

Abstract
Alzheimer Disease (AD) is a terminal age-associated dementia characterized by early synaptic dysfunction and late neurodegeneration. While the presence of plaques of fibrillar aggregates of the amyloid beta peptide (Aβ) is a signature of AD, evidence suggests that the pre-plaque small oligomeric Aβ promotes both synaptic dysfunction and neuronal death. We found that young Tg2576 transgenic mice, which accumulate Aβ and develop cognitive impairments prior to plaque deposition, have high CNS activity of calcineurin (CaN), a phosphatase involved in negative regulation of memory function via inactivation of the transcription factor CREB, and display CaN-dependent memory deficits. These results thus suggested the involvement of pre-fibrillary forms of Aβ. To investigate this issue, we compared the effect of monomeric, oligomeric and fibrillar Aβ on CaN activity, CaN-dependent pCREB and pBAD levels and cell death in SY5Y cells and in rat brain slices and determined the role of CaN on CREB phosphorylation in the CNS of Tg2576 mice. Our results show that oligomeric Aβ specifically induces CaN activity and promotes CaN-dependent CREB and BAD dephosphorylation and cell death. Furthermore, Tg2576 mice display Aβ oligomers and reduced pCREB in the CNS, which is normalized by CaN inhibition. These findings suggest a role for CaN in mediating effects of oligomeric Aβ on neural cells. Since elevated CaN levels have been reported in the CNS of cognitively impaired aged rodents, our results further suggest that abnormal CaN hyper-activity may be a common event exacerbating the cognitive and neurodegenerative impact of oligomeric Aβ in the ageing CNS.
Keywords: Amyloid beta, oligomer, calcineurin, CREB, BAD, Alzheimer's Disease
INTRODUCTION
The deposition in the CNS of senile plaques primarily formed by fibrillar aggregates of misfolded amyloid beta (Aβ) peptide is one pathological hallmark of Alzheimer Disease (AD), an age-associated terminal neurodegenerative dementia for which there is currently no cure. The symptomatic presentation of AD begins with initial synaptic dysfunction and cognitive impairment and progresses to massive neuronal death in the latest stages of the disease (Selkoe, 2002). Although the etiology of AD is still elusive, there is ample consensus that the CNS presence of Aβ plays a central neuropathologic role (Golde, 2005; Haass and Selkoe, 2007). The process of plaque formation begins with the overproduction of monomers of β-sheet misfolded Aβ, a 40- or 42-aminoacid long peptide resulting from atypical cleavage of the larger amylolid precursor protein (APP) (Van Gassen and Annaert, 2003). Aβ monomers then assemble into structurally-defined oligomers (mostly trimers to dodecamers) and higher molecular weight protofibrils, which are the composing units of the macromolecular insoluble fibrils mostly found deposited in amyloid senile plaques (Chiti and Dobson, 2006).
Recent evidence implicates small oligomeric assemblies of Aβ as the main offending species, whereas monomers and fibrils may be relatively innocuous (Glabe and Kayed, 2006; Lansbury and Lashuel, 2006; Haass and Selkoe, 2007). Notably, in AD patients the number of amyloid plaques does not correlate with the degree of disease severity (McKee et al., 1991; Bennett et al., 1993), whereas the levels of soluble Aβ do so both in humans (McLean et al., 1999) and laboratory animals (Moechars et al., 1999; Oddo et al., 2003). Transgenic mouse models of AD develop cognitive impairments months before CNS plaque deposition (Gordon et al., 2002) and Aβ oligomers isolated from the CNS of plaque-free cognitively-impaired APP transgenic mice produce memory deficits when injected intacerebroventricularly (icv) in adult rats (Lesne et al., 2006). Furthermore, Aβ oligomers, but not monomers or fibrils, are cytotoxic (Demuro et al., 2005) and disrupt long-term potentiation (LTP, the cellular basis of learning and memory) both in vitro and in vivo (Walsh et al., 2002; Wang et al., 2002). Most notably, it has been recently shown that a specific Aβ dimer extracted from cerebrospinal fluid (CSF) obtained from AD patients inhibits hippocampal LTP in vivo, whereas Aβ monomers extracted from the same specimens are ineffective (Klyubin et al., 2008).
The Ca2+/calmodulin dependent protein phosphatase calcineurin (CaN) is abundant in CNS neurons and plays a central role in the regulation of synaptic plasticity, learning and memory, and neuronal apoptosis. Through modulating crucial signaling mechanisms including direct or indirect dephosphorylative inactivation of the transcription factor CREB, CaN opposes LTP and memory function (Groth et al., 2003; Mansuy, 2003). Additionally, CaN is capable of promoting neuronal cell death through direct dephosphorylative activation of the pro-apoptotic BAD (Wang et al., 1999; Springer et al., 2000). Increased CaN activity has been found in the CNS of aging rats where it correlates with the degree of age-related cognitive impairments (Foster et al., 2001). Alteration of CaN has also been demonstrated in the AD brain (Brion et al., 1995; Hata et al., 2001; Norris et al., 2005) and in APP transgenic mice where Aβ-promoted neurite degeneration in the CNS is triggered by a Ca2+/CaN-dependent mechanism (Kuchibhotla et al., 2008). Furthermore, in vitro evidence indicates that Aβ induces neuronal apoptosis through a CaN-dependent mechanism (Agostinho and Oliveira, 2003). In addition, Aβ has been found to decrease NMDA receptor trafficking at the cell membrane (a mechanism relevant to synaptic plasticity and memory) in a CaN-dependent fashion (Chen et al., 2002; Snyder et al., 2005).
We recently reported that 5 month old APP transgenic mice (Tg2576) display abnormally high levels of CaN activity in the CNS, coincident with the occurrence of Aβ-promoted memory deficits, but prior to the appearance of amyloid deposits (Dineley et al., 2007). We further showed that pharmacological normalization of CaN activity acutely reversed the deficits that these animals display in the fear conditioned memory task. These results suggested that high CaN activity, and consequently CaN-dependent memory deficits, could be a downstream effect of the presence of pre-fibrillar Aβ species in the CNS. In order to test this hypothesis, we utilized a reproducible in vitro system of human neuroblastoma SY5Y cell lines, ex vivo rat brain slices and an in vivo model of AD in transgenic rodents to determine the effect of monomeric, oligomeric and fibrillar Aβ on CREB and BAD phosphorylation and cell death as a function of CaN activity.
