

The crystal structure of a putative NTP pyrophosphohydrolase, YP_001813558.1 from E. sibiricum, reveals a novel segment-swapped linked-dimer assembly.
Keywords: structural genomics, putative NTP pyrophosphohydrolase, MazG nucleotide pyrophosphohydrolase, dUTPases
Abstract
The crystal structure of a putative NTPase, YP_001813558.1 from Exiguobacterium sibiricum 255-15 (PF09934, DUF2166) was determined to 1.78 Å resolution. YP_001813558.1 and its homologs (dimeric dUTPases, MazG proteins and HisE-encoded phosphoribosyl ATP pyrophosphohydrolases) form a superfamily of all-α-helical NTP pyrophosphatases. In dimeric dUTPase-like proteins, a central four-helix bundle forms the active site. However, in YP_001813558.1, an unexpected intertwined swapping of two of the helices that compose the conserved helix bundle results in a ‘linked dimer’ that has not previously been observed for this family. Interestingly, despite this novel mode of dimerization, the metal-binding site for divalent cations, such as magnesium, that are essential for NTPase activity is still conserved. Furthermore, the active-site residues that are involved in sugar binding of the NTPs are also conserved when compared with other α-helical NTPases, but those that recognize the nucleotide bases are not conserved, suggesting a different substrate specificity.
1. Introduction
Nucleoside triphosphate pyrophosphatases (or pyrophosphohydrolases; NTPases) perform the important function of hydrolyzing the α–β phosphodiester bond of nucleoside triphosphates (NTPs) and are often involved in removing noncanonical nucleotide triphosphates to prevent their incorporation into DNA or RNA (Bessman et al., 1996 ▶; Wu et al., 2007 ▶; Hwang et al., 1999 ▶; Minasov et al., 2000 ▶). dUTP pyrophosphatase (dUTPase; EC 3.6.1.23) catalyzes the hydrolysis of dUTP to dUMP and pyrophosphate. The available dUTPase structures are classified into three distinct groups based on their oligomeric state: trimeric, dimeric and monomeric. The crystal structures of trimeric dUTPases from Escherichia coli (Cedergren-Zeppezauer et al., 1992 ▶; Larsson et al., 1996 ▶), human (Mol et al., 1996 ▶) and two mammalian retroviruses (Prasad et al., 1996 ▶; Dauter et al., 1999 ▶) possess an all-β fold. Monomeric dUTPases contain all five of the characteristic sequence motifs present in trimeric dUTPases, but they are arranged in a different order. The monomeric enzyme from Epstein–Barr virus (EVB; Tarbouriech et al., 2005 ▶) also adopts an all-β fold and contains three domains and an active site that is very similar to those of trimeric dUTPases. Dimeric dUTPases, such as those from Trypanosoma cruzi (Harkiolaki et al., 2004 ▶) and Campylobacter jejuni (Moroz et al., 2004 ▶), differ from the monomeric and trimeric forms and adopt an all-α topology, indicating a different evolutionary origin.
Dimeric dUTPase and MazG proteins are members of the all-α-helical NTP pyrophosphatase SCOP superfamily (Murzin et al., 1995 ▶; Andreeva et al., 2008 ▶), which also contains the HisE-encoded phosphoribosyl ATP pyrophosphohydolase (PRATP-PH) family (Moroz et al., 2005 ▶; Javid-Majd et al., 2008 ▶). The α-helical NTP pyrophosphatases share a highly conserved four-helix bundle, one face of which forms the active site, while the other participates in oligomer assembly (Harkiolaki et al., 2004 ▶; Moroz et al., 2004 ▶). In some cases, the four-helix bundle forms upon dimerization (Harkiolaki et al., 2004 ▶) while, in others, it is contained within a single protomer (Moroz et al., 2004 ▶).
Here, we report the crystal structure of NTPase YP_001813558.1 from the extremophile Exiguobacterium sibiricum 255-15 (PF09934, DUF2166), which was originally isolated from the Siberian permafrost (Vishnivetskaya et al., 2000 ▶). The structure reveals an interesting variant of the all-α-helical NTP pyrophosphatase fold family that contains an unusual intertwined swapping of helical segments, resulting in an obligatory dimer that cannot dissociate without unfolding of the monomers. This novel ‘linked dimer’ defines a new subfamily of the α-helical NTP pyrophosphatase fold and is distinct from other previously observed domain-swapped dimers. The YP_001813558.1 gene of E. sibiricum 255-15 encodes a protein with a molecular weight of 19.1 kDa (residues 2–170) and a calculated isoelectric point of 4.93. The structure was determined using the semiautomated high-throughput pipeline of the Joint Center for Structural Genomics (JCSG; Lesley et al., 2002 ▶) as part of the NIGMS Protein Structure Initiative (PSI).
