

The crystal structure of BT2081 from B. thetaiotaomicron reveals a two-domain protein with a putative carbohydrate-binding site in the C-terminal domain.
Keywords: gut microbiome, sugars, structural genomics, immunoglobulin-like fold, jelly-roll fold
Abstract
BT2081 from Bacteroides thetaiotaomicron (GenBank accession code NP_810994.1) is a member of a novel protein family consisting of over 160 members, most of which are found in the different classes of Bacteroidetes. Genome-context analysis lends support to the involvement of this family in carbohydrate metabolism, which plays a key role in B. thetaiotaomicron as a predominant bacterial symbiont in the human distal gut microbiome. The crystal structure of BT2081 at 2.05 Å resolution represents the first structure from this new protein family. BT2081 consists of an N-terminal domain, which adopts a β-sandwich immunoglobulin-like fold, and a larger C-terminal domain with a β-sandwich jelly-roll fold. Structural analyses reveal that both domains are similar to those found in various carbohydrate-active enzymes. The C-terminal β-jelly-roll domain contains a potential carbohydrate-binding site that is highly conserved among BT2081 homologs and is situated in the same location as the carbohydrate-binding sites that are found in structurally similar glycoside hydrolases (GHs). However, in BT2081 this site is partially occluded by surrounding loops, which results in a deep solvent-accessible pocket rather than a shallower solvent-exposed cleft.
1. Introduction
Bacteroides thetaiotaomicron is a Gram-negative anaerobic bacterium that is a dominant member of the human intestinal tract microbiome. This bacterial species is essential for the metabolism and uptake of dietary plant polysaccharides by the human host (Comstock & Coyne, 2003 ▶; Xu et al., 2003 ▶; Zocco et al., 2007 ▶). To utilize these polysaccharides, B. thetaiotaomicron expresses a large number of carbohydrate-processing enzymes. Indeed, it is estimated that ∼6.6% of the B. thetaiotaomicron genome encodes such proteins, many of which are novel and have not been functionally or structurally characterized (Davies et al., 2005 ▶).
The BT2081 gene product of B. thetaiotaomicron encodes a protein with a molecular weight of 37 198 Da (residues 1–341) and a calculated isoelectric point of 4.9. It contains a predicted N-terminal signalling peptide, suggesting that it is secreted from the cytoplasm. Sequence and genomic context analysis suggest that it is a putative carbohydrate-processing protein that is part of the extracellular polysaccharide-processing machinery. Its N-terminal region (residues 1–114) shares high sequence similarity to the carbohydrate-binding domains of endo-1,4-β-xylanase from Streptomyces griseoflavus (35% sequence identity), β-xylosidase from Magnetospirillum magnetotacticum (29% sequence identity) and a putative β-fructosidase from Sarcoptes scabiei (39% sequence identity), all of which are glycoside hydrolases (GHs), which catalyze the cleavage of the glycosidic bonds in monosaccharide, disaccharides and polysaccharides into their constituent sugar units. Moreover, genome-context analysis shows that BT2081 and several of its paralogs belong to characteristic PULs (polysaccharide-utilization loci) of B. thetaiotaomicron and are associated with transmembrane porin domains that are homologous to the B. thetaiotaomicron outer membrane protein transporter SusC. SusC is part of the well characterized eight-component sus (starch-utilization system) operon used by B. thetaiotaomicron in carbohydrate metabolism. Furthermore, in the sus operon two other BT2081 paralogs (BT0450 and BT1761) are immediately followed by GHs. To further investigate the role that BT2081 may play in carbohydrate metabolism, we have determined its crystal structure using the semi-automated high-throughput pipeline of the Joint Center for Structural Genomics (JCSG; Lesley et al., 2002 ▶) as part of the Protein Structure Initiative (PSI) of the National Institute of General Medical Sciences, NIH.
