

Abstract
Chemokine receptors control leukocyte chemotaxis and cell-cell communication but have also been associated with pathogen entry. GPR33, an orphan member of the chemokine-like receptor family, is a pseudogene in most humans. After the appearance of GPR33 in first mammalian genomes, this receptor underwent independent pseudogenization in humans, other hominoids and some rodent species. It was speculated that a likely cause of GPR33 inactivation was its interplay with a rodent–hominoid-specific pathogen. Simultaneous pseudogenization in several unrelated species within the last 1 million years (myr) caused by neutral drift appears to be very unlikely suggesting selection on the GPR33 null-allele. Although there are no signatures of recent selection on human GPR33 we found a significant increase in the pseudogene allele frequency in European populations when compared with African and Asian populations. Because its role in the immune system was still hypothetical expression analysis revealed that GPR33 is highly expressed in dendritic cells (DC). Murine GPR33 expression is regulated by the activity of toll-like receptors (TLR) and AP-1/NF-κB signaling pathways in cell culture and in vivo. Our data indicate an important role of GPR33 function in innate immunity which became dispensable during human evolution most likely due to past or balancing selection.
Keywords: GPR33, GPCR, Innate immunity, Pseudogene, Dendritic cells, Population genetics
1. Introduction
Recent genome sequencing revealed that vertebrate genomes including the human genome contain more than 900 genes for G protein-coupled receptors (GPCR) which constitute the largest family among transmembrane receptors [1]. Most of those genes code for odorant receptors, whereas non-odorant receptors account for approximately one third of the GPCR repertoire. Although more than 200 non-odorant GPCR were assigned to specific agonists and functions about 155, so-called ‘orphan’ GPCR [2], await classification of their physiological relevance. Not all GPCR genes encode for functional proteins. Pseudogenization is particularly frequent in odorant GPCR [3], while signatures from only 30 non-olfactory rhodopsin-like GPCR pseudogenes have been detected in the human genome to date [4]. The recent availability of large scale genomic information also initiated attempts to identify the origin(s) and to follow the evolutionary history of these receptor genes and families. Since GPCR control almost every physiological process, one can assume that several receptor variants were involved in adaptation to environmental changes and niches during evolution. Consistently, genomic scans for signatures of selection revealed a number of such loci containing genes and pseudogenes of GPCR [5]. This raises the question whether inactive GPCR variants may provide advantage and are, therefore, selected as shown for other genes such as caspase12 [3]. Indeed, there are several examples where receptor inactivation may provide an advantage under distinct environmental circumstances. For example, the FY*O allele at the Duffy locus is at or near fixation in sub-Saharan Africa but rare in other parts of the world [6]. The Duffy antigen which is a chemokine receptor acts as a co-receptor for the cell entry of Plasmodium vivax. Mutation studies have shown that inactivation of Duffy leads to resistance to infections by P. vivax [7].
Selection on the null-allele is probably involved in the evolution of the chemokine-like receptor GPR33 [8]. After the appearance of GPR33 in the mammalian genome more than 125–190 myr ago, this receptor underwent independent pseudogenization in humans, other hominoids and some rodent species including rats and gerbils. In the human GPR33, a premature stop codon terminated the open-reading frame after the third transmembrane domain of the receptor but the mRNA is still detectable in various tissues. Simultaneous pseudogenization in several unrelated species within the last 1 myr caused by neutral drift appears to be very unlikely. GPR33 inactivation may have resulted from a rodent-hominoid-specific pathogen, where selection of the pseudogene would have been advantageous because rats and gerbils are frequently the host of zoonotic pathogens like hanta viruses and Yersinia pestis, respectively, and both species share their habitat with humans [8].
Herein we show that GPR33 allele frequency is significantly different in different cohorts. In contrast to African and Asian populations the derived null-allele of GPR33 is near fixation in the European population. Detailed analyses of cellular expression and expression regulation project the functional relevance of GPR33 in the innate immune response of DC. Our data are consistent with a scenario that fixation of an inactive GPR33 allele is driven by an advantage to an unknown pathogen.