2. Materials and methods
2.1. Protein production and crystallization
Clones were generated using the Polymerase Incomplete Primer Extension (PIPE) cloning method (Klock et al., 2008 ▶). The gene encoding YP_001813558.1 (gi|172057098; UniProt B1YMF4) was amplified by polymerase chain reaction (PCR) from E. sibiricum 255-15 genomic DNA using PfuTurbo DNA polymerase (Stratagene) and I-PIPE (Insert) primers (forward primer, 5′-ctgtacttccagggcATGAAACAACCGAACTACTATCAGGACG-3′; reverse primer, 5′-aattaagtcgcgttaTGCTTTTTCTTTCATTTGGCGCACTAC-3′; target sequence in upper case) that included sequences for the predicted 5′ and 3′ ends. The expression vector pSpeedET, which encodes an amino-terminal tobacco etch virus (TEV) protease-cleavable expression and purification tag (MGSDKIHHHHHHENLYFQ/G), was PCR-amplified with V-PIPE (Vector) primers (forward primer, 5′-taacgcgacttaattaactcgtttaaacggtctccagc-3′; reverse primer, 5′-gccctggaagtacaggttttcgtgatgatgatgatgatg-3′). The V-PIPE and I-PIPE PCR products were mixed to anneal the amplified DNA fragments together. Escherichia coli GeneHogs (Invitrogen) competent cells were transformed with the V-PIPE/I-PIPE mixture and dispensed onto selective LB–agar plates. The cloning junctions were confirmed by DNA sequencing. Expression was performed in a selenomethionine-containing medium with suppression of normal methionine synthesis (Van Duyne et al., 1993 ▶). At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 µg ml−1 and the cells were harvested and frozen. After one freeze–thaw cycle, the cells were homogenized in lysis buffer [50 mM HEPES pH 8.0, 50 mM NaCl, 10 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine–HCl (TCEP)] and the lysate was clarified by centrifugation at 32 500g for 30 min. The soluble fraction was passed over nickel-chelating resin (GE Healthcare) pre-equilibrated with lysis buffer, the resin was washed with wash buffer [50 mM HEPES pH 8.0, 300 mM NaCl, 40 mM imidazole, 10%(v/v) glycerol, 1 mM TCEP] and the protein was eluted with elution buffer [20 mM HEPES pH 8.0, 300 mM imidazole, 10%(v/v) glycerol, 1 mM TCEP]. The eluate was buffer-exchanged with TEV buffer (20 mM HEPES pH 8.0, 200 mM NaCl, 40 mM imidazole, 1 mM TCEP) using a PD-10 column (GE Healthcare) and incubated with 1 mg TEV protease per 15 mg eluted protein for 2 h at 295 K and 18 h at 277 K. The protease-treated eluate was run over nickel-chelating resin (GE Healthcare) pre-equilibrated with HEPES crystallization buffer (20 mM HEPES pH 8.0, 200 mM NaCl, 40 mM imidazole, 1 mM TCEP) and the resin was washed with the same buffer. The flowthrough and wash fractions were combined and concentrated to 16.5 mg ml−1 by centrifugal ultrafiltration (Millipore) for crystallization trials. YP_001813558.1 was crystallized using the nanodroplet vapor-diffusion method (Santarsiero et al., 2002 ▶) with standard JCSG crystallization protocols (Lesley et al., 2002 ▶). Sitting drops composed of 200 nl protein mixed with 200 nl crystallization solution were equilibrated against a 50 µl reservoir at 277 K for 29 d prior to harvest. The crystallization reagent that produced the YP_001813558.1 crystal used for structure solution consisting of 1.4 M trisodium citrate and 0.1 M HEPES pH 7.5. For crystal diffraction screening and data collection, 1,2-ethanediol (ethylene glycol) was diluted to 20%(v/v) using reservoir solution and then added to the crystal drop in a 1:1 ratio as a cryoprotectant. Initial screening for diffraction was carried out using the Stanford Automated Mounting (SAM; Cohen et al., 2002 ▶) system and an X-ray microsource (Miller & Deacon, 2007 ▶) installed at the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, California, USA). The data were indexed in the monoclinic space group C2. The oligomeric state of YP_001813558.1 was determined using a 1 × 30 cm Superdex 200 column (GE Healthcare) coupled with miniDAWN static light-scattering (SEC/SLS) and Optilab differential refractive-index detectors (Wyatt Technology). The mobile phase consisted of 20 mM Tris pH 8.0, 150 mM NaCl and 0.02%(w/v) sodium azide. The molecular weight was calculated using ASTRA v.5.1.5 software (Wyatt Technology).
2.2. Data collection, structure solution and refinement
Multiple-wavelength anomalous diffraction (MAD) data at wavelengths corresponding to the low-energy remote (λ1) and inflection point (λ2) of a selenium MAD experiment were collected on beamline 8.2.2 at Advanced Light Source (ALS, Berkeley, California, USA). The data were collected at 100 K using an ADSC Q315 CCD detector. Collection of the two wavelengths was interleaved using a 10° wedge size. The MAD data were integrated and reduced using MOSFLM (Leslie, 1992 ▶) and scaled with the program SCALA (Collaborative Computational Project, Number 4, 1994 ▶). The diffraction data were anisotropic, with a faster falloff along a*. The selenium substructure solution, phasing and density modification were performed with SHELXD (Sheldrick, 2008 ▶) and autoSHARP (Vonrhein et al., 2007 ▶), resulting in a mean figure of merit of 0.30 with four selenium sites. Automatic model building was performed with ARP/wARP (Cohen et al., 2004 ▶), which traced and built side chains for 161 residues (94% of the structure). Model adjustments and completion were performed with Coot (Emsley & Cowtan, 2004 ▶). Structure refinement was carried out using REFMAC v.5.5.0110 and included one TLS group and experimental phase restraints in the form of Hendrickson–Lattman coefficients from SHARP (Pannu et al., 1998 ▶; Winn et al., 2003 ▶). Data-reduction and refinement statistics for YP_001813558.1 are summarized in Table 1 ▶.