RESULTS
Consistent with previous reports (Demuro et al., 2005), oligomeric Aβ induced cytotoxicity in SY5Y neuroblastoma cells in our experimental conditions, while monomeric or fibrillar Aβ were ineffective. Figure 1 shows the results from a representative cytotoxicity experiment in SY5Y cells treated for 12 hrs with increasing concentrations of monomeric, oligomeric and fibrillar Aβ. The purity of the Aβ preparations was checked by dot-blot employing the conformation-specific A-11 antibody (which specifically recognizes oligomeric Aβ) and the 6E10 antibody (which recognizes Aβ regardless of its tertiary or quaternary structure) and the oligomeric species obtained identified by Western blot. Representative examples of such dot-blots and Western blots are shown in Fig. 1A. Figure 1B shows that the release of LDH from SY5Y cells (an index of cell death) was significantly increased by treatment of cells with oligomeric Aβ in a dose-dependent fashion, with the minimum cytotoxic dose being >0.5 μM. In the same experiment, monomeric and fibrillar Aβ failed to induce significant cell death at any of the concentrations tested. Next, we compared the effect of monomeric, oligomeric and fibrillar Aβ on CaN activity (Fig. 2A), and found that CaN activity in SY5Y cells was significantly increased by oligomeric Aβ, but not by monomeric or fibrillar Aβ. Pre-incubation of test solutions of Aβ with the A-11 antibody that selectively interacts with and neutralizes oligomeric Aβ (Kayed and Glabe, 2006), completely abolished CaN activation induced by Aβ oligomers and had no effect on monomeric or fibrillar Aβ-treated cells. On the other hand, no effect was observed on the combined activity of PP1/PP2A, two other protein phosphatases that at variance with CaN are Ca2+/calmodulin insensitive (Mansuy and Shenolikar, 2006) in SY5Y cells treated with monomeric, oligomeric or fibrillar Aβ, either in the presence or absence of pre-incubation with the A-11 antibody (Fig. 2B). In the same experiment, oligomeric Aβ promoted cell death (Fig. 2C), which was also abolished by pre-treatment of the test solution with the A-11 antibody.
Figure 1. Oligomeric Aβ, but not monomeric or fibrillar Aβ, induces cell death SY5Y cells.

(A) Left: Representative dot blot analysis detecting monomeric, oligomeric and fibrillar human Aβ1–42 from standard preparations spotted onto nitrocellulose membranes. Membranes were probed with the oligomer-specific A-11 antibody (top) or the 6E10 antibody (which recognizes Aβ sequence, regardless of conformation). Right: representative western blot analysis employing the A-11 antibody to detect oligomers in a typical preparation of oligomeric Aβ1–42. Dimers, trimers and dodecamers are shown to be the most prevalent oligomeric species.
(B) LDH release cytotoxocity assay in human SY5Y neuroblastoma cells treated for 12 hr with increasing concentrations of monomerc, oligomeric or fibrillar Aβ. Dashed line represents LDH release value in control cells. Values are mean ± SD of 4 independent replicates. Experiment was repeated twice with similar results. *: p<0.001 vs. control cells (ANOVA).
Figure 2. Oligomeric Aβ, but not monomeric or fibrillary Aβ, selectively induces CaN activity and cell death in SY5Y cells.

Calcineurin activity (A), combined PP-1/PP-2A activity (B) and LDH release (C) in SY5Y neuroblastoma cells treated for 3 hr with 1 μM of monomeric, oligomeric and fibrillary human Aβ1–42. Black bars represent cells treated with Aβ that was pre-incubated with the oligomer-neutralizing antibody A-11. N=4 independent replicates per group; *: p<0.001 vs. control (ANOVA).
Activation of CaN is known to initiate an apoptotic program of cell death through dephosphorylation and subsequent activation of the pro-apoptotic protein BAD (Wang et al., 1999; Springer et al., 2000). Consistent with this literature evidence, we found that treating SY5Y cells with increasing concentrations of oligomeric Aβ resulted in BAD dephosphorylation (Fig. 3). Please note that while βactin levels did not change, there was a concomitant reduction of pBAD and total BAD in cells treated with the highest (and most toxic) concentration of oligomeric Aβ (5 μM). This phenomenon could be explained by postulating that only those cells devoid of substantial BAD expression survived the most toxic concentrations of Aβ oligomers. We therefore asked whether induction of CaN and cytotoxicity induced by oligomeric Aβ in SY5Y cells were causally linked. Figure 4A shows the result from an LDH release assay in SY5Y cells treated with increasing concentrations of oligomeric Aβ in the presence of the CaN inhibitor FK506 or Rapamycin, an FK506 immunosuppressant analog that does not inhibit CaN (Abraham and Wiederrecht, 1996) and that was thus used here as control treatment. We found that FK506 completely abolished LDH release induced by oligomeric Aβ in SY5Y cells whereas Rapamycin was ineffective. This result was consistent with the differential effect of Rapamycin and FK506 in blocking the oligomeric Aβ-induced increase of CaN activity in SY5Y cells (Fig. 4B).
Figure 3. Oligomeric Aβ induces BAD dephosphorylation in SY5Y cells.
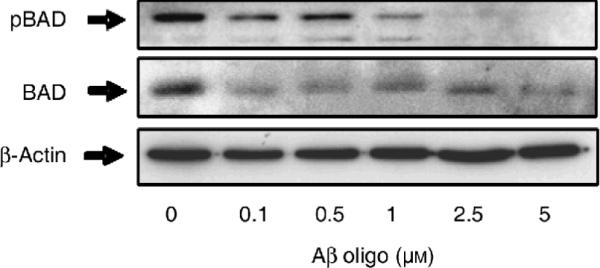
Representative Western blot detecting phosphorylated (pSer112) BAD in total protein extracts from SY5Y cells treated for 12 hr with increasing concentrations of oligomeric Aβ as shown. The blot was stripped and re-probed for total BAD (middle) and β-actin (bottom) to control for equal protein loading in each lane.