2. Materials and methods
2.1. Protein production and crystallization
Clones were generated using the Polymerase Incomplete Primer Extension (PIPE) cloning method (Klock et al., 2008 ▶). The gene encoding BT2081 (GenBank NP_810994, Swiss-Prot Q8A605) was amplified by polymerase chain reaction (PCR) from B. thetaiotaomicron VPI-5482 genomic DNA using PfuTurbo DNA polymerase (Stratagene) and I-PIPE (Insert) primers (forward primer, 5′-ctgtacttccagggcCGCGAAGAAGCTCCCAATGCAGAGGCAG-3′; reverse primer, 5′-aattaagtcgcgttaGTCTTCCGAGCGATAGATTAGTTCGACT-3′; target sequences in upper case) that included sequences for the predicted 5′ and 3′ ends. The expression vector pSpeedET, which encodes an amino-terminal tobacco etch virus (TEV) protease-cleavable expression and purification tag (MGSDKIHHHHHHENLYFQ/G), was PCR-amplified with V-PIPE (Vector) primers (forward primer, 5′-taacgcgacttaattaactcgtttaaacggtctccagc-3′; reverse primer, 5′-gccctggaagtacaggttttcgtgatgatgatgatgatg-3′). V-PIPE and I-PIPE PCR products were mixed to anneal the amplified DNA fragments together. Escherichia coli GeneHogs (Invitrogen) competent cells were transformed with the I-PIPE/V-PIPE mixture and dispensed onto selective LB–agar plates. The cloning junctions were confirmed by DNA sequencing. Using the PIPE method, the gene segment encoding Met1–Ile20 was deleted because it was predicted to be a signal peptide. Expression was performed in selenomethionine-containing medium at 310 K with suppression of normal methionine synthesis. At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 µg ml−1 and the cells were harvested and frozen. After one freeze–thaw cycle, the cells were sonicated in lysis buffer [50 mM HEPES pH 8.0, 50 mM NaCl, 10 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine–HCl (TCEP)] and the lysate was clarified by centrifugation at 32 500g for 30 min. The soluble fraction was passed over nickel-chelating resin (GE Healthcare) pre-equilibrated with lysis buffer, the resin was washed with wash buffer [50 mM HEPES pH 8.0, 300 mM NaCl, 40 mM imidazole, 10%(v/v) glycerol, 1 mM TCEP] and the protein was eluted with elution buffer [20 mM HEPES pH 8.0, 300 mM imidazole, 10%(v/v) glycerol, 1 mM TCEP]. The eluate was buffer-exchanged with TEV buffer (20 mM HEPES pH 8.0, 200 mM NaCl, 40 mM imidazole, 1 mM TCEP) using a PD-10 column (GE Healthcare) and incubated with 1 mg TEV protease per 15 mg eluted protein. The protease-treated eluate was passed over nickel-chelating resin (GE Healthcare) pre-equilibrated with HEPES crystallization buffer (20 mM HEPES pH 8.0, 200 mM NaCl, 40 mM imidazole, 1 mM TCEP) and the resin was washed with the same buffer. The flowthrough and wash fractions were combined and concentrated to 13.5 mg ml−1 by centrifugal ultrafiltration (Millipore) for crystallization trials. BT2081 was crystallized by mixing 100 nl protein solution with 100 nl crystallization solution in a sitting drop over a 50 µl reservoir volume using the nanodroplet vapor-diffusion method (Santarsiero et al., 2002 ▶) with standard JCSG crystallization protocols (Lesley et al., 2002 ▶). The crystallization reagent consisted of 42%(v/v) polyethylene glycol 600, 0.25 M calcium acetate and 0.1 M sodium cacodylate pH 6.33. PEG 400 was added to the crystal as a cryoprotectant to a final concentration of 5%(v/v). A triangular prism-shaped crystal of approximately 200 × 200 × 200 µm in size was harvested after 43 d at 277 K for data collection. Initial screening for diffraction was carried out using the Stanford Automated Mounting system (SAM; Cohen et al., 2002 ▶) at the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, California, USA). The diffraction data were indexed in the trigonal space group P3221. The oligomeric state of BT2081 in solution was determined using a 1 × 30 cm Superdex 200 size-exclusion column (GE Healthcare). The mobile phase consisted of 20 mM Tris pH 8.0, 150 mM NaCl and 0.02%(w/v) sodium azide. The molecular weight was calculated using ASTRA v.5.1.5 software (Wyatt Technology). Protein concentrations were determined using the Coomassie Plus assay (Pierce).
