.001 compared to control. B: Total protein lysates of BxPC-3 pancreatic cancer cells treated with either NO-ASA 30 µM or dexamethasone 4 µM were immunoprecipitated using either an antibody against the p65 subunit of NF-κB or a nonspecific isotypic IgG antibody (control) and immunoblotted (IB) as indicated. C: The same study as in B was performed on HT-29 colon cancer cells giving similar results. D: Left panel: EMSA from BxPC-3 cells treated with NO-ASA up to 30 µM for 3 h. NO-ASA inhibited the binding of NF-κB to the DNA probe. Right panel: In this EMSA, the nuclear protein extract was reacted with an anti-ANXA1 mAb or with nonspecific isotypic IgG (control) prior to being reacted with the κB probe. ns: non-specific binding.
To clarify this finding, we immunoprecipitated the nuclear protein fraction of BxPC-3 cells treated with NO-ASA as above using an anti-p65 mAb. Immunoblotting with the anti-ANXA1 mAb revealed the presence of markedly increased amounts of ANXA1 in the NO-ASA treated cells compared to controls. Immunoprecipitation with an isotypic non-specific mAb failed to precipitate ANXA1 (Fig. 4B). HT-29 cells gave similar results (Fig. 4C). Finally, we performed an EMSA using nuclear extracts from BxPC-3 cells treated with NO-ASA (Fig. 4D). As expected (3), NO-ASA markedly suppressed the binding of NF-κB to the κB probe. When, however, the nuclear extract from NO-ASA treated cells was reacted with an anti-ANXA1 mAb during the nuclear protein extract–κB probe binding step, the binding of NF-κB to the κB oligomer was restored, as evidenced by a strong NF-κB band in the EMSA. A nonspecific control IgG antibody had no such effect. This finding suggests that ANXA1 is associated with the NF-κB dimer and prevents its binding to the κB binding site. The induction of ANXA1 by NO-ASA and its physical association with NF-κB were also demonstrated in HT-29 human colon cancer cells (data not shown).
Confocal microscopy studies indicated that ANXA1 physically associated with the NF-κB p65 subunit. In BxPC-3 cells we examined whether p65 and ANXA1 co-localized following treatment with NO-ASA. In untreated cells, the two proteins co-localized minimally, if at all (Fig 5A). In response to a 2 h treatment with NO-ASA, there was marked concentration-dependent co-localization of p65 and ANXA1. When cells were treated with 20 µM NO-ASA, co-localization was more pronounced in the nuclei (Fig. 5A), consistent with the enhanced nuclear ANXA1 levels detected by immunoblotting (Fig. 1). Dexamethasone generated similar results (Suppl. Fig. 2). We were unable to document binding of ANXA1 to p50, the other subunit of the NF-κB dimer in these cells. Whether such an interaction occurs below the detection power of our methods remains unclear.
Fig. 5. ANXA1 co-localizes with the p65 subunit of NF-κB in response to NO-ASA treatment.

A: BxPC-3 cells were treated for 2 h with NO-ASA 10 µM (NO-ASA10) or 20 µM (NO-ASA20), fixed with 4% paraformaldehyde and reacted with mouse anti-ANXA1 mAb and anti-p65 rabbit mAb, which do not cross-react with each other, followed by Alexa 555 (anti-mouse; red fluorescence)- and Alexa 488 (anti-rabbit; green fluorescence)-conjugated secondary antibodies and examined by confocal microscopy. Co-localization of the two proteins generates yellow fluorescence. Score graph panels show the fluorescence intensity of p65 alone (a); ANXA1 alone (b); or of both when co-localized (c). B: Min mice (n= 6) and C57BL/6J wild type (WT) mice (n=6) were treated with vehicle (Veh), or NO-ASA 100 mg/kg or dexamethasone 10 mg/kg intraperitoneally, 26 and 2 h before sacrifice. NF-κB activity was determined by ELISA in protein extracts from small intestinal epithelial cells of these mice. Values are mean±SD (*P<0.05 and **P<0.01). C: Protein cell lysates from small intestinal epithelial cells from each animal were immunoprecipitated using an anti-p65 mAb and immunoblotted using an anti-ANXA1 mAb. Representative examples from wild type and Min mice are shown. D: Single small intestinal epithelial cells from Min mice were analyzed by confocal microscopy. Yellow color indicates co-localization of ANXA1 and p65.