Figure 4. The CaN inhibitor FK506, but not its non CaN-inhibiting analog rapamycin, prevents cell death induced by oligomeric Aβ in SY5Y cells.

(A) LDH release in the culture medium of SY5Y neuroblastoma cells treated for 12 hr with oligomeric Aβ at the concentrations shown. Cells were additionally treated with FK506 (10 μM) or rapamycin (10 μM).
(B) CaN activity in SY5Y cells treated for 12 hr with 2.5 μM oligomeric Aβ in the presence or absence of 10 μM FK506 or 10 μM rapamycin.
N=3–6 replicates per group; *: p<0.01 vs. control cells (ANOVA). No effect of Aβ, FK506 or rapamycin was observed on the combined PP1/PP-2A activity (not shown).
CaN negatively regulates phosphorylative activation of CREB, a transcription factor essential for synaptic plasticity and LTP (Josselyn et al., 2004; Hotte et al., 2007), both of which are known to be perturbed by oligomeric Aβ (Chen et al., 2002; Walsh et al., 2002; Xie, 2004). Indeed, in SY5Y cells treated with increasing concentrations of oligomeric Aβ we found that CREB phosphorylation was dramatically reduced, with the minimum effective dose of Aβ oligomers being >0.5 μM (Fig. 5). As previously noted for total BAD (Fig. 3), also total CREB (but not βactin) levels were decreased in cells treated with 5 μM oligomeric Aβ, further suggesting a selective toxicity in responsive cells. Oligomeric Aβ's negative regulation of phosphorylated CREB was mirrored by a reduction of pCREB-driven transcriptional activity. In these experiments (Fig. 6A), SY5Y cells were transiently transfected with a secreted alkaline phosphatase protein (SEAP) reporter gene vector construct driven by a CREB-sensitive promoter (pCRE-SEAP). Control cells were transfected with a similar construct driven however by a CREB-insensitive promoter (pTal-SEAP). All cells were additionally transfected with a second vector encoding renilla luciferase to control for homogeneous transfection efficiency among individual samples (not shown). Twenty-four hr after transfection cells were treated with 0.75 μM each of monomeric, oligomeric and fibrillar Aβ, in the presence or absence of the adenylate cyclase (AC) activator forskolin (10 μM), and the release of SEAP in the culture medium (reflecting CREB-driven transcription) assayed 12 hr thereafter (Fig. 6A). In basal condition, CRE-SEAP activity was noticeably, albeit not significantly, reduced by treatment of cells with oligomeric Aβ; this is not surprising if one considers that basal CRE-SEAP activity in SY5Y cells was very low and close to background values. No changes were observed in cells treated with monomeric or fibrillar Aβ. As expected, treatment of cells with the AC activator forskolin greatly induced PKA-promoted CREB transcriptional activity as reflected by increased SEAP release in the culture medium of CRE-SEAP-transfected SY5Y cells. Additional treatment of these cells with oligomeric Aβ significantly opposed the AC-stimulated release of SEAP; however, no effect on AC-stimulated SEAP release was induced by monomeric of fibrillar Aβ treatment. In any instance, treatment with monomeric, oligomeric, fibrillar Aβ or forskolin did not affect SEAP release from cells transfected with the CREB-insensitive SEAP reporter gene construct pTAL-SEAP. To determine whether induction of CaN may play a role in oligomeric Aβ-promoted decrease of active CREB, SY5Y cells were exposed to oligomeric Aβ in the presence or absence of the CaN inhibitor FK506. The results illustrated in Fig. 6B show that opposing effect of oligomeric Aβ on forskolin-induced CREB-driven transcription in cells transfected with the CRE-SEAP reporter gene was partially but significantly rescued by treatment of cells with FK506.
Figure 5. Oligomeric Aβ induces CREB dephosphorylation in SY5Y cells.
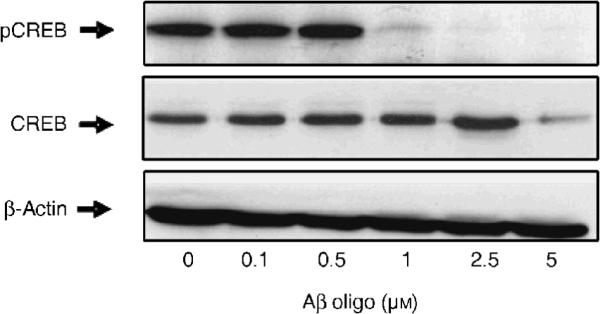
Representative Western blot detecting phosphorylated (pSer133) CREB in total protein extracts from SY5Y cells treated for 12 hr with increasing concentrations of oligomeric Aβ as shown. The blot was stripped and re-probed for total CREB (middle) and β-actin (bottom) to control for equal protein loading in each lane.
Figure 6. Oligomeric Aβ, but not monomeric or fibrillar Aβ, decreases CREB-driven transcriptional activity in SY5Y cells in a CaN-dependent fashion.

(A) SEAP activity assay in the culture medium of SY5Y cells transiently transfected with CREB-sensitive pCRE-SEAP or the CREB-insensitive pTAL-SEAP reporter gene and treated for 3 hr with 0.75 μM monomeric, oligomeric or fibrillar Aβ in the presence or absence of the adenilate cyclase activator forskolin (10 μM). N=4 independent replicates per group; *: p<0.01 vs. forskolin-treated vehicle cells (ANOVA).
(B) SEAP activity assay in the culture medium of SY5Y cells transiently transfected with pCRE-SEAP reporter gene and treated for 3 hr with 0.75 μM oligomeric Aβ in the presence of 10 μM forskolin (FRSK). Parallel cells were additionally treated with 1 or 5 μM FK506. N=3–4 independent replicates per group; *: p<0.05 vs. forskolin+oligomeric Aβ-treated cells (ANOVA).