Table 1. Summary of crystal parameters, data-collection and refinement statistics for YP_001813558.1 (PDB code 3nl9).
Values in parentheses are for the highest resolution shell.
| λ1 MADSe | λ2 MADSe | |
|---|---|---|
| Crystal parameters | ||
| Space group | C2 | |
| Unit-cell parameters (Å, °) | a = 52.09, b = 69.04, c = 50.21, β = 111.8 | |
| Mosaicity (°) | 0.91 | |
| Data collection | ||
| Wavelength (Å) | 1.0000 | 0.9798 |
| Resolution range (Å) | 39.6–1.78 (1.83–1.78) | 39.6–1.78 (1.83–1.78) |
| No. of observations | 43073 | 43110 |
| No. of unique reflections | 15531 | 15528 |
| Completeness (%) | 98.1 (97.9) | 98.1 (97.3) |
| Mean I/σ(I) | 9.8 (2.1) | 8.6 (1.8) |
| Rmerge on I† | 0.069 (0.555) | 0.082 (0.563) |
| Rmeas on I‡ | 0.086 (0.687) | 0.102 (0.698) |
| Rp.i.m. on I§ | 0.050 (0.401) | 0.059 (0.408) |
| Overall B factor from Wilson plot (Å2) | 21.3 | 21.0 |
| Model and refinement statistics | ||
| Data set used in refinement | λ1 MADSe | |
| Resolution range (Å) | 39.6–1.78 | |
| No. of reflections (total) | 15531 | |
| No. of reflections (test) | 788 | |
| Completeness (%) | 97.8 | |
| Cutoff criterion | |F| > 0 | |
| Rcryst¶ | 0.177 | |
| Rfree†† | 0.222 | |
| Stereochemical parameters | ||
| Restraints (r.m.s.d. observed) | ||
| Bond angles (°) | 1.30 | |
| Bond lengths (Å) | 0.015 | |
| Average protein isotropic B value (Å2) | 26.0‡‡ | |
| Average solvent isotropic B value (Å2) | 33.6 | |
| ESU§§ based on Rfree (Å) | 0.14 | |
| Protein residues/atoms | 169/1340 | |
| Water/cryoprotectant molecules | 141/2 | |
R
merge = 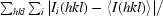
 .
.
The redundancy-independent (multiplicity-weighted) merging R factor, R
meas = 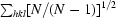
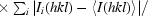 (Diederichs & Karplus, 1997 ▶).
(Diederichs & Karplus, 1997 ▶).
The precision-indicating merging R factor, R
p.i.m. = 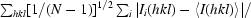 (Weiss & Hilgenfeld, 1997 ▶; Weiss et al., 1998 ▶).
(Weiss & Hilgenfeld, 1997 ▶; Weiss et al., 1998 ▶).
R
cryst = 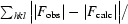
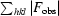 , where F
calc and F
obs are the calculated and observed structure-factor amplitudes, respectively,
, where F
calc and F
obs are the calculated and observed structure-factor amplitudes, respectively,
R free is the same as R cryst but for 5.1% of the total reflections chosen at random and omitted from refinement
This value represents the total B and includes both TLS and residual B components.
2.3. Validation and deposition
The quality of the crystal structure was analyzed using the JCSG Quality Control server (http://smb.slac.stanford.edu/jcsg/QC). This server processes the coordinates and data through a variety of validation tools including AutoDepInputTool (Yang et al., 2004 ▶), MolProbity (Chen et al., 2010 ▶), WHAT IF v.5.0 (Vriend, 1990 ▶), RESOLVE (Terwilliger, 2003 ▶), MOLEMAN2 (Kleywegt, 2000 ▶) as well as several in-house scripts and summarizes the results. Protein quaternary structure analysis used the PISA server (Krissinel & Henrick, 2007 ▶). Fig. 1 ▶(c) was adapted from an analysis using PDBsum (Laskowski et al., 2005 ▶); all others were prepared with PyMOL (DeLano Scientific). Atomic coordinates and experimental structure factors for YP_001813558.1 have been deposited in the PDB (PDB code 3nl9).
Figure 1.

Crystal structure of YP_001813558.1 from E. sibiricum 255-15. (a) Ribbon diagram of the YP_001813558.1 protomer in the asymmetric unit, color-coded from the N-terminus (blue) to the C-terminus (red). Helices H1–H6 are indicated. (b) The novel dimeric assembly of YP_001813558.1 generated by helical segment swapping. Green and red tracings represent chain A and the symmetry-related chain A′ that form the dimer. The N- and C-termini are labeled. (c) Diagram showing the secondary-structure elements of YP_001813558.1 superimposed on its primary sequence. The α-helices (H1–H6), β-turns (β) and γ-turn (γ) are indicated.