2. Materials and methods
If not stated otherwise all standard substances were purchased from Sigma Aldrich (Taufkirchen, Germany), Merck (Darmstadt, Germany) or C. Roth GmbH (Karlsruhe, Germany). Poly I:C, R-848 and LPS was obtained from Enzo Life Sciences (Lörrach, Germany). PMA was from Tocris Bioscience (Bristol, UK). Cayman Chemicals (Ann Arbor, USA) supplied celastrol, BAY-11-7082 (BAY), nordihydroguaiaretic acid (NDGA) and U-0126.
2.1. Determination of GPR33 allele frequency in human populations and population genetic studies
Individuals of several populations (see Suppl. Methods) were genotyped at the SNP rs17097921 (c.418T > C polymorphism, representing TGA (pseudogene)-CGA (intact gene) allelic variants of the GPR33) using the TaqMan® allelic discrimination assay (Custom TaqMan® SNP Genotyping Assay; Applied Biosystems, Foster City, CA). Additionally, populations from the HapMap SNP data collections and from the Perlegen’s reference genotype data (AFD panels) were analyzed at SNP rs17097921 (Genome Variation Server http://gvs-p.gs.washington.edu/GVS/).
To determine whether selection was acting on the GPR33 locus, tests of neutrality (Tajimas’s D, Fu and Li’s D* and F*) were performed with SNP data retrieved from the HapMap Phase 2 collection (for details see Suppl. Methods) and the DnaSP 4.90.1 software package [9] using sliding window analysis. Analogous analyses were applied to the population of Sorbs (see Suppl. Methods).
Additionally we investigated evidence for natural selection on the GPR33 locus by examining the iHS (a measure of recent positive selection), Fay and Wu’s H, Tajima’s D and Fst in HapMap Phase 2 data using Haplotter (http://hg-wen.uchicago.edu/selection/haplotter.htm, [5]).
2.2. Animals
C57BL/6 mice were bred and kept as described previously [10]. All animal experiments were conducted in accord with accepted standards of humane animal care and approved by the respective regional government agency of Saxony.
For in vivo studies C57BL/6 mice at the age of 9 weeks were treated with poly I:C (100 μg intraperitoneal and alternatively intranasal), R-848 (10μg intraperitoneal and alternatively nasal) or 0.9% NaCl solution as control. After 9 h all animals were sacrificed and the total RNA was isolated from lung and the major lymphoid tissues. The mRNA levels of the GPR33 transcripts were analyzed by quantitative real-time PCR (see below).
2.3. Primary cell culture
Bone marrow (for isolation procedure see [11]) of 8–16 week old C57BL/6 mice was cultivated for 7 days at 37 °C in a humidified atmosphere containing 7% CO2 in a 1:1 mixture of RPMI 1640 supplemented with 10% heat-inactivated FBS, 2mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM (β-mercaptoethanol and a supernatant from a cell line stably transfected with the murine GM-CSF gene (kindly provided by Dr. A. Steinkasserer, Department of Dermatology, University Hospital Erlangen, Germany) at 4 × 106 cells in a 75 cm2 tissue culture flask.
2.4. Quantitative polymerase chain reaction (qPCR)
Total RNA was isolated from tissues and cultured DC using TRI REAGENT™ (Sigma Aldrich) according to the manufacturer’s instructions. Reverse transcription and qPCR were performed as described previously [10], except cDNA from 50 ng total RNA was subjected to qPCR and the house keeping gene clathrin was used for normalization. For oligonucleotide primers see Supplementary Materials Table S1.
Because of low expression levels of receptor protein (which is common to most GPCR) and the lack of a specific anti-GPR33 antibody we could not perform FACS and Western blot analyses to quantify GPR33 expression at the protein level.
2.5. Stimulation and inhibition of DC
For stimulation or inhibition assays, DC were harvested on day 7 and seeded (3 × 106 cells in 2.5 ml serum-free RPMI 1640) in 6-cm dishes. At day 8 DC were incubated with different compounds for 8 h: chloroquine (200 μM), MnCl2 (300 μM), poly I:C (40 μg/ml), R-848 (1 μg/ml), LPS (5 μg/ml), PMA (25 ng/ml) and zymosan A (50 μg/ml). For inhibition assays, DC were preincubated for 30 min with different NF-κB inhibitors (celastrol (250 nM) and (BAY 100 μM)), AP-1 inhibitors (NDGA (5 μM) and U-0126 (50 μM)) and the protein synthesis inhibitor cycloheximide (50 μg/ml). GPR33 mRNA levels were quantified by qPCR.