While neuroblastoma cell lines have been used extensively and successfully as a model of CNS neural cells, it is prudent to confirm in neural tissue results obtained using transformed and/or proliferating cell systems. In order to achieve this, we employed an ex vivo model of acutely cultured rat brain slices and determined the effect of treatment with monomeric, oligomeric and fibrillar Aβ on CaN activity and CREB and BAD phosphorylation (Fig. 7). As with SY5Y cells, treatment of brain slices with oligomeric Aβ, but not monomeric or fibrillar Aβ, significantly increased CaN activity and this effect was expectedly prevented by the additional treatment with FK506 (Fig. 7A). When pCREB and pBAD levels were measured by Western blot in such brain slices (Fig. 7B), there was a significant reduction of both pCREB and pBAD induced by oligomeric Aβ, whereas monomeric and fibrillar Aβ were ineffective. This effect of oligomeric Aβ was reversed by treatment with FK506, thus indicating that reduced levels of pCREB and pBAD were likely dependent on oligomeric Aβ-induced CaN hyper-activation.
Figure 7. Oligomeric Aβ, but not monomeric or fibrillar Aβ, increases CaN activity and induces CaN-dependent decrease of pCREB and pBAD in rat brain slices.

Rat brain coronal slices (500 mm thick) were maintained in ACSF and treated with monomeric, oligomeric and fibrillar Aβ (0.4 μM) for 3 hrs. A separated set of oligomeric Aβ-treated slices were additionally treated with 10 μM FK506. At the end of the experiment, slices were collected and prepared for measurement of CaN activity (A) and detection of pBAD, BAD, pCREB and CREB by Western blot (B, top). Intensity of specific bands on western blot was analyzed and the result expressed as ratio between phosphorylated protein and the corresponding total protein in the same sample (B, bottom). The experiment was repeated twice with similar results. N=6 in (A) and n=4 (2 for each experiment) in (B). *: p<0.05 vs. vehicle (ANOVA)
Based on the results described above, we then asked whether a relationship between the presence of oligomeric Aβ species and reduced pCREB as a function of CaN activity could also be observed in vivo. We used an immunohistochemical approach to detect the presence of pCREB and Aβ oligomers in the hippocampus of 5 month old Tg2576 mice treated with FK506 or Rapamycin (Fig. 8A). Consistent with previously published evidence (Dineley et al., 2007), we found a robust reduction of immunoreactive pCREB in the hippocampus of Tg2576 mice as compared to wild type littermate controls. pCREB expression was predominantly neuronal as it co-localized with the neuron-specific antigen NeuN. Reduced pCREB was also coincidental with the presence of oligomeric Aβ, as revealed by co-staining of consecutive sections with the oligomer-specific antibody A-11 and NeuN. Western blot analysis on soluble protein extracts from the hippocampus of Tg2576 and WT mice (Fig. 8B) revealed that these Aβ oligomers were predominantly trimers, consistent with findings previously reported in Tg2576 mice of the same age (Lesne et al., 2006). Using the same pharmacological treatment of CaN inhibitor FK506 previously shown to normalize CaN hyper-activation and rescue cognitive deficits in Tg2576 mice (Dineley et al., 2007), effectively reversed the reduction of pCREB immunoreactivity but did not affect apparent levels of Aβ oligomers. Hippocampal pCREB immunoreactivity did not increase in Tg2576 mice treated with the non-CaN inhibiting FK506 analog Rapamycin. This indicates that the observed reduction of pCREB expression was dependent on CaN hyper-activity in these mice.
Figure 8. Inhibition of CaN rescues decreased pCREB immunoreactivity in the hippocampus of oligomeric Aβ-bearing Tg2576 mice.

(A) Representative photomicrographs of the CA3/Dentate Gyrus area from 5 month old wild type (WT) and Tg2576 littermate mice treated with FK506 (10 mg/kg ip) or Rapamycin (10 mg/kg ip) 6 hr prior to sacrifice. Consecutive sections were co-stained for pCREB and the neuron-specific antigen NeuN (top two rows) or for Aβ oligomers (using the oligomer-specific A-11 antibody) and NeuN (bottom two rows). (B) Representative Western blot detecting Aβ (using the sequence-specific antibody 6E10) in soluble protein-enriched fractions from the hippocampus of 5 month old Tg2576 and WT littermate mice. Arrows indicate the presence of secereted amyloid precursor protein alpha (sAPPa) and a trimeric Aβ oligomer (based on molecular weight) selectively in Tg2576 mice. Hippocampi from 3 animals were pooled together in each sample (6 mice per group). Three-hundred μg of protein were loaded onto each lane.
DISCUSSION
Employing both SY5Y human neuroblastoma cells and acutely cultured ex vivo rat brain slices, our results show that while oligomeric Aβ increased CaN activity, decreased BAD and CREB phosphorylation and induced cell death, monomeric or fibrillar Aβ were ineffective. All three structural forms of Aβ were obtained starting from human Aβ1–42 using established biochemistry methods that utilize identical buffers and solvents (Kayed et al., 2003). Therefore, it is unlikely that the observed effects elicited by oligomeric Aβ but not by monomeric or fibrillar Aβ are sequence-dependent or attributable to contaminants in the different Aβ preparations. Rather, these effects appear to be specifically associated with the oligomeric quaternary structure of Aβ. Indeed, our results show that pre-incubation of test compounds with the oligomeric structure-specific antibody A-11, which recognizes and neutralizes oligomeric Aβ but not monomeric or fibrillar Aβ (Kayed and Glabe, 2006), completely abolished the increase in CaN activity brought about by oligomeric Aβ in SY5Y cells.