In Min mice, NO-ASA and dexamethasone induce ANXA1, which binds to the p65 subunit of NF-κB and inhibits its activation
To assess whether the changes in cancer cell lines occur in vivo, we evaluated the effect of NO-ASA and dexamethasone on NF-κB activity in the intestinal mucosa of Min mice and the corresponding wild type mice C57BL/6J. Heterozygous Min mutants spontaneously develop tumors in the intestine and represent a useful model of intestinal carcinogenesis (20). As previously reported NO-ASA inhibits intestinal carcinogenesis in Min mice (21). Mice were treated with NO-ASA 100 mg/kg or dexamethasone 10 mg/kg intraperitoneally once a day for 1 week when they were euthanized. Protein lysates from the scraped intestinal mucosa were evaluated for NF-κB activity.
As shown in Fig. 5B, NO-ASA and dexamethasone suppressed NF-κB activity in Min mice by 64.3% and 60.8%, respectively (p<0.01). There was a modest (35.6% and 38.8%) but significant (p<0.05) reduction of NF-κB activity in wild type mice in response to these agents. To confirm the physical association between ANXA1 and p65 we used an anti-p65 mAb to precipitate protein lysates from the intestinal mucosa. Immunoblotting established the presence of ANXA1 in the precipitates (Fig. 5C). In addition, confocal microscopy demonstrated co-localization of ANXA1 and p65 in mucosal cells dispersed from intestinal mucosal scrapings, confirming their physically association in the intestinal mucosa of these mice (Fig. 5D).
ANXA1 derived peptides: Inhibition of NF-κB activity and suppression of xenograft tumor growth
The N-terminal sequence of ANXA1 can reproduce the anti-inflammatory actions of the full-length protein (22). Therefore, we treated BxPC-3 cells for 3 h with two commercially available N-terminal fragments of ANXA1, Ac2-26 or Ac2-12, 30 µM each. They decreased NF-κB activity by 25% and 30%, respectively (p<0.05 for both, data not shown). Similar results were obtained in SW480 cells (data not shown). Consequently, we synthesized a series of peptides based on the N-terminal sequence of the ANXA1 protein. As shown in Fig. 6, three of six such peptides inhibited NF-κB activity in SW480 cells; the most potent was the tripeptide Ac-Gln-Ala-Trp (Ac=acetyl), designated QW-3. Their NF-κB inhibitory activity was accompanied by enhanced apoptosis. For example, treatment of SW480 cells with QW-3 30 µM for 3h decreased NF-κB activity by 40 % and enhanced apoptosis 1.7 fold over untreated controls (data not shown). A similar effect was observed in BxPC-3 pancreatic and MCF-7 breast cancer cells (data not shown).
Fig. 6. N-terminal peptides of ANXA1 inhibit NF-κB and the growth of SW480 human colon cancer cell xenografts in nude mice.

A: Six peptides, based on the N-terminal sequence of ANXA1, each 30 µM for 3 h, inhibited NF-κB activity in SW480 cells. The amino acid sequence of the peptides are shown in the box (Ac= acetyl); the numbers correspond to those in the abscissa. *, p<0.01 compared to control. B: Peptide QW-3 (#1 in box) inhibited NF-κB activity in BxPC-3 cells. *, p<0.01 compared to control. C: QW-3, suppressed the growth of subcutaneous xenografts of SW480 human colon cancer cells in nude mice. Starting on day 0, mice (n=18) were treated with QW-3 80 µg (dissolved in 0.5% DMSO in PBS; injected volume 100 µl) daily intraperitoneally. Control mice (n=18) were given vehicle (identical to test peptide solvent). Values are mean±SEM. *p<0.05 and ** p<0.01.