2.6. Characterization of GPR33 transcripts
The transcriptional start sites of the murine and porcine GPR33 were determined with the GeneRacer™ kit (Invitrogen) for full-length, RNA ligase-mediated rapid amplification of 5′ cDNA ends (RACE) according to the manufacturer’s instructions using mRNA isolated from murine bone marrow-derived DC and from porcine lung lavage cells. mRNA was isolated from total RNA with the Oligotex® mRNA isolation system (Qiagen) according to the manufacturer’s instructions. Gene-specific primers used are listed in Supplementary Materials Table S1. Reaction products were cloned into the pCR®4Blunt-TOPO® vector (Invitrogen) and sequenced (42 murine and 36 porcine clones).
3. Results and discussion
3.1. Population genetic analysis of worldwide distribution of the GPR33 null-allele
We have shown that the inactive GPR33 null-allele is near to fixation in the human population and selection on this derived allele was hypothesized [8]. To follow this idea, we first analyzed the public HapMap dataset at the GPR33 locus for signatures of recent selection of Neither iHS top scores nor Fst values [12] met statistical significance (empirical p in the top 1% for each population). Next, we evaluated the HapMap and Sorb data with DnaSP (see Methods) and found no significant signatures of selection in Tajimas’s D, Fu and Li’s D* and F* tests. These results make recent selection (last 10–50 kyr) at the human GPR33 null-allele unlikely.
3.2. Fixation of the derived GPR33 allele in European populations
The inactive, derived GPR33 stop allele (null-allele, pseudo-gene) is predominant in the human population but the intact allele still exists in a reasonable number of humans [8]. It was speculated that selection may have increased the frequency of the GPR33 null-allele near to fixation. The recent availability of large panels of SNP data from different human populations [13] has given new momentum to the search for signatures of recent selection. Population genetic models predict that rapid selection should leave ‘footprints’ in closely linked genomic regions (selective sweeps). Complete or near complete fixation of an allele may be indicative of adaptive changes in populations of one species, especially when accompanied by signatures of a selective sweep.
Variations of allele frequencies between populations may also be indicative for positive selection [14]. Therefore, we determined GPR33 allele frequencies in geographically different populations. We found a tendency of elevated frequency of the ancestral CGA allele (intact GPR33) towards Asia (Fig. 1, Supplementary Materials Table S2). Whereas the frequency of the ancestral allele is between 1% and 11% in African and Asian populations, the derived GPR33 null-allele is near fixation in European cohorts (mean frequency ancestral allele 0.16%). This difference is highly significant (Fisher’s exact test, p < 0.0001) (Fig. 1). In Pima Indians the derived null-allele is completely fixed (Supplementary Materials Table S2). Although bottle neck, founder effects and genetic drift may also cause the observed geographic distribution of different allele frequencies, an allele distribution with very similar frequencies in Africa and Asia but an almost complete loss of one allele in European populations is quite unusual considering coexistence of the ancestral and the derived allele for 50–800 kyr [8].
Fig. 1.

The human ancestral GPR33 allele shows significant differences in its geographical frequency. The frequency of the intact ancestral CGA allele was determined in different human populations (see also Supplementary Materials Table S2). Information from additional populations (e.g. CEPH-panel) was taken from [8]. In sum, data from 4954 individuals were analyzed. The different populations were grouped according to their geographic origin into America (North/Middle/South), Europe, Africa (Sub-Saharan, Africa), Middle East, Southern Asia, Northern Asia, Central/Eastern Asia, and Southeastern Asia (from left to right). The corresponding mean allele frequencies of the ancestral allele were calculated and are indicated in the grey circles. The significance of frequency differences were calculated with Fisher’s exact test and shown along the connecting lines (p-values).
In sum, population genetic analyses do not support recent (last 10–50 kyr) selection of GPR33 alleles, which is consistent with our previous analysis estimating the age of the human null-allele to be older than 50 kyr [8]. Significant differences in the geographic allele distribution may be indicative for past selection or balancing selection but may also reflect the current status of genetic drift. Currently available methods and data do not allow for support of one evolutionary scenario as compared to another. Therefore, other evidence, specifically information about the physiological relevance, is needed to elucidate the reasons for the obviously synchronized inactivation of GPR33 in unrelated rodent and primate species.