3. Results and discussion
3.1. Overall structure
The crystal structure of YP_001813558.1 was determined to 1.78 Å resolution using the MAD method (Fig. 1 ▶ a). Data-collection, model and refinement statistics for the YP_001813558.1 structure are summarized in Table 1 ▶. The asymmetric unit contains one YP_001813558.1 molecule (residues 2–170), i.e. one half of the linked crystallographic dimer (Fig. 1b ▶), two 1,2-ethanediol molecules and 141 water molecules. Residues Gly0 (remaining after cleavage of the expression and purification tag) and SeMet1 and side-chain atoms of Lys2, Gln72, Lys76, Lys79, Glu140 and Ser141 had poorly defined or no electron density and were omitted from the model. The Matthews coefficient (V M) for YP_001813558.1 was 2.2 Å3 Da−1, with an estimated solvent content of 44.0% (Matthews, 1968 ▶). The Ramachandran plot produced by MolProbity (Chen et al., 2010 ▶) indicated that 98.8% of the residues are in favored regions, with no outliers. YP_001813558.1 is an all-α structure containing six α-helices (H1–H6, Fig. 1 ▶ a), with a total α-helical content of 64.5%.
PSI-BLAST (Altschul et al., 1997 ▶) and FFAS (Jaroszewski et al., 2000 ▶) searches both detected similarities between YP_001813558.1 and other NTPases, such as the PRATP-PH family NTPases [PDB codes 1yvw (J. Benach, A. P. Kuzin, F. Forouhar, M. Abashidze, S. M. Vorobiev, R. Shastry, X. Rong, T. B. Acton, G. T. Montelione & J. F. Hunt, unpublished work), 2a7w (J. Benach, F. Forouhar, A. P. Kuzin, M. Abashidze, S. M. Vorobiev, X. Rong, T. B. Acton, G. T. Montelione & J. F. Hunt, unpublished work), 1y6x (Javid-Majd et al., 2008 ▶), 1yxb (J. Benach, A. P. Kuzin, F. Forouhar, M. Abashidze, S. M. Vorobiev, X. Rong, T. B. Acton, G. T. Montelione & J. F. Hunt, unpublished work)], MazG NTPases [PDB codes 1vmg (Joint Center for Structural Genomics, unpublished work), 2a3q (Center for Eukaryotic Structural Genomics, unpublished work), 2q5z and 2q73 (Robinson et al., 2007 ▶)], Bacillus subtilis NTPase YPJD (PDB code 2gta; S. M. Vorobiev, W. Zhou, J. Seetharaman, D. Wang, L. C. Ma, T. Acton, R. Xio, G. T. Montelione, L. Tong & J. F. Hunt, unpublished work) and the RS21-C6 core segment RSCUT, which has been reported to have NTPase activity (PDB code 2oie; Wu et al., 2007 ▶). Superimposition of these structures onto the YP_001813558.1 crystallographic dimer shows that the general topology of the four-helix bundle is conserved; for example, the equivalent secondary elements of PRATP-PH from B. cereus (PDB code 1yvw) can be aligned with an r.m.s.d. of 2.5 Å (for 81 of 90 Cα atoms). Similarly, MazG from Sulfolobus solfataricus (PDB code 1vmg) can be superimposed onto YP_001813558.1 with an r.m.s.d. of 2.3 Å for 77 of 80 Cα atoms. MazG from E. coli can hydrolyze all eight of the canonical ribonucleoside and deoxynucleoside triphosphates to their respective monophosphates and PPi, with a preference for deoxynucleotides (Zhang & Inouye, 2002 ▶). YP_001813558.1 (170 residues) is significantly larger than MazG (PDB code 1vmg; 83 residues) and PRATP-PH (PDB code 1yvw; 95 residues) primarily owing to the presence of two additional helices, H3 located at the top of the four-helix bundle and H6 located at the C-terminus, and a long mostly unstructured loop between H1 and H2 (residues 17–29) that is α-helical and significantly shorter in both MazG (PDB code 1vmg; residues 23–33) and PRATP-PH (PDB code 1yvw; residues 23–32) (see Figs. 2 ▶ a and 2 ▶ c).
Figure 2.

Superposition of the YP_001813558.1 biological dimer (gray) with other α-helical NTPases (blue): (a) S. solfataricus MazG (PDB code 1vmg; biological dimer), (b) C. jejuni dUTPase (PDB code 1w2y; single protomer, i.e. half of the biological dimer), (c) B. cereus PRATP-PH (PDB code 1yvw; dimer, i.e. half of the biological tetramer).
An initial DALI (Holm et al., 2008 ▶) search for homologues of YP_001813558.1 did not identify any significant matches owing to the unusual segment swapping; however, a search with the MazG dimer (PDB code 1vmg) revealed structural similarities to the dUTPases 2cic (Z score 10.4; r.m.s.d. 3.3 Å; 139 Cα atoms aligned; O. V. Moroz, M. J. Fogg, D. Gonzalez-Pacanowska & K. S. Wilson, unpublished work), 1w2y (Z score 10.1, r.m.s.d. 3.4 Å, 139 Cα atoms aligned; Moroz et al., 2004 ▶) and 2cje (Z score 8.2; r.m.s.d. 2.9 Å; 121 Cα atoms aligned; O. V. Moroz, M. J. Fogg, D. Gonzalez-Pacanowska & K. S. Wilson, unpublished work) and, of course, to other MazG NTP proteins, 2q73 (Z score 8.2; r.m.s.d. 1.5 Å; 77 Cα atoms aligned; Robinson et al., 2007 ▶) and 2q5z (Z score 7.8, r.m.s.d. 1.7 Å, 78 Cα atoms aligned; Robinson et al., 2007 ▶). Comparison of the superimposed YP_001813558.1 and C. jejuni dUTPase (PDB code 1w2y) structures shows that the H3 helix of YP_001813558.1 is absent in the 1w2y structure and the loops between helices in the two structures are very different. In addition, 1w2y contains an additional helix at the C-terminus (Fig. 2 ▶ b) that is not found in YP_001813558.1.