Activation of CaN is dependent on calmodulin, a Ca2+-sensitive modulatory protein which is activated by increasing intracellular Ca2+ levels (Mansuy, 2003). It has been shown that one early event induced in SY5Y cells by oligomeric Aβ, but not monomeric or fibrillar Aβ, is the increase of intracellular Ca2+ via a permeabilization of the cell membrane (Demuro et al., 2005). We repeated these original observations by performing intracellular Ca2+ imaging in Fluo-3-loaded SY5Y cells treated with our preparations of monomeric, oligomeric and fibrillar Aβ and confirmed that under our experimental conditions only oligomeric Aβ induced intracellular Ca2+, whereas monomeric or fibrillar Aβ were ineffective (Fig. S1 in Supplementary Results). Thus, it is prudent to propose that activation of the Ca2+/calmodulin-dependent CaN and CaN-dependent CREB and BAD de-phosphorylation resulted from oligomeric Aβ-induced elevation of intracellular Ca2+ levels. To lend further support to this hypothesis, we exposed SY5Y cells to depolarizing concentrations of KCl (100 mM), which are known to induce increases in intracellular Ca2+ and CaN activity in these cells (Lee et al., 2005), and observed decreased pCREB and pBAD levels coincident with elevated CaN activity (Fig. S2 in Supplementary Results). In addition, in our experiments we did not observe any oligomeric Aβ- or KCl-induced increase of the combined activity of PP1/PP-2A (Fig. 2B and Fig. S2), which are two other phosphatases that, at variance with CaN, are Ca2+/calmudulin independent (Mansuy and Shenolikar, 2006).
Our current data do not allow us to argue as to the exact oligomeric species of Aβ that elicited the effects reported here. Our oligomeric preparation appears to contain dimers, trimers, dodecamers and possibly heavier oligomeric Aβ species (Fig. 1A) and therefore we currently do not know which of these species may be primarily responsible for increase of intracellular Ca2+ levels and activation of CaN activity and signaling in SY5Y cells and brain slices. As evidence indicates that Aβ dimers (Klyubin et al., 2008), trimers (Cleary et al., 2005; Townsend et al., 2006; Shankar et al., 2007) and dodecamers (Lesne et al., 2006) all exert similar effects on synaptic function and memory, it is tempting to speculate that these different oligomeric species share common mechanism of action involving activation of CaN. Further studies, however, are needed to clarify this issue.
In neurons, CaN has been reported to be involved in regulating both synaptic plasticity and cell death through modulation of two distinct pathways. Either directly or indirectly (through activation of PP-1), CaN promotes dephosphorylative inactivation of CREB, a transcription factor reportedly essential for long term potentiation (LTP), synaptic plasticity and memory function (Bito et al., 1996; Lin et al., 2003; Snyder et al., 2005). In addition, CaN promotes dephosphorylative activation of the pro-apoptotic Bcl-2 family member BAD, which is then released from binding to cytosolic 14-3-3 scaffolding proteins and translocates to the mitochondrion where it interacts with and negates the anti-apoptotic function of Bcl-2, thereby initiating a program of cell death (Chao and Korsmeyer, 1998; Wang et al., 1999; Springer et al., 2000; Yang et al., 2004). Indeed, pharmacological or transgenic inhibition of CaN has been reported to protect neurons from apoptotic cell death and to increase memory function (Wang et al., 1999; Mansuy, 2003). Our results show that in SY5Y cells and rat brain slice cultures, oligomeric Aβ decreased CREB phosphorylation and CREB-dependent transcriptional activity, decreased BAD phosphorylation and induced cell death, whereas monomeric or fibrillar Aβ were ineffective, consistent with their respective effects on CaN activity. Additional treatment with the CaN inhibitor FK506 abolished these effects of oligomeric Aβ, whereas treatment with rapamycin was ineffective. Both FK506 and rapamycin are macrolide immunosuppressants that act indirectly in a very similar fashion, initially by binding to the immunophyllin FKBP12. The FK506-FKBP12 complex binds to and inhibits CaN, thereby inhibiting IL-2 production and the subsequent immune cascade (Liu et al., 1991). Alternatively, the rapamycin-FKBP12 complex exerts action through the inhibition of mTOR, a kinase essential to mediate IL-2 action, thereby blocking the IL-2-mediated initiation of the immune cascade (Abraham and Wiederrecht, 1996). Therefore, any action exerted by FK506 but not by rapamycin is likely to be mediated by inhibition of CaN. We found this to be the case for FK506-induced reversal of the decrease of pCREB immunoreactivity in the CNS of Tg2576 mice (Fig. 8A) and FK506-promoted rescue of 5YSY cells from Aβ oligomer-induced cell death (Fig. 4A). More directly, we found that in SY5Y cells FK506 but not rapamycin inhibited the increase in CaN activity induced by oligomeric Aβ (Fig. 4B) and that in rat brain slices FK506 inhibited CaN hyper-activity and reversed the decrease in pCREB and pBAD elicited by Aβ oligomers (Fig. 7B). In addition, we found that oligomeric Aβ decreased CREB phosphorylation and CREB-driven transcription both in basal and forskolin-stimulated conditions. Forskolin is a pharmacological activator of AC that results in strong PKA-mediated phosphorylation/activation of CREB. Therefore, any decrease of CREB phosphorylation under forskolin-stimulated conditions is likely to results because of increased CREB dephosphorylation, an event directly or indirectly regulated by increased CaN activity (Bito et al., 1996; Lin et al., 2003).