3.3. GPR33 is highly expressed in DC
GPR33 is an orphan GPCR structurally related to ChemR23, a chemokine receptor for chemerin and SIV co-receptor [15], formyl peptide receptors and other chemokine receptors. We have shown that tissue expression of GPR33 overlaps with those of several chemokine receptors [8] but detailed information about quantitative and cell-specific expression is missing. Therefore, we analyzed the murine and porcine GPR33 transcript structure by RACE PCR (see Methods) and found that GPR33 mRNA is transcribed from an intron-containing gene although the GPR33 coding sequence is intronless (Supplementary Materials Fig. S1). There were no evidences for splice variants and exon–intron boundaries are well conserved among mammals (Supplementary Materials Fig. S2).
We next quantified mRNA levels in different murine tissues and found highest expression in lymphoid organs such as lymph nodes, spleen and thymus (Fig. 2). In subfractionated murine spleens highest GPR33 expression levels were found in DC (Supplementary Materials Fig. S3A). These findings are supported by public available microarray data for gene expression in different cell types (Supplementary Materials Fig. S3B).
Fig. 2.

GPR33 is expressed in different lymphoid mouse tissues. Mice were sacrificed and the total RNA was isolated from all major tissues. The GPR33 mRNA levels were quantified by qPCR assays. GPR33 mRNA expression is presented as x-fold over GPR33 expression in heart (ΔCt 14.9 ± 0.17). Data are means ± SEM of four independent experiments performed in duplicates. *p < 0.05; **p < 0.01; ***p < 0.001.
3.4. GPR33 expression in DC is induced by stimulation of TLR
To verify that DC constitutively express GPR33 mRNA and to analyze expression regulation, DC were differentiated from mouse bone marrow cells. The authenticity of these derived DC was verified by detection of CD11c in FACS analysis. Then, DC were incubated with different compounds known to have effects on gene regulation (Supplementary Materials Table S3). As shown in Fig. 3, compounds having an activating effect on GPR33 mRNA expression are known to stimulate TLR (LPS, poly I:C, R-848, zymosan A) [16] and/or AP-1 and NF-κB signaling cascades (chloroquine, PMA) [17,18]. Consistently, TLR signal via both, NF-κB and AP-1 signaling cascades [16,19], and bioinformatic analysis of the putative GPR33 promoter region revealed several AP-1 and NF-κB transcription factor binding sites (Supplementary Materials Fig. S4). GPR33 mRNA expression was measured after 8 h because of the observed maximum expression level at this time point when stimulated with poly I:C (see time course: Supplementary Materials Fig. S5A).
Fig. 3.

GPR33 mRNA expression in mouse DC is elevated by TLR agonists and AP-1 and NF-κB activators. Bone marrow-derived DC were incubated for 8h with different compounds (Supplementary Materials Table S3) and GPR33 mRNA levels were quantified by qPCR assays. Several compounds, MnCl2. chloroquine, PMA and four TLR agonists; LPS, poly 1:C, R-848 and zymosan A, significantly increased GPR33 mRNA expression. Data are presented as GPR33 mRNA expression x-fold over control (RPMI 1640 medium, ΔCt 10.4±0.12) as mean±SEM of n ≥ 6 independent experiments performed in duplicates. ***p < 0.001.
Several metal ions induce transcription of immune response-involved proteins and inflammatory cytokines for example, manganese and other metal ions mediate transcriptional effects via the NF-κB signaling cascade [20,21]. Similarly, an increase of GPR33 mRNA levels was shown for manganese ions and found to be highly specific whereas other metal ions known to modulate transcription were ineffective (Supplementary Materials Table S3). The mechanism for the specific regulation of GPR33 by manganese ions is currently unknown.
For control purposes the DC activation marker CCR7 and CD80 were quantified and showed significant increases in mRNA levels upon incubation with LPS, poly I:C, zymosan A and chloroquine (Supplementary Materials Table S4). Further, several inhibitors of NF-κB signaling, celastrol and BAY, and inhibitors of AP-1 signaling, U-0126 and NDGA, blocked the activation of GPR33 mRNA transcription induced by the substances above (Supplementary Materials Table S5). Therefore, our results clearly showed that GPR33 transcription is regulated by the activity of TLR and NF-κB and AP-1 signaling cascades. Interestingly, poly I:C-induced increase in GPR33 mRNA levels was inhibited already after short incubation with cycloheximide, a blocker of the protein synthesis (Supplementary Materials Fig. S5B). This implicates that GPR33 expression via activation of TLR-NF-κB/AP-1 signaling cascades involves additional protein components, most probably “early transcripts” induced by TLR activation and required for GPR33 transcription.