3.2. Linked dimer
The crystallographic structure of YP_001813558.1 displays a very unusual interlaced segment-swapped dimer, which implies that this obligatory dimer assembly is important for its function (Fig. 3 ▶). Size-exclusion chromatography combined with static light scattering confirmed that the dimer is the major oligomeric state in solution. Initial concerns that the segment-swapped dimer may have arisen from incorrect tracing of the model were eliminated by independent tracing of a SAD data set collected from a different crystal, which also resulted in a segment-swapped dimer. Interestingly, this intertwined dimer does not result in a knotted protein. In other words, the polypeptide chain would not form a knot if the C-terminus of chain A were joined to the N-terminus of chain B and the N- and C-termini of the resulting structure were pulled apart. This is notable because some knotted proteins are believed to have evolved by gene duplication and fusion of intertwined dimers (Bolinger et al., 2010 ▶). In the present case, such a duplication would not lead to a knotted structure, despite the highly intertwined nature of the chains.
Figure 3.

Simplified traces of the YP_001813558.1 linked dimer. Stereoview of the crystallographic dimer with the same orientation and color scheme as in Fig. 1 ▶(b) showing the inter-linked dimer. Note that in this representation the N- and C-termini of each monomer are joined in order to highlight the linked dimer. The linked N- and C-termini are marked with an asterisk. Smoothed curves were calculated as described previously (Norcross & Yeates, 2006 ▶).
A surface area of 5104 Å2 per monomer is buried upon dimer formation. The conserved central four helices that form part of the active site are helices H2 (residues 30–52) and H4 (residues 86–111) from chain A and the equivalent helices from its symmetry-related partner (chain A′) and are assembled in a down–up–down–up topology (Fig. 4 ▶ a). The core of the S. solfataricus MazG (PDB code 1vmg) structure also consists of a dimeric four-helix bundle with each monomer contributing two helices (Fig. 4 ▶ b), but in a different arrangement that appears to represent a minimal functional unit for dUTPases (Moroz et al., 2004 ▶). The four-helix bundle of the C. jejuni dUTPase (PDB code 1w2y) is contained within a single protomer (Fig. 4 ▶ c); thus, dUTPases are thought to have evolved from MazG-like ancestors by gene duplication (Moroz et al., 2005 ▶). The central core four-helix bundle from PRATP-PH also reveals a similar down–up–down–up topology, as shown in Fig. 4 ▶(c).
Figure 4.

Comparison of the core four-helix bundles from the α-helical NTPase superfamily. These four-helix bundles either assemble upon dimerization or are present in a single monomer, resulting in the same down–up–down–up topology. White numbers are closest to the viewer and black numbers are farthest away. (a) Ribbon diagram showing the dimer of YP_001813558.1. (b) Ribbon diagram showing the central four helices of S. solfataricus MazG. (c) Ribbon diagram showing the central four helices from a single protomer of C. jejuni dUTPase.
3.3. Putative metal-binding site predicted from the homolog structures
The location of the potential metal-binding site in YP_001813558.1 and MazG was deduced based on homology with the structure of C. jejuni dUTPase with a substrate analog bound to the active site. Divalent cations, preferably magnesium, are essential for NTPase activity (Nyman, 2001 ▶). Interestingly, although the YP_001813558.1 active site assembles quite differently from those of the other NTPases, the putative metal-binding sites in all three proteins are absolutely conserved, except for a one-residue offset of Asp95 in YP_001813558.1. This potential metal-binding site is formed by Glu43 and Glu47 in H2 of chain A and by Asp95 and Asp99 in H4 of the symmetry-related chain in the dimer (Fig. 5 ▶ a). A symmetry-related site is obviously formed on the opposite side of the dimer from the twofold operation. The metal-binding residues in S. solfataricus MazG (PDB code 1vmg) are Glu35, Glu38, Glu54 and Asp57 (Fig. 5 ▶ b). In dUTPase (PDB code 1w2y), which is related to MazG (PDB code 1vmg) by an ancestral duplication, the metal-binding residues are Glu46, Glu49, Glu74 and Asp77 (Fig. 5 ▶ c). The metal-binding residues, 2′-deoxyuridine 5′-α,β-imidodiphosphate (DUN) and waters participate in the octahedral coordination of Mg ions with distances that range from 1.86 to 2.25 Å.
Figure 5.