Finally, we investigated the effect of QW-3 on the growth of subcutaneous xenografts of SW480 human colon cancer cells in nude mice. Starting when the average tumor volume was about 750 mm3, nude mice (n=18) were treated with QW-3 80 µg once a day intraperitoneally for 12 days. Compared to the control group (n=18; vehicle alone), QW-3 suppressed tumor growth, its effect being statistically significant on days 8 (p<0.05) and 12 (p<0.01), reducing tumor volume by 48.0% and 58.1%, respectively (Fig. 6C). The mice tolerated this treatment well without any evidence of distress or toxicity, including no change to their body weight compared to controls.
DISCUSSION
Our study provides a novel mechanism for the regulation of NF-κB. This mechanism integrates three seemingly disparate components: a) NF-κB, the master regulator of multiple cellular phenomena, some of which are associated with inflammation and cancer; b) ANXA1, a member of a superfamily whose members regulate several functions in the cell; and c) anti-inflammatory drugs, including gulcocorticoids and the newer modified NSAIDs; glucocorticoids are used clinically as strong anti-inflammatory compounds and modified NSAIDs hold promise as chemopreventive agents.
The mechanism that our results document is the following: most gulcocorticoids and modified NSAIDs induce the expression of ANXA1, which associates physically with NF-κB and suppresses its transcriptional activity by rendering the NF-κB dimer incapable of binding to DNA. These steps have been clearly documented. The induction of ANXA1 is selective, occurring only in response to gulcocorticoids and modified NSAIDs such as NO-ASA and phosphoaspirin, but not in response to conventional NSAIDs. A second feature of this induction is that it is proportional to the pharmacological potency of the inducing agent. This was clearly established in the case of gulcocorticoids, the class of ANXA1 inducers for which such analysis was possible (their anti-inflammatory potency and ANXA1 induction were almost perfectly correlated). This may also be true of NSAIDs, if we include the two modified aspirins, which are much more potent than conventional aspirin (16, 18, 19).
The induction of ANXA1 is accompanied by its physical association with NF-κB, in particular with its p65 subunit. The binding of ANXA1 to NF-κB has been documented amply in vitro (cell protein extracts), in cultured cells and in intestinal epithelial cells of mice. It is noteworthy that our data indicate that ANXA1 binds only to the p65 subunit of NF-κB; we were unable to document an interaction with the p50 subunit. Ongoing studies are attempting to address the reason for such preferential interaction and to identify the region critical for the physical association of the two molecules. It is unclear where in the cell the association of ANXA1 and NF-κB occurs. Based mainly on our confocal microscopy studies, it appears that this binding occurs in the cytoplasm and that the complex translocates to the nucleus; such translocation, however, appears quite limited in dexamethasone-treated cells. Thus, it is conceivable that the cellular distribution of the ANXA1/ NF-κB complex may differ depending on the agent inducing it.
The binding of ANXA1 to NF-κB inhibits the activation of NF-κB. It is, however, unclear how the biding of ANXA1 to p65 inhibits the activation of NF-κB. It is conceivable that the presence of ANXA1 in the NF-κB dimer prevents the binding of NF-κB to its target DNA sequence either through steric hindrance or through a conformational change in p65 that alters its DNA binding site. Whatever the mechanism, this inhibition, although not complete under our experimental conditions, is, nevertheless, functionally important. This was documented by in vitro and in vivo cellular changes. For example, the induction of ANXA1 and its interaction with NF-κB in cultured cells derived from two human cancers (colon and pancreatic) enhanced apoptotic cell death; disruption of any of these steps abrogated cell death and rescued cell growth. Furthermore, the inhibition of NF-κB activation by ANXA1 changed several NF-κB-dependent signaling molecules (survivin, Bcl-2, etc), confirming the involvement of the expected pathway (23).
Our findings extend previous observations that the N-terminal portion of ANXA1 mediates some of its known biological effects. Indeed, not only did we demonstrate that a 12- and a 22-amino acid peptide inhibited NF-κB activation, we were also able to design even shorter effective peptides, with a tripeptide (QW-3) being the most potent. Several tripeptides are known to be very effective biologically, with perhaps the best known examples being glutathione and the thyrotpropin releasing hormone. QW-3 inhibited the activation of NF-κB in cultured cells (as well as their growth).