2.2. Data collection, structure solution and refinement
Single-wavelength anomalous diffraction (SAD) data were collected on beamline 11-1 at the SSRL at a wavelength corresponding to the peak (λ1) of a selenium SAD experiment. The data set was collected at 100 K using a MAR Mosaic 325 mm CCD detector (Rayonix) using the Blu-Ice data-collection environment (McPhillips et al., 2002 ▶). The SAD data were integrated and reduced using MOSFLM (Leslie, 1992 ▶) and then scaled with the program SCALA (Collaborative Computational Project, Number 4, 1994 ▶). Phasing was performed with SHELXD (Sheldrick, 2008 ▶) and autoSHARP [mean figure-of-merit (acentric/centric) of 0.28/0.09 with 11 anomalous scatterers per asymmetric unit (eight SeMet, two cacodylate ions and one calcium ion); Vonrhein et al., 2007 ▶]. Automatic model building was performed with ARP/wARP (Cohen et al., 2004 ▶). Model completion was performed with Coot (Emsley & Cowtan, 2004 ▶) and TLS refinement with REFMAC5 (Winn et al., 2003 ▶). The refinement included experimental phase restraints in the form of Hendrickson–Lattman coefficients and two TLS groups per chain, with the TLS groups being assigned with the aid of the TLSMD server (Painter & Merritt, 2006 ▶). Data-collection and refinement statistics are summarized in Table 1 ▶. X-ray fluorescence emission peaks for selenium, arsenic, nickel and calcium were observed when the crystal was scanned on SSRL beamline 1-5 with X-rays above (500 eV) the Se K edge. Calcium was assigned based on binding geometry and its presence in the crystallization conditions. Additional diffraction data were collected above and below the arsenic and nickel edges, with the resultant anomalous electron-density maps revealing the presence of arsenic in the form of cacodylate (which was present in the crystallization condition) and the absence of nickel.
Table 1. Summary of crystal parameters, data-collection and refinement statistics for BT2081 (PDB code 3hbz).
Values in parentheses are for the highest resolution shell.
| Space group | P3221 |
| Unit-cell parameters (Å) | a = 94.55, b = 94.55, c = 107.81 |
| Data collection | |
| Wavelength (Å) | 0.9785 |
| Resolution range (Å) | 29.8–2.05 (2.10–2.05) |
| No. of observations | 268747 |
| No. of unique reflections | 35437 |
| Completeness (%) | 99.9 (99.9) |
| Mean I/σ(I) | 16.7 (1.8) |
| Rmerge on I† (%) | 0.091 (0.75) |
| Model and refinement statistics | |
| Resolution range (Å) | 29.8–2.05 |
| No. of reflections (total) | 35400‡ |
| No. of reflections (test) | 1773 |
| Completeness (%) | 99.9 |
| Cutoff criterion | |F| > 0 |
| Rcryst§ | 0.159 |
| Rfree¶ | 0.191 |
| Stereochemical parameters | |
| Restraints (r.m.s.d. observed) | |
| Bond angles (°) | 1.60 |
| Bond lengths (Å) | 0.018 |
| Average isotropic B value (Å2) | 42.9†† |
| ESU‡‡ based on Rfree (Å) | 0.121 |
R
merge = 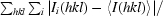
 .
.
Typically, the number of unique reflections used in refinement is slightly less than the total number that were integrated and scaled. Reflections are excluded owing to systematic absences, negative intensities and rounding errors in the resolution limits and unit-cell parameters.
R
cryst = 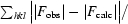
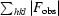 , where F
calc and F
obs are the calculated and observed structure-factor amplitudes, respectively.