To further address the mechanism of transcriptional regulation of mouse GPR33 we generated different promoter constructs by cloning up to 4 kbp genomic sequence upstream the transcription start (see Supplementary Fig. S1) into the luciferase reporter vector pGL4.10 (Promega). Because all our attempts failed to transfect DC, vectors were transiently transfected into COS-7, HEK293, CHO-K1 and NIH/3T3 cells. Because these cell lines contain the components of the NF-κB/AP-1 signaling cascades we stimulated with PMA and MnCl2. Although a positive vector control carrying a CMV promoter produced high luciferase activity all mouse GPR33 promoter constructs showed neither basal nor compound-induced luciferase expression (data not shown). These findings indicate that the promoter constructs and/or the cell lines used do not meet all requirements for cell-specific and regulated GPR33 expression.
3.5. In vivo expression of GPR33 is induced by stimulation of TLR
Bacterial cell-surface LPS, double-stranded RNA of viruses, the unmethylated CpG islands of bacterial and viral DNA and other conserved features of pathogens provoke innate immune response via activation of TLR. To verify that TLR activation not only increases transcription of GPR33 in isolated and differentiated DC but also in lymphoid tissues spleens were disintegrated (see Supplementary Methods) and incubated with TLR activators. As shown in Supplementary Materials Fig. S6 all TLR activators increased GPR33 mRNA transcription to a reasonable extent which encouraged us to perform animal experiments. Poly I:C, a TLR3 activator and commonly used to mimic viral infections [22] was administered to mice and RNA was isolated from spleen, thymus, lymph nodes and lungs after 9 h. GPR33 expression was significantly increased in these tissues (Fig. 4A) with highest levels in spleen and lungs depending on the intraperitoneal and nasal application routes, respectively. Consistently, R-848, an activator of TLR7 which recognizes single-stranded viral RNA [16], strongly increased GPR33 expression in lymphoid organs after in vivo application in mice (Fig. 6B). These results indicate that TLR activation elevates GPR33 expression under in vivo conditions and, therefore, exposes GPR33 as an early transcriptional target of the innate immune response.
Fig. 4.
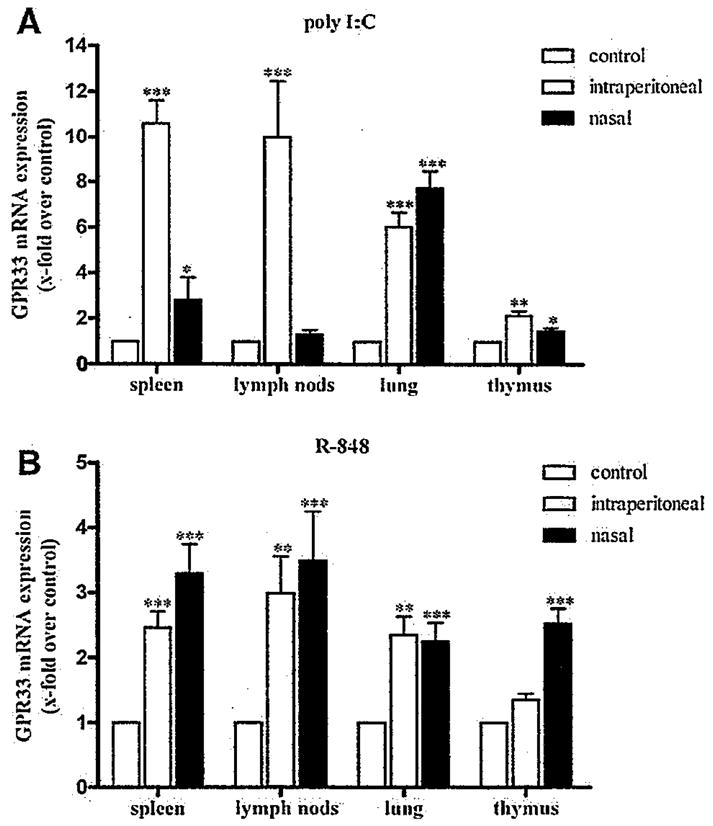
Poly I:C and R-848 elevate GPR33 mRNA expression in vivo. C57BL/6 mice at the age of 9 weeks were treated intraperitoneal and alternatively nasal with A: poly I:C and B: R-848 or 0.9#NaCl solution as control. After 9 h mice were sacrificed and total RNA was isolated from lung and the major lymphoid tissues. The mRNA levels of the GPR33 transcripts were quantified by qPCR assays. Data are presented as GPR33 mRNA expression x-fold over negative control (organs from the 0.9% NaCl solution group) as mean ± SEM of n ≥ 4 independent experiments performed in duplicates. Control ΔCts: spleen (i.p. 8.39±0.16, nasal 8.87±0.23), lymph nodes (i.p. 8.22±0.25, nasal 8.55±0.15), lung (i.p. 9.70±0.26, nasal 9.62±0.11) and thymus (i.p. 8.50± 0.19, nasal 9.22±0.12). *p < 0.05; **p < 0.01; ***p < 0.001.