The novel interaction between ANXA1 with NF-κB may help explain several prior observations on the effect of glucocorticoids and ANXA1 derived peptides in aspects inflammation and related phenomena. For example, it is known that NF-κB activity is persistently increased during neutrophil-mediated inflammatory disorders (24). On the other hand ANXA1, peptides based on its N-terminal sequence, and glucocorticoids (but not conventional NSAIDs) inhibit various neutrophil mediated phenomena such as inflammation and ischemia reperfusion injury (25); and also glucocorticoids suppress NF-κB in neutrophils (26). It is conceivable that the sequence anti-inflammatory agent → induction of ANXA1 → inhibition of NF-κB may explain these effects (and the lack of effect by conventional NSAIDs).
Besides its inhibitory effect on cell growth, QW-3 inhibited the growth of colon cancer cells grown as xenografts, achieving in effect cytostasis (xenograft volume remained stable during the period of observation). Although the overall significance of this observation for cancer control is at present unclear, it nonetheless suggests that the link between ANXA1, NF-κB and tumor growth is worth exploring further. Indeed, annexins have long been considered significant players in tumor development and progression (12). Our findings suggest the potential for drug development not only for cancer but also for the control of inflammation related diseases. ANXA1-derived peptides or small molecules with similar activities may bypass the considerable side effects of conventional anti-inflammatory agents, including glucocorticoids or may have significant anticancer properties.
While ANXA1 mediates some of the effects of potent glucocorticoids and of the potent modified NSAIDs, it is uncertain whether its inhibitory action on NF-κB is the main (or only) mediator of their pharmacological effects. Our data, limited as they are to cell culture studies, establish a very strong association between the anti-inflammatory potency of glucocorticoids, induction of ANXA1 and NF-κB inhibition. It is thus tempting to speculate that the ability of a compound to induce ANXA1 may determine its anti-inflammatory potency. If this is proven to be the case, one may predict that defects in ANXA1 may be responsible for some cases of steroid resistance, a well described and clinically significant phenomenon (27).
In conclusion, our findings reveal that ANXA1 is an endogenous inhibitor of NF-κB that can be induced in human cancer cells and mice by potent anti-inflammatory glucocorticoids and modified NSAIDs. ANXA1 inhibits the activation of NF-κB by binding to its p65 subunit. Oligopeptides based on the N-terminal sequence of ANXA1 have similar effects and one of them inhibits strongly the growth of tumor xenografts. This novel molecular mechanism for the action of anti-inflammatory agents suggests an area for mechanism-driven drug development.
Supplementary Material
Acknowledgments
Financial support: NIH grants CA92423 and CA101019
Footnotes
Potential conflicts of interest: None
REFERENCES
- 1.Baron JA, Cole BF, Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 2.Rigas B. The use of nitric oxide-donating nonsteroidal anti-inflammatory drugs in the chemoprevention of colorectal neoplasia. Curr Opin Gastroenterol. 2007;23:55–59. doi: 10.1097/MOG.0b013e32801145b0. [DOI] [PubMed] [Google Scholar]
- 3.Williams JL, Ji P, Ouyang N, Liu X, Rigas B. NO-donating aspirin inhibits the activation of NF-kappaB in human cancer cell lines and Min mice. Carcinogenesis. 2008;29:390–397. doi: 10.1093/carcin/bgm275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Rigas B. NF-kappaB, inflammation and pancreatic carcinogenesis: NF-kappaB as a chemoprevention target (review) Int J Oncol. 2006;29:185–192. [PubMed] [Google Scholar]
- 6.Wong ET, Tergaonkar V. Roles of NF-kappaB in health and disease: mechanisms and therapeutic potential. Clin Sci (Lond) 2009;116:451–465. doi: 10.1042/CS20080502. [DOI] [PubMed] [Google Scholar]