, where F
calc and F
obs are the calculated and observed structure-factor amplitudes, respectively.
R free is the same as R cryst but for 5.0% of the total reflections chosen at random and omitted from refinement.
This value represents the total B that includes TLS and residual B components.
2.3. Validation and deposition
The quality of the crystal structure was analyzed using the JCSG Quality Control server (http://smb.slac.stanford.edu/jcsg/QC). This server processes the coordinates and data through a variety of validation tools including AutoDepInputTool (Yang et al., 2004 ▶), MolProbity (Chen et al., 2010 ▶), WHAT IF v.5.0 (Vriend, 1990 ▶), RESOLVE (Terwilliger, 2004 ▶), MOLEMAN2 (Kleywegt, 2000 ▶) as well as several in-house scripts and summarizes the results. Protein quaternary-structure analysis used the PISA server (Krissinel & Henrick, 2007 ▶). Fig. 1 ▶(b) was adapted from PDBsum (Laskowski, 2009 ▶) and all other figures were prepared with PyMOL (DeLano Scientific). Atomic coordinates and experimental structure factors for BT2081 from B. thetaiotaomicron at 2.05 Å resolution were deposited in the PDB (http://www.pdb.org) under code 3hbz.
Figure 1.

Crystal structure of BT2081 from B. thetaiotaomicron. (a) Ribbon diagram of the BT2081 monomer, showing the distinct N-terminal (green) and C-terminal (purple) domains. Helices (H1–H7) and β-strands (β1–β28) are indicated. (b) Diagram showing the secondary-structure elements of BT2081 superimposed on its primary sequence. The labeling of secondary-structure elements [colored by domain as in (a)] is in accord with PDBsum (http://www.ebi.ac.uk/pdbsum), in which α-helices (H1, H3, H5 and H6), 310-helices (H2, H4 and H7) and β-strands (β1–β28) are labeled sequentially, β-turns and γ-turns are designated by Greek letters (β, γ) and β-hairpins by red loops.
3. Results and discussion
3.1. Overall structure
The crystal structure of BT2081 (Fig. 1 ▶) was determined to 2.05 Å resolution using the SAD method. Data-collection, model and refinement statistics are summarized in Table 1 ▶. The final model includes one BT2081 molecule (residues 21–360), two calcium ions, two sodium ions, two cacodylate anions, two acetate anions, 19 polyethylene glycol molecules and 251 water molecules in the asymmetric unit. Gly0, which is part of the expression construct and remained after cleavage of the N-terminal purification tag, is also part of the final model. The nucleotide sequence corresponding to residues 1–20 was omitted from the expression construct as this region was predicted to encode either a lipoprotein signal peptide or, more likely, a single transmembrane helix anchoring BT2081 in the outer membrane of the cell. Electron density was not observed for the C-terminal Asp361 or for some of the side-chain atoms of Arg21, Glu23, Glu139, Lys239, Lys249, Glu299, Lys317 and Glu318. The Matthews coefficient (V M; Matthews, 1968 ▶) for BT2081 is 3.69 Å3 Da−1 and the estimated solvent content is 67%. The Ramachandran plot produced by MolProbity (Chen et al., 2010 ▶) shows that 97.9% and 99.7% of the residues are in the favored and allowed regions, respectively. The single residue in the disallowed region, Ala27, is in a section of poorly defined electron density.
BT2081 is composed of 28 β-strands (β1–β28), four α-helices (H1, H3, H5 and H6) and three 310-helices (H2, H4 and H7) (Fig. 1 ▶). The total β-sheet, α-helical and 310-helical content is 39.0, 6.2 and 2.6%, respectively. Crystallographic packing, as well as PISA analyses (Krissinel & Henrick, 2007 ▶) of BT2081, suggest that a monomer is likely to be the biologically relevant form of the protein, which is consistent with results from analytical size-exclusion chromatography (anSEC).