In summary, our in vitro and ex vivo results indicate that in SY5Y human neuroblastoma cells and acutely cultured rat brain slices, oligomeric Aβ increases CaN activity to promote a dual signaling pathway, which can lead to either decreased CREB activity, or to BAD-promoted cell death, as graphically depicted in Fig. 9. According to this model, one could speculate that oligomeric Aβ can affect synaptic plasticity and memory independently of neuronal death, via a signaling pathway involving increased CaN activity and decreased pCREB. Indeed, our present in vivo results corroborate this hypothesis by showing co-localization of Aβ oligomers and CaN-dependent reduced pCREB levels in the hippocampus of cognitively impaired young-adult Tg2576 mice (Fig. 8). We further propose that as the impact of oligomeric Aβ is protracted in time and the neurons become dyshomeostatic, a second pathway leading to cell death, further contributes to deterioration of cognition. While the existence and relevance of this putative CaN-dependent dual pathway stimulated by oligomeric Aβ awaits further confirmation in vivo, several published in vivo and in vitro studies are consistent with its predictions. Transgenic mouse models of CNS hyper-amyloidosis develop cognitive impairments before the appearance of insoluble amyloid plaques and in the absence of overt neuronal death, but consistent with the presence of soluble Aβ species (Moechars et al., 1999; Oddo et al., 2003). Notably, we showed that PS1/APP doubly transgenic mice have elevated levels of neuroprotective Bcl-2 in their brains, an event consistent with the reported resistance of these mice to an Aβ-promoted neuronal death (Karlnoski et al., 2007). Other reports showed that a specific oligomeric Aβ species from the CNS of cognitively-impaired Tg2576 APP transgenic mice induces transient memory deficits when injected icv in young-adult rats (Lesne et al., 2006), whereas naturally occurring Aβ oligomers decrease synaptic plasticity in vivo (Walsh et al., 2002; Klyubin et al., 2008). Furthermore, Aβ oligomers in vitro decrease NMDA receptor trafficking (an event relevant to synaptic plasticity and memory) through a mechanism involving CaN activation (Snyder et al., 2005) and “aggregated” Aβ (consisting of a mixture of monomeric, oligomeric and fibrillar Aβ species) has been shown in vitro to depress LTP and induce neuronal cell death in a CaN-dependent fashion (Chen et al., 2002). Lastly, we have recently shown that memory deficits in amyloid plaque-free young-adult Tg2576 APP transgenic mice parallel increased CaN activity and elevated soluble Aβ levels in the CNS (Dineley et al., 2007). In the same study, we further showed that acute pharmacological normalization of elevated CaN activity restores memory in these mice without affecting CNS Aβ levels. Consistent with these findings, our present data show that a similar acute treatment with the CaN inhibitor FK506 increased pCREB levels in the hippocampus of Tg2576 mice without affecting the apparent levels of co-localized Aβ oligomers (Fig. 8). However, despite this compelling literature evidence, a causal link between the presence of Aβ oligomers and elevated CaN activity in the CNS still awaits ultimate confirmation, for example through in vivo immune-neutralization of Aβ oligomers and assessment of CaN activity levels in the CNS of APP transgenic mice. Such studies are currently ongoing in our laboratory and will hopefully clarify this issue.
Figure 9. A schematic illustrating the putative role of calcineurin in mediating both the dysfunctional synaptic and apoptotic consequences of oligomeric Aβ.

Oligomeric Aβ intermediates increase intacellular Ca2+ levels, possibly through cell membrane destabilization (Demuro et al., 2005). Increased intracellular Ca2+ promoted by oligomeric Aβ induces the activity of the Ca2+/calmodulin (CaM)-dependent phosphatase calcineurin. Active calcineurin dephosphorylates/inactivates CREB, thus leading to reversible impaired synaptic plasticity and decreased memory function; through prolonged exposure to oligomeric amyloids neurons become dyshomeostatic and eventually a CaN-dependent apoptotic pathway involving dephosphorylation/activation of the pro-apoptotic BAD is allowed to progress and contributes neurodegenerative-dependent cognitive deficits.
Up-regulation of CaN has been reported in the CNS of aged rats and found to correlate with the degree of age-related cognitive impairments (Foster et al., 2001). More recently, a similar dual pathway initiated by CaN and leading to synaptic plasticity/memory deficits and to neuronal death in response to intracellular Ca2+ dysregulation has been proposed in the aged brain (Foster, 2007). Furthermore, in preliminary experiments we found decreased levels of pCREB and pBAD in brain specimens from both AD patients and age-matched non-demented subjects, with some indication that in the former such reduction may be even more pronounced in neurons than non-neuronal brain cells (Supplementary Figure 3). It is therefore tempting to speculate that CaN hyper-activity may be a dysfunctional event induced by oligomeric Aβ and shared by the CNS aging process, thus providing one possible mechanism leading to the exacerbated impact displayed by Aβ in the ageing brain.
In conclusions, the data presented here illustrate molecular events indicating that CaN may play a central role in mediating the impact on neural cells of oligomeric Aβ, the Aβ species that reportedly is a prime offender in AD. Whether CaN may represent a useful target to both ameliorate synaptic plasticity/memory and decrease neuronal death, two seemingly independent and yet fundamental events characterizing the devastating impact of Aβ in the ageing AD brain, remains to be established.
EXPERIMENTAL PROCEDURES
Antibodies
Antibodies against CREB, p(Ser133)CREB, BAD and p(Ser112)BAD were from Cell Signaling Technology, Boston, MA. The anti-oligomer A-11 antibody was produced as previously described (Kayed and Glabe, 2006) and the anti-Aβ antibody 6E10 was from Convence, France.
Cell cultures
Human neuroblastoma SY5Y cells were from American Type Culture Collection (ATCC, Manassas, VA). Stock cell cultures were maintained at 37°C in a humidified cell incubator under 5% CO2 atmosphere in T-125 culture flasks containing 10 ml of DMEM/F12 supplemented with 10% heat-inactivated fetal bovine serum (FBS, Sigma Chem., St. Louis, MO). Half of the medium was replaced every two days and at confluence cells were dislodged by vigorous shaking and seeded into two new flasks.
Preparation of Aβ
Human Aβ1–42 peptide was purchased from Keck, Yale University, New Heaven, CT. Monomeric, oligomeric and fibrillar Aβ to be employed in the same experiment were always prepared from aliquots of the same batch of Aβ. To prepare oligomeric Aβ, lyophilized Aβ aliquots (0.3 mg) were dissolved in 0.2 ml of 1,1,1,3,3,3-Hexafluoro-2-propanol (HFP) and then added to 0.7 ml of H2O. Samples were loosely capped and stirred on a magnetic stirrer under a fume hood for 48 hr and then used within 36 hr. Fibrillar Aβ was prepared following the same procedure as for oligomeric Aβ, except that the samples were stirred tightly capped for 7 days and then loosely capped for 48 hr. Monomeric Aβ was prepared immediately before use by rapidly evaporating the HFP via gently bubbling nitrogen gas in the solution. Quality of Aβ preparations so obtained were routinely checked by dot-blot and western blot employing both A-11 and 6E10 antibodies. Occasionally, the structural appearance of oligomeric and fibrillar Aβ preparations was checked by electron microscopy to visually exclude the presence of contaminant fibrils or oligomers, respectively.