- 7.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 8.Holcomb B, Yip-Schneider M, Schmidt CM. The role of nuclear factor kappaB in pancreatic cancer and the clinical applications of targeted therapy. Pancreas. 2008;36:225–235. doi: 10.1097/MPA.0b013e31815b3207. [DOI] [PubMed] [Google Scholar]
- 9.Schmitz R, Stanelle J, Hansmann ML, Kuppers R. Pathogenesis of classical and lymphocyte-predominant Hodgkin lymphoma. Annu Rev Pathol. 2009;4:151–174. doi: 10.1146/annurev.pathol.4.110807.092209. [DOI] [PubMed] [Google Scholar]
- 10.D'Acquisto F, Perretti M, Flower RJ. Annexin-A1: a pivotal regulator of the innate and adaptive immune systems. Br J Pharmacol. 2008;155:152–169. doi: 10.1038/bjp.2008.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamal AM, Flower RJ, Perretti M. An overview of the effects of annexin 1 on cells involved in the inflammatory process. Mem Inst Oswaldo Cruz. 2005;100 Suppl 1:39–47. doi: 10.1590/s0074-02762005000900008. [DOI] [PubMed] [Google Scholar]
- 12.Mussunoor S, Murray GI. The role of annexins in tumour development and progression. J Pathol. 2008;216:131–140. doi: 10.1002/path.2400. [DOI] [PubMed] [Google Scholar]
- 13.Buckingham JC, John CD, Solito E, et al. Annexin 1, glucocorticoids, and the neuroendocrine-immune interface. Ann N Y Acad Sci. 2006;1088:396–409. doi: 10.1196/annals.1366.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9(1):62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 15.Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rigas B, Kozoni V. The novel phenylester anticancer compounds: Study of a derivative of aspirin (phoshoaspirin) Int J Oncol. 2008;32:97–100. [PubMed] [Google Scholar]
- 17.Schimmer BP, Parker KL. Adrenocortical steroids and their synthetic analogs. In: Hardman JG, Limbird LE, editors. Goodman & Gillman's The pharmacological basis of therapeutics. New York: McGraw Hill; 2001. pp. 1649–1677. [Google Scholar]
- 18.Zhao W, Mackenzie GG, Murray OT, Zhang Z, Rigas B. Phosphoaspirin (MDC-43), a novel benzyl ester of aspirin, inhibits the growth of human cancer cell lines more potently than aspirin: a redox-dependent effect. Carcinogenesis. 2009;30:512–519. doi: 10.1093/carcin/bgp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams JL, Borgo S, Hasan I, Castillo E, Traganos F, Rigas B. Nitric oxide-releasing nonsteroidal anti-inflammatory drugs (NSAIDs) alter the kinetics of human colon cancer cell lines more effectively than traditional NSAIDs: implications for colon cancer chemoprevention. Cancer Res. 2001;61:3285–3289. [PubMed] [Google Scholar]
- 20.Lipkin M, Yang K, Edelmann W, et al. Preclinical mouse models for cancer chemoprevention studies. Ann N Y Acad Sci. 1999;889:14–19. doi: 10.1111/j.1749-6632.1999.tb08719.x. [DOI] [PubMed] [Google Scholar]
- 21.Williams JL, Kashfi K, Ouyang N, del Soldato P, Kopelovich L, Rigas B. NO-donating aspirin inhibits intestinal carcinogenesis in Min (APC(Min/+)) mice. Biochem Biophys Res Commun. 2004;313:784–788. doi: 10.1016/j.bbrc.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 22.Scannell M, Flanagan MB, deStefani A, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178:4595–4605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- 23.Shen HM, Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009;14:348–363. doi: 10.1007/s10495-009-0315-0. [DOI] [PubMed] [Google Scholar]
- 24.Miskolci V, Rollins J, Vu HY, Ghosh CC, Davidson D, Vancurova I. NFkappaB is persistently activated in continuously stimulated human neutrophils. Mol Med. 2007;13:134–142. doi: 10.2119/2006-00072.Miskolci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.La M, D'Amico M, Bandiera S, et al. Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: analysis of their mechanism of action. Faseb J. 2001;15:2247–2256. doi: 10.1096/fj.01-0196com. [DOI] [PubMed] [Google Scholar]
- 26.Vancurova I, Bellani P, Davidson D. Activation of nuclear factor-kappaB and its suppression by dexamethasone in polymorphonuclear leukocytes: newborn versus adult. Pediatr Res. 2001;49:257–262. doi: 10.1203/00006450-200102000-00021. [DOI] [PubMed] [Google Scholar]
- 27.Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373:1905–1917. doi: 10.1016/S0140-6736(09)60326-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.