The BT2081 monomer consists of two structural domains (Fig. 1 ▶). The N-terminal domain (residues 21–114) adopts a β-sandwich fold consisting of two-stranded (β1 and β4) and five-stranded (β2, β3 and β5–β7) β-sheets, which concurs with its classification into the immunoglobulin (Ig)-like fold superfamily of SCOP (Andreeva et al., 2004 ▶). The C-terminal domain (residues 115–361) also adopts a β-sandwich fold, the core of which comprises two five-stranded antiparallel β-sheets that form a concave (β15/β18, β22, β23, β25 and β26) and a convex (β8/β12, β11, β19, β24 and β28) surface. This domain adopts a β-jelly-roll topology, consistent with its assignment to the galactose-binding domain-like superfamily of SCOP.
3.2. Similarity to other proteins
Carbohydrate-active enzymes are quite modular, often containing various carbohydrate-binding and catalytic domains in different combinations. Glycoside hydrolases (GHs), for example, often contain both catalytic and noncatalytic carbohydrate-binding modules (CBMs) which can be assembled in different orders (Davies et al., 2005 ▶; Henrissat & Davies, 1997 ▶; Cantarel et al., 2009 ▶). CBMs, which can bind a range of different polysaccharides, function to increase the catalytic efficiency of GHs by bringing the catalytic module into closer proximity with its substrate (Bolam et al., 1998 ▶; Tomme et al., 1995 ▶).
The Ig-like and jelly-roll domains of BT2081 have previously been observed as modules in carbohydrate-active enzymes. CtCel9D-Cel44A, a multi-enzyme GH complex from Clostridium thermocellum, for example, contains polycystic kidney disease (PKD) and CBM family 44 (CBM44) domains (PDB code 2c26; Najmudin et al., 2006 ▶) that also adopt Ig-like and β-jelly-roll folds, respectively. However, the relative orientation of the two domains differs significantly and as a result the superposition of these two structures using FATCAT (http://fatcat.burnham.org; Ye & Godzik, 2004 ▶) required two rotations or twists around the linker region that connects the two domains in order to obtain optimal alignment of the full-length structures. When BT2081 and PKD-CBM44 are structurally aligned in this way, the r.m.s.d. between 202 Cα atoms is 3.15 Å despite only 6.7% sequence identity (Fig. 2 ▶). An important distinction between these two proteins, which highlights the modularity of carbohydrate-active enzymes, is that, in addition to the PKD and CBM44 domains, CtCel9D-Cel44A contains additional domains (Ig-like, CBM30, GH9 and GH44) that are absent from BT2081.
Figure 2.

Structural comparison of BT2081 with PKD-CBM44 of C. thermocellum CtCel9D-Cel44A. Superposition of BT2081 (orange/yellow) with PKD-CBM44 (lilac/pale blue; PDB code 2c26), which also contain both the Ig-like and β-jelly-roll domains. Calcium ions are represented as spheres. Protein regions which were used for alignment by FATCAT are shown in darker shades (orange for BT2081 and lilac for PKD-CBM44) to highlight similarities in the protein cores. Please note that two twists have been introduced by FATCAT into the structure of PKD-CBM44 in order to obtain optimal full-length alignments of the structures.
Similarly, some bacterial sialidases also contain both Ig-like and β-jelly-roll domains. NedA, a sialidase from Micromonospora viridifaciens (PDB code 1euu; Gaskell et al., 1995 ▶), contains these two domains, but also has a third GH family 33 (GH33) catalytic domain at its N-terminus that adopts a six-bladed β-propeller fold. The C-terminal β-jelly-roll domain of NedA belongs to CBM family 32 (CBM32), while the middle Ig-like domain is thought to act as a linker region between the CBM32 and GH33 domains. Similar to CtCel9D-Cel44A, the relative orientation of the Ig-like and β-jelly-roll domains differs significantly between BT2081 and NedA and, as a result, four consecutive twists in the region between these domains were required to obtain an optimal full-length alignment of the structures by FATCAT. The resultant r.m.s.d. between 184 Cα atoms, which share only 4.8% sequence identity, is 3.11 Å.