(a–c) Comparison of the active sites of YP_001813558.1, S. solfataricus MazG and C. jejuni dUTPase. The putative conserved active-site metal-binding residues are shown as stick models. Note that Asp95 in YP_001813558.1 is offset by one residue when compared with the other two structures. No metal was found in YP_001813558.1. One Li+ ion (red ball) is bound in MazG based on the crystallization conditions. Three Mg2+ ions (red balls) are bound in the C. jejuni dUTPase structure. The nucleotide-binding sites contain either a 1,2-ethanediol (EDO) molecule (YP_001813558.1), an unknown ligand (UNL; S. solfataricus MazG) or 2′-deoxyuridine 5′-α,β-imidodiphosphate (DUN; dUTPase; PDB code 1w2y) and are represented in red. (d) Comparison of the nucleotide-recognition site in YP_001813558.1 (green), S. solfataricus MazG (light blue) and C. jejuni dUTPase (pink) as a stereoview. The EDO molecule from YP_001813558.1 (red sticks), UNL from S. solfataricus MazG (blue balls) and DUN from C. jejuni dUTPase (purple sticks) are shown. Mse12 is modeled as three conformations in the MazG structure. (e) Stereoview of the superposition of the substrate analogs DUN (purple) from C. jejuni dUTPase and 2-deoxy-5-methylcytidine-5′-(tetrahydrogen triphosphate (yellow) from M. musculus RS21-C6 and the EDO (red) molecule bound to the YP_001813558.1 structure. Hydrogen bonds are shown as dotted lines. The key residues from YP_001813558.1 that are predicted to be involved in substrate binding are presented as a green stick model.
3.4. Nucleotide-binding site
In C. jejuni dUTPase, Asp77 plays a central role in substrate binding. In addition to coordinating the Mg2+ ion and binding the terminal phosphate of the substrate analog 2′-deoxyuridine 5′-α,β-imidodiphosphate (DUN), Asp77 also binds the ribosyl 3′-OH group (Moroz et al., 2004 ▶). In Mus musculus RS21-C6, the binding mode of the terminal phosphate is significantly different compared with that of C. jejuni dUTPase, presumably owing to the absence of magnesium ions. However, Asp98 (equivalent to Asp77) is located close to the bound 2-deoxy-5-methylcytidine-5′-(tetrahydrogen triphosphate) and binds to the ribosyl 3′-OH group of the nucleoside moiety via a water-mediated interaction (Wu et al., 2007 ▶). Therefore, it is thought that the corresponding conserved residues, Asp99 in YP_001813558.1 and Asp57 in S. solfataricus MazG, perform similar roles in these enzymes. Another important residue for recognition of the substrate ribosyl 3′-OH in C. jejuni dUTPase is Asn179. This residue is conserved in both YP_001813558.1 (Asn126) and M. musculus RS21-C6 (Asn125), but not in S. solfataricus MazG.
In YP_001813558.1, the putative sugar-binding residues are Tyr102 and Phe103, between which the sugar moiety is sandwiched, and Asn126, which discriminates between deoxyribose and ribose (Fig. 5 ▶ e). The latter is conserved in most members of the all-α-helical NTP pyrophosphatase superfamily that have been shown to have a preference for dNTP (the dUTPase, dCTPase and RS21-C6 families), but is not conserved in the ribonucleosidetriphosphate-hydrolyzing HisE and EcMazG families (Nonaka et al., 2009 ▶; Robinson et al., 2007 ▶). Neither the YP_001813558.1 nor the S. solfataricus MazG structures have known biological ligands in their nucleotide binding sites (Fig. 5 ▶ d). The YP_001813558.1 structure contains a 1,2-ethanediol molecule and the S. solfataricus MazG structures contain an unidentified ligand (UNL) in the nucleotide-binding site. Since those ligands could mimic nucleotide substrates (Fig. 5 ▶ d), we speculate that both YP_001813558.1 and S. solfataricus MazG enzymes can function as dNTPases.
The uracil-recognition site of C. jejuni dUTPase is formed by Gln14 N∊2, Asn18 Oδ1 and Asn22 Nδ2 and is not conserved in YP_001813558.1 or S. solfataricus MazG. The corresponding residues in YP_001813558.1 are Val10, His14 and His19; His14 N∊2 is hydrogen bonded to the O2 atom of a 1,2-ethanediol molecule in the ligand-binding site. The corresponding region in the S. solfataricus MazG structure contains Mse12, which adopts three side-chain conformations, Tyr16 and Asp20, where Asp20 Oδ1 and Asp20 Oδ2 interact with the O7 and O9 atoms of the UNL ligand, respectively. Thus, it appears that YP_001813558.1 and S. solfataricus MazG may not bind uracil (Fig. 5 ▶ d). The major determinant of the substrate specificity involved in base recognition in YP_001813558.1 would be Arg36, where Arg36 Nη1 and Arg36 Nη2 interact with the O1 and O2 atoms of the 1,2-ethanediol molecule, respectively (Fig. 5 ▶ e). Arg36 provides two hydrogen-bond donors that could interact with two adjacent acceptors on the base. Of the canonical bases, only cytosine would satisfy these conditions for making two hydrogen bonds. Therefore, potential substrates for YP_001813558.1 include dCTP and its derivatives (e.g. 5-methyl or 5-hydroxymethyl dCTP). In addition, the two modified bases O 4-methylthymine and 8-hydroxyguanine are also predicted to interact in the same manner as cytosine. These modified bases could provide additional hydrogen bonds from O4 of O 4-methylthymine (or O6 of 8-hydroxyguanine) to His19 and/or Arg32 of YP_001813558.1.