4. Conclusion
DC provide a functional link between innate and acquired immunity, sample foreign structures and orchestrate the interplay between T- and B lymphocytes [23]. DC are equipped with TLR which, upon activation with foreign DNA, RNA and LPS, mediate de-repression and subsequent activation of inflammatory response genes that play essential roles in innate and acquired immunity [16,19]. TLR3 and TLR7 are expressed in DC and recognize double-and single-stranded RNA, respectively. The chemokine-like receptor GPR33 is one of those genes expressed in activated murine DC. Its expression in lymphoid tissues is regulated by local and systemic activation of TLR and AP-1/NF-κB signaling pathways. It is therefore reasonable to assume that GPR33 takes part in the cellular signaling network of the innate immunity in response to cellular (mimicked by LPS) and viral (mimicked by Poly I:C and R-848) pathogen challenge. Although functional in most mammals this chemokine-like receptor underwent independent pseudogenization in humans, other hominoids and some rodent species [8]. Simultaneous pseudogenization in several unrelated species within the last 1 Myr caused by neutral drift appears to be very unlikely. Hitchhiking of membrane receptors for pathogen entry and subsequent inactivation have been demonstrated for several chemokine receptors [7,15]. Such a scenario may have also favored an inactive allele of GPR33 in humans. Our present population genetic data exclude recent selection on GPR33 locus. If past selection caused the increase in frequency of the derived null-allele (pseudogene), the significant differences in the allele frequency in human populations and the fact that the ancestral intact allele is still present in the population may suggest balancing selection where both alleles were kept depending on the periodical occurrence of the selective force e.g. a pathogen. Unless the natural agonist of GPR33 is identified we can not exclude that the intact ancestral allele in humans is inactivated due to non-obvious inactivating mutations in promoter and coding regions. In any case, independent GPR33 pseudogenization in several unrelated species in a comparable time period likely has a biologic significance related to the function of the GPR33 in innate immune system.
We are very grateful to Otgonbayar Ishdorj, Institute of Medical Sciences, Ulaanbaatar, Mongolia; and Sevjidmaa Baasanjav, Institute of Medical Genetics, Charité, Berlin, for their help in providing the Mongolian DNA samples. We are also very thankful to Marian Mokan, Department of Medicine, Jessenius School of Medicine, Martin, Slovakia and to Iwar Klimes, Institute of the Experimental Endocrinology SAS, Bratislava, Slovakia for their help in providing the Slovak DNA samples. We thank Lusine Danielyan, Department of Clinical Pharmacology, University Hospital of Tübingen, Germany for her help with the Armenian DNA samples. The work was supported by the Deutsche Forschungsgemeinschaft (RO 3619/1-1) and the Leipzig Interdisciplinary Research Cluster of Genetic Factors, Clinical Phenotypes and Environment.
Abbreviations
- BAY
BAY-11-7082
- Ct
threshold cycle
- DC
dendritic cells
- GPCR
G protein-coupled receptors
- kyr
thousand years
- myr
million years
- NDGA
nordihydroguaiaretic acid
- poly I:C
polyinosinic-polycytidylic acid
- qPCR
quantitative real-time PCR analysis
- SNP
single nucleotide polymorphism
- TLR
toll-like receptor
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbrc.2010.04.077.