The final concentration of the oligomeric and fibrillar Aβ preparation was nominally calculated based on the concentration of the starting Aβ monomer. In all of our experiments we always observe effects in SY5Y cells for concentrations of such prepared Aβ oligomers >0.5 μM (>0.1 μM in rat brain slices). The timing and dosage for each experiment reported here were carefully chosen based on preliminary experiments so as to maximize the efficiency of the measurement of the signaling element studied (e.g., CaN activity, pCREB, pBAD) while reducing any biasing effect due to oligomeric Aβ-induced cell death (which in our hands occurs substantially in SY5Y cells at >12–16 hrs treatment time for a dosage of Aβ of 1–2.5 μM).
LDH cytotoxicity assay
The release of lactate dehydrogenase (LDH) from cultured cells was assayed in the culture medium employing a commercially available kit (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions.
Phosphatase activity assay. The activity of CaN (PP-2B) and combined activity of PP1/PP2A were assayed in cytosolic cell extracts using a commercially available colorimetric kit (EMD Biosciences, San Diego, CA) employing R2 phosphopeptide as substrate according to the manufacturer's instructions.
Western blot
Cells were lysed in SDS lysis buffer containing 5mM EDTA, 50mM Tris, 2% SDS, 1mM DTT, 1mM PMSF and 1% protease cocktail inhibitors (Sigma, St. Louis, MO). Following lysis, cells were sonicated for 15 seconds, and then centrifuged at 20,000g for 5 minutes and the supernatants collected. Protein samples containing 30 μg of proteins as determined by the BCA assay (Pierce, Rcokford, IL), were subjected to SDS-PAGE using 12% gels, followed by electrophoretic transfer to nitrocellulose backed membranes (Biorad, Hercules, CA). Membranes were then incubated with appropriate dilutions (usually 1:1,000 v:v) of the primary antibody and this initial incubation was then followed by a horseradish peroxidase (HRP) conjugated secondary antibody (Biorad, Hercules, CA) against rabbit IgG (for polyclonal primaries) or mouse IgG (for monoclonal primaries). Detection was achieved using ECL (Pierce, Rockford, IL).
Cell transfection and SEAP assay
All transfections were performed using a liposomal mediated plasmid introduction. The liposome used was DMRIE-C (Invitrogen, Carlsbad, CA). Cells at 40 to 50% confluence received a total of 1.2 pmoles/ml of DNA coupled to DMRIE-C at a ratio of 1:3 and diluted in OptiMEM (Invitrogen, Carlsbad, CA). 3 hours post transfection, the liposome/DNA mix was replaced with fresh culture medium and the cells were allowed at least 48 hours of recovery prior to use.
The secreted alkaline phosphatase assay (SEAP) (Clontech, Palo Alto, CA) was used as a measure of transcription factor activity. Plasmids containing the SEAP gene only (pTAL-SEAP) or the SEAP gene downstream of enhancer sequences specific to binding CRE (CRE-SEAP) were transiently transfected as described above. SEAP activity was assayed directly from the culture medium using the Great Escape Chemiluminescent Detection Kit according to manufacturer's instruction (Clontech, Palo Alto, CA). SEAP levels are a direct measurement of the effect of test compounds on the enhancer activity.
Animals
For brain slice preparations, male Sprague Dawley rats (150–250 g) were used, while Tg2576 and wild type littermates were bred in the UTMB animal care facility. Animals were maintained under USDA standards (12 hr light/dark cycle, food and water ad libitum) according to IACUC-approved protocols. For mouse breeding protocols, dams were C6/SJL F1 females purchased from Jackson Laboratories and sires were Tg2576 (C6/SJL) males from our colony. Both males and female mice at 5 months of age were used for experimentation.
Brain slice preparation
According to IACUC-approved protocols, rats were decapitated and brains quickly removed and blocked in cold (4°C) artificial CSF (ACSF) prepared as follows: 117 mM NaCl, 4.7 mM KCl, 1.2 mM Na2PO4, 2.5 mM CaCl2, 1.2 mM MgCl2, 25 mM NaHCO3, 11 mM glucose, oxygenated and equilibrated to pH 7.4 with 95% O2/5% CO2. Five-hundred μM tick coronal brain slices comprising the hippocampus were prepared using a Vibroslice (Camden Instruments, London, UK) and equilibrated in ACSF at room temperature for at least 30 min prior to adding test compounds. At the end of the experiments slices were snap frozen in liquid nitrogen and stored at −80°C until further analyses were performed.
Immunohistochemistry
According to IACUC-approved animal protocols and procedures, mice were deeply anesthetized with ketamine/xylazine and perfused transcardially with 0.9% saline followed by 4% paraformaldehyde. Brains were carefully removed, postfixed in 4% paraformaldehyde and then transferred in 30% sucrose overnight at 4°C prior to freezing for storage at −80°C. For immunohistochemistry staining using a free-floating technique, thirty μm-thick sections were cut using a cryostat, washed with 0.1 M PBS and blocked for 40 min in 5% NGST/BSA (5% normal goat serum in 0.1 M PBS containing 0.05% Triton X-100 and 0.3% Bovine Serum Albumin) followed by incubation with primary antibodies in 1% NGST/BSA overnight at room temperature. Antibodies used were: rabbit anti-pCREB (Cell Signaling Tech., Danvers, MA), rabbit anti-oligomer A-11 [produced in Dr. Kayed's laboratory as previously described (Kayed and Glabe, 2006)], mouse anti-NeuN (Chemicon, Temecula, CA). Section were then washed with PBS and incubated with the appropriate secondary antibody (goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 568, Molecular Probes, Carlsbad, CA) for 1 hr at room temperature. After incubation with the secondary antibodies, section were washed three times with PBS, mounted onto microscope slides and observed with a fluorescence microscope equipped with computer-assisted CCD camera for image capturing and processing. Microscope intensity level settings of fluorescence image capturing (for both A-11 and anti NeuN antibodies) were optimized on sections from wt mice and then the microscope settings maintained constant for all sections observed (wt and Tg2576 mice). Four rostral to caudal sections comprising the hippocampus were observed for each animal (n=3 mice per group) and compared within and across treatment groups with similar results.