The pyrophosphate-recognition residues of C. jejuni dUTPase are mostly conserved in YP_001813558.1 and S. solfataricus MazG, except for the C-terminal region. Lys175 of C. jejuni dUTPase is structurally equivalent to Val122 of YP_001813558.1 and Lys80 of MazG. This residue is located in the loop region near the C-terminus of C. jejuni dUTPase, which also contains the pyrophosphate-recognition residues Arg182, Tyr187, Lys194 and Asn202. This region does not superimpose well in YP_001813558.1 and is absent in MazG. The corresponding pyrophosphate-recognition loop in YP_001813558.1 is located between H5 and H6. This loop and the two neighboring C-terminal helices (H5 and H6) of YP_001813558.1 are in an open conformation and are more exposed to solvent compared with the equivalent region in C. jejuni dUTPase, which may suggest an induced-fit mechanism for substrate binding involving movement of the C-terminal region.
4. Conclusions
We report a very unusual segment-swapped linked-dimer structure of a dUTPase from E. sibiricum 255-15, which implies that this obligatory dimer assembly is important for its function of adaptation to an extreme cold environment. Unusual, covalently interlinked dimeric structures have been implicated previously in stabilizing proteins (Boutz et al., 2007 ▶; Duff et al., 2003 ▶). Structural analysis and comparisons indicate that YP_001813558.1 is a dNTPase that potentially prefers dCTPs or its derivatives. Further biochemical analyses are needed to confirm these predictions. The availability of further sequences and structures of NTP pyrophosphohydrolases should shed light on the evolutionary history of this intriguing protein family. The information presented here, in combination with further biochemical and biophysical studies, should yield valuable insights into the functional role of YP_001813558.1. Additional information about YP_001813558.1 is available from TOPSAN (Krishna et al., 2010 ▶) at http://www.topsan.org/explore?PDBid=3nl9.
Supplementary Material
PDB reference: putative NTP pyrophosphohydrolase, 3nl9
Acknowledgments
This work was supported by the NIH, National Institute of General Medical Sciences, Protein Structure Initiative grant No. U54 GM074898. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL) and the Advanced Light Source (ALS). The SSRL is a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health (National Center for Research Resources, Biomedical Technology Program and the National Institute of General Medical Sciences). The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231. We acknowledge Drs Robyn L. Stanfield and Sung-il Yoon at The Scripps Research Institute for valuable discussions. E. sibiricum 255-15 was a gift from Drs Tamara Cole and Jim Tiedje, Michigan State University, East Lansing, Michigan, USA. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
- Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Nucleic Acids Res.25, 3389–3402. [DOI] [PMC free article] [PubMed]
- Andreeva, A., Howorth, D., Chandonia, J.-M., Brenner, S. E., Hubbard, T. J. P., Chothia, C. & Murzin, A. G. (2008). Nucleic Acids Res.36, D419–D425. [DOI] [PMC free article] [PubMed]
- Bessman, M. J., Frick, D. N. & O’Handley, S. F. (1996). J. Biol. Chem.271, 25059–25062. [DOI] [PubMed]
- Bolinger, D., Sulkowska, J. I., Hsu, H. P., Mirny, L. A., Kardar, M., Onuchic, J. N. & Virnau, P. (2010). PLoS Comput. Biol.6, e1000731. [DOI] [PMC free article] [PubMed]
- Boutz, D. R., Cascio, D., Whitelegge, J., Perry, L. J. & Yeates, T. O. (2007). J. Mol. Biol.368, 1332–1344. [DOI] [PMC free article] [PubMed]
- Cedergren-Zeppezauer, E. S., Larsson, G., Nyman, P. O., Dauter, Z. & Wilson, K. S. (1992). Nature (London), 355, 740–743. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cohen, A. E., Ellis, P. J., Miller, M. D., Deacon, A. M. & Phizackerley, R. P. (2002). J. Appl. Cryst.35, 720–726. [DOI] [PMC free article] [PubMed]
- Cohen, S. X., Morris, R. J., Fernandez, F. J., Ben Jelloul, M., Kakaris, M., Parthasarathy, V., Lamzin, V. S., Kleywegt, G. J. & Perrakis, A. (2004). Acta Cryst. D60, 2222–2229. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cruickshank, D. W. J. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed]
- Dauter, Z., Persson, R., Rosengren, A. M., Nyman, P. O., Wilson, K. S. & Cedergren-Zeppezauer, E. S. (1999). J. Mol. Biol.285, 655–673. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed]
- Duff, A. P., Cohen, A. E., Ellis, P. J., Kuchar, J. A., Langley, D. B., Shepard, E. M., Dooley, D. M., Freeman, H. C. & Guss, J. M. (2003). Biochemistry, 42, 15148–15157. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Harkiolaki, M., Dodson, E. J., Bernier-Villamor, V., Turkenburg, J. P., Gonzalez-Pacanowska, D. & Wilson, K. S. (2004). Structure, 12, 41–53. [DOI] [PubMed]
- Holm, L., Kaariainen, S., Rosenstrom, P. & Schenkel, A. (2008). Bioinformatics, 24, 2780–2781. [DOI] [PMC free article] [PubMed]
- Hwang, K. Y., Chung, J. H., Kim, S. H., Han, Y. S. & Cho, Y. (1999). Nature Struct. Biol.6, 691–696. [DOI] [PubMed]
- Jaroszewski, L., Rychlewski, L. & Godzik, A. (2000). Protein Sci.9, 1487–1496. [DOI] [PMC free article] [PubMed]
- Javid-Majd, F., Yang, D., Ioerger, T. R. & Sacchettini, J. C. (2008). Acta Cryst. D64, 627–635. [DOI] [PMC free article] [PubMed]
- Kleywegt, G. J. (2000). Acta Cryst. D56, 249–265. [DOI] [PubMed]
- Klock, H. E., Koesema, E. J., Knuth, M. W. & Lesley, S. A. (2008). Proteins, 71, 982–994. [DOI] [PubMed]
- Krishna, S. S., Weekes, D., Bakolitsa, C., Elsliger, M.-A., Wilson, I. A., Godzik, A. & Wooley, J. (2010). Acta Cryst. F66, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- Larsson, G., Svensson, L. A. & Nyman, P. O. (1996). Nature Struct. Biol.3, 532–538. [DOI] [PubMed]
- Laskowski, R. A., Chistyakov, V. V. & Thornton, J. M. (2005). Nucleic Acids Res.33, D266–D268. [DOI] [PMC free article] [PubMed]
- Lesley, S. A. et al. (2002). Proc. Natl Acad. Sci. USA, 99, 11664–11669.