Footnotes
The sequences reported in this paper have been deposited in the GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) database (Accession No. GQ981321-GQ981327).
References
- 1.Fredriksson R, Lagerstrom MC, Schioth HB. Expansion of the superfamily of G-protein-coupled receptors in chordates. Ann N Y Acad Sci. 2005;1040:89–94. doi: 10.1196/annals.1327.011. [DOI] [PubMed] [Google Scholar]
- 2.Wigglesworth MJ, Wolfe LA, Wise A. Orphan seven transmembrane receptor screening. Ernst Schering Found Symp Proc. 2006:105–143. doi: 10.1007/2789_2006_006. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Grus WE, Zhang J. Gene losses during human origins. PLoS Biol. 2006;4:e52. doi: 10.1371/journal.pbio.0040052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rompler H, Staubert C, Thor D, et al. G protein-coupled time travel: evolutionary aspects of GPCR research. Mol Interv. 2007;7:17–25. doi: 10.1124/mi.7.1.5. [DOI] [PubMed] [Google Scholar]
- 5.Voight BF, Kudaravalli S, Wen X, et al. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamblin MT, Di Rienzo A. Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am J Hum Genet. 2000;66:1669–1679. doi: 10.1086/302879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hadley TJ, Peiper SC. From malaria to chemokine receptor: the emerging physiologic role of the Duffy blood group antigen. Blood. 1997;89:3077–3091. [PubMed] [Google Scholar]
- 8.Rompler H, Schulz A, Pitra C, et al. The rise and fall of the chemoattractant receptor GPR33. J Biol Chem. 2005;280:31068–31075. doi: 10.1074/jbc.M503586200. [DOI] [PubMed] [Google Scholar]
- 9.Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSPDNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- 10.Schliebe N, Strotmann R, Busse K, et al. V2 vasopressin receptor deficiency causes changes in expression and function of renal and hypothalamic components involved in electrolyte and water homeostasis. Am J Physiol Renal Physiol. 2008;295:F1177–1190. doi: 10.1152/ajprenal.00465.2007. [DOI] [PubMed] [Google Scholar]
- 11.Lutz MB, Kukutsch N, Ogilvie AL, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 12.Akey JM, Zhang G, Zhang K, et al. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frazer KA, Ballinger DG, Cox DR, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabeti PC, Schaffner SF, Fry B, et al. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 15.Samson M, Edinger AL, Stordeur P, et al. ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur J Immunol. 1998;28:1689–1700. doi: 10.1002/(SICI)1521-4141(199805)28:05<1689::AID-IMMU1689>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 16.Kawai T, Akira S. The roles of TLRs RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park J, Choi K, Jeong E, et al. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia. 2004;47:9–20. doi: 10.1002/glia.20017. [DOI] [PubMed] [Google Scholar]
- 18.Liese AM, Siddiqi MQ, Siegel JH, et al. Augmented TNF-alpha and IL-10 production by primed human monocytes following interaction with oxidatively modified autologous erythrocytes. J Leukoc Biol. 2001;70:289–296. [PubMed] [Google Scholar]
- 19.Huang W, Ghisletti S, Perissi V, et al. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint Mol. Cell. 2009;35:48–57. doi: 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis JB, Randol TM, Lockwood PE, et al. Effect of subtoxic concentrations of metal ions on NFkappaB activation in THP-1 human monocytes. J Biomed Mater Res A. 2003;64:217–224. doi: 10.1002/jbm.a.10352. [DOI] [PubMed] [Google Scholar]
- 21.Ramesh GT, Ghosh D, Gunasekar PG. Activation of early signaling transcription factor NF-kappaB following low-level manganese exposure. Toxicol Lett. 2002;136:151–158. doi: 10.1016/s0378-4274(02)00332-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fortier ME, Kent S, Ashdown H, et al. The viral mimic, polyinosinic:polycytidylic acid, induces fever in rats via an interleukin-1-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. 2004;287:R759–766. doi: 10.1152/ajpregu.00293.2004. [DOI] [PubMed] [Google Scholar]
- 23.Joffre O, Nolte MA, Sporri R, et al. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. 2009;227:234–247. doi: 10.1111/j.1600-065X.2008.00718.x. [DOI] [PubMed] [Google Scholar]