Supplementary Material
ACNOWLEDGEMENTS
The authors wish to that Drs. Volker Neugebauer and Yu Fu of UTMB at Galveston, TX, for providing expertise and valuable help in preparing acute organotypic rat brain slice cultures. This work was supported by NIH/NINDS grant NS053986 and by the Alzheimer Association grant IIRG 90755 to GT; LCR is a recipient of a predoctoral scholarship from the NIH/NIEHS Environmental Toxicology Training Program T32ES007254-16 and currently supported by a predoctoral F31 award NS062558 from NIH/NINDS.
REFERENCES
- Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol. 1996;14:483–510. doi: 10.1146/annurev.immunol.14.1.483. [DOI] [PubMed] [Google Scholar]
- Agostinho P, Oliveira CR. Involvement of calcineurin in the neurotoxic effects induced by amyloid-beta and prion peptides. Eur J Neurosci. 2003;17:1189–1196. doi: 10.1046/j.1460-9568.2003.02546.x. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Cochran EJ, Saper CB, Leverenz JB, Gilley DW, Wilson RS. Pathological changes in frontal cortex from biopsy to autopsy in Alzheimer's disease. Neurobiol Aging. 1993;14:589–596. doi: 10.1016/0197-4580(93)90043-b. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Brion JP, Couck AM, Conreur JL. Calcineurin (phosphatase 2B) is present in neurons containing neurofibrillary tangles and in a subset of senile plaques in Alzheimer's disease. Neurodegeneration. 1995;4:13–21. doi: 10.1006/neur.1995.0002. [DOI] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annual Review of Immunology. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002;77:354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-protein specifically disrupt cognitive function. Nature Neuroscience. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007 doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007;6:319–325. doi: 10.1111/j.1474-9726.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- Foster TC, Sharrow KM, Masse JR, Norris CM, Kumar A. Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci. 2001;21:4066–4073. doi: 10.1523/JNEUROSCI.21-11-04066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- Golde TE. The A beta hypothesis: Leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathology. 2005;15:84–87. doi: 10.1111/j.1750-3639.2005.tb00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O'Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol. 2002;173:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- Groth RD, Dunbar RL, Mermelstein PG. Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun. 2003;311:1159–1171. doi: 10.1016/j.bbrc.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hata R, Masumura M, Akatsu H, Li F, Fujita H, Nagai Y, Yamamoto T, Okada H, Kosaka K, Sakanaka M, Sawada T. Up-regulation of calcineurin Abeta mRNA in the Alzheimer's disease brain: assessment by cDNA microarray. Biochem Biophys Res Commun. 2001;284:310–316. doi: 10.1006/bbrc.2001.4968. [DOI] [PubMed] [Google Scholar]
- Hotte M, Thuault S, Dineley KT, Hemmings HC, Nairn AC, Jay TM. Phosphorylation of CREB and DARPP-32 during late LTP at hippocampal to prefrontal cortex synapses in vivo. Synapse. 2007;61:24–28. doi: 10.1002/syn.20339. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Kida S, Silva AJ. Inducible repression of CREB function disrupts amygdala-dependent memory. Neurobiol Learn Mem. 2004;82:159–163. doi: 10.1016/j.nlm.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Karlnoski R, Wilcock D, Dickey C, Ronan V, Gordon MN, Zhang W, Morgan D, Taglialatela G. Up-regulation of Bcl-2 in APP transgenic mice is associated with neuroprotection. Neurobiol Dis. 2007;25:179–188. doi: 10.1016/j.nbd.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Glabe CG. Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol. 2006;413:326–344. doi: 10.1016/S0076-6879(06)13017-7. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- Lee YI, Seo M, Kim Y, Kim SY, Kang UG, Kim YS, Juhnn YS. Membrane depolarization induces the undulating phosphorylation/dephosphorylation of glycogen synthase kinase 3beta, and this dephosphorylation involves protein phosphatases 2A and 2B in SH-SY5Y human neuroblastoma cells. J Biol Chem. 2005;280:22044–22052. doi: 10.1074/jbc.M413987200. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu HY, Gean PW. The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J Neurosci. 2003;23:8310–8317. doi: 10.1523/JNEUROSCI.23-23-08310.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Mansuy IM. Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun. 2003;311:1195–1208. doi: 10.1016/j.bbrc.2003.10.046. [DOI] [PubMed] [Google Scholar]
- Mansuy IM, Shenolikar S. Protein serine/threonine phosphatases in neuronal plasticity and disorders of learning and memory. Trends Neurosci. 2006;29:679–686. doi: 10.1016/j.tins.2006.10.004. [DOI] [PubMed] [Google Scholar]
- McKee AC, Kosik KS, Kowall NW. Neuritic pathology and dementia in Alzheimer's disease. Ann Neurol. 1991;30:156–165. doi: 10.1002/ana.410300206. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Nottingham SA, Kennedy SE. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J Neurosci. 2000;20:7246–7251. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen G, Annaert W. Amyloid, presenilins, and Alzheimer's disease. Neuroscientist. 2003;9:117–126. doi: 10.1177/1073858403252227. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Xie CW. Calcium-regulated signaling pathways: role in amyloid beta-induced synaptic dysfunction. Neuromolecular Med. 2004;6:53–64. doi: 10.1385/NMM:6:1:053. [DOI] [PubMed] [Google Scholar]
- Yang L, Omori K, Suzukawa J, Inagaki C. Calcineurin-mediated BAD Ser155 dephosphorylation in ammonia-induced apoptosis of cultured rat hippocampal neurons. Neurosci Lett. 2004;357:73–75. doi: 10.1016/j.neulet.2003.12.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.