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Miller, M. D. & Deacon, A. M. (2007). Nucl. Instrum. Methods Phys. Res. A, 582, 233–235. [DOI] [PMC free article] [PubMed]
- Minasov, G., Teplova, M., Stewart, G. C., Koonin, E. V., Anderson, W. F. & Egli, M. (2000). Proc. Natl Acad. Sci. USA, 97, 6328–6333. [DOI] [PMC free article] [PubMed]
- Mol, C. D., Harris, J. M., McIntosh, E. M. & Tainer, J. A. (1996). Structure, 4, 1077–1092. [DOI] [PubMed]
- Moroz, O. V., Harkiolaki, M., Galperin, M. Y., Vagin, A. A., Gonzalez-Pacanowska, D. & Wilson, K. S. (2004). J. Mol. Biol.342, 1583–1597. [DOI] [PubMed]
- Moroz, O. V., Murzin, A. G., Makarova, K. S., Koonin, E. V., Wilson, K. S. & Galperin, M. Y. (2005). J. Mol. Biol.347, 243–255. [DOI] [PubMed]
- Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia, C. (1995). J. Mol. Biol.247, 536–540. [DOI] [PubMed]
- Nonaka, M., Tsuchimoto, D., Sakumi, K. & Nakabeppu, Y. (2009). FEBS J.276, 1654–1666. [DOI] [PubMed]
- Norcross, T. S. & Yeates, T. O. (2006). J. Mol. Biol.362, 605–621. [DOI] [PubMed]
- Nyman, P. O. (2001). Curr. Protein Pept. Sci.2, 277–285. [DOI] [PubMed]
- Pannu, N. S., Murshudov, G. N., Dodson, E. J. & Read, R. J. (1998). Acta Cryst. D54, 1285–1294. [DOI] [PubMed]
- Prasad, G. S., Stura, E. A., McRee, D. E., Laco, G. S., Hasselkus-Light, C., Elder, J. H. & Stout, C. D. (1996). Protein Sci.5, 2429–2437. [DOI] [PMC free article] [PubMed]
- Robinson, A., Guilfoyle, A. P., Harrop, S. J., Boucher, Y., Stokes, H. W., Curmi, P. M. & Mabbutt, B. C. (2007). Mol. Microbiol.66, 610–621. [DOI] [PubMed]
- Santarsiero, B. D., Yegian, D. T., Lee, C. C., Spraggon, G., Gu, J., Scheibe, D., Uber, D. C., Cornell, E. W., Nordmeyer, R. A., Kolbe, W. F., Jin, J., Jones, A. L., Jaklevic, J. M., Schultz, P. G. & Stevens, R. C. (2002). J. Appl. Cryst.35, 278–281.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Tarbouriech, N., Buisson, M., Seigneurin, J. M., Cusack, S. & Burmeister, W. P. (2005). Structure, 13, 1299–1310. [DOI] [PubMed]
- Terwilliger, T. C. (2003). Acta Cryst. D59, 1174–1182. [DOI] [PMC free article] [PubMed]
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol.229, 105–124. [DOI] [PubMed]
- Vishnivetskaya, T., Kathariou, S., McGrath, J., Gilichinsky, D. & Tiedje, J. M. (2000). Extremophiles, 4, 165–173. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2007). Methods Mol. Biol.364, 215–230. [DOI] [PubMed]
- Vriend, G. (1990). J. Mol. Graph.8, 52–56. [DOI] [PubMed]
- Weiss, M. S. & Hilgenfeld, R. (1997). J. Appl. Cryst.30, 203–205.
- Weiss, M. S., Metzner, H. J. & Hilgenfeld, R. (1998). FEBS Lett.423, 291–296. [DOI] [PubMed]
- Winn, M. D., Murshudov, G. N. & Papiz, M. Z. (2003). Methods Enzymol.374, 300–321. [DOI] [PubMed]
- Wu, B., Liu, Y., Zhao, Q., Liao, S., Zhang, J., Bartlam, M., Chen, W. & Rao, Z. (2007). J. Mol. Biol.367, 1405–1412. [DOI] [PubMed]
- Yang, H., Guranovic, V., Dutta, S., Feng, Z., Berman, H. M. & Westbrook, J. D. (2004). Acta Cryst. D60, 1833–1839. [DOI] [PubMed]
- Zhang, J. & Inouye, M. (2002). J. Bacteriol.184, 5323–5329. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: putative NTP pyrophosphohydrolase, 3nl9