3.3. BT2081 N-terminal domain
A structural similarity search of the N-terminal domain of BT2081 only performed using the FATCAT server, revealed many proteins that belong to the Ig-like fold family. The closest match was to the soluble upper-middle domain (UMD; residues 232–320) of the outer membrane protein usher from Yersinia pestis (PDB code 3fcg; Yu et al., 2009 ▶), with an r.m.s.d. of 2.65 Å between 49 Cα atoms and a sequence identity of 7.6%. The second and third closest structural neighbors were SoxY from Paracoccus pantotrophus (PDB code 2oxg; Sauve et al., 2007 ▶), with an r.m.s.d. of 3.08 Å between 64 Cα atoms and a sequence identity of 5.6%, and the I-set domain (residues 3537–3630) of human obscurin (PDB code 2edw; R. Sano, F. Hayashi, M. Yoshida & S. Yokoyama, unpublished work), with an r.m.s.d. of 3.07 Å between 59 Cα atoms and a sequence identity of 2.5%. Although none of the top structural neighbors from this search matched carbohydrate-active enzymes, the Ig-like fold has been observed in a number of these proteins. In addition to CtCel9D-Cel44A and NedA, structures from CBM families 9, 20, 25, 26, 31, 33 and 34 have also been found to adopt Ig-like folds (Hashimoto, 2006 ▶). Despite their structural similarity, the BT2081 N-terminal domain shares little sequence similarity with these CBMs (see Table 2 ▶ a for superposition statistics).
Table 2. Analysis of the N- and C-terminal domains of BT2081 using FATCAT .
(a).
Superposition of the BT2081 N-terminal domain with CBM families that adopt Ig-like folds.
| PDB code | CBM family | Optimized r.m.s.d. (Å) | Equivalent positions (No. of Cα atoms) | Sequence identity (%) | P-value† |
|---|---|---|---|---|---|
| 1i82 | CBM9 | 3.83 | 67 | 3.3 | 2.94 × 10−1 |
| 1b90 | CBM20 | 3.39 | 43 | 9.1 | 8.34 × 10−1 |
| 2c3v | CBM25 | 3.01 | 50 | 6.5 | 1.29 × 10−1 |
| 2c3g | CBM26 | 3.45 | 56 | 3.6 | 2.05 × 10−1 |
| 2cov | CBM31 | 3.43 | 65 | 5.5 | 7.31 × 10−2 |
| 2bem | CBM33 | 3.09 | 72 | 4.7 | 2.71 × 10−1 |
| 1bvz | CBM34 | 3.14 | 59 | 2.6 | 7.30 × 10−1 |
(b).
Representative closest structural neighbors of the BT2081 C-terminal domain.
| PDB code | CBM/GH family | Optimized r.m.s.d. (Å) | Equivalent positions (No. of Cα atoms) | Sequence identity (%) | P-value† |
|---|---|---|---|---|---|
| 1gwk | CBM29 | 3.08 | 128 | 6.8 | 3.22 × 10−4 |
| 2zew | CBM16 | 3.01 | 136 | 10.4 | 9.93 × 10−4 |
| 1v0a | CBM11 | 3.05 | 130 | 9.8 | 1.78 × 10−3 |
| 1byh | GH16 | 3.04 | 137 | 5.0 | 1.79 × 10−3 |
| 1gmm | CBM6 | 3.05 | 115 | 4.6 | 1.95 × 10−3 |
The FATCAT P-value measures the probability of obtaining a similar result between two random structures. This P-value is calculated based on empirical fitting of the extreme value distribution to the FATCAT similarity score (Ye & Godzik, 2004 ▶). The smaller the P-value, the more statistically significant the similarity between corresponding structures (P-values of <0.05 are considered to be significant).
Sequence analysis of the BT2081 N-terminal domain with BLAST revealed significant sequence similarity to portions of several GHs, including endo-1,4-β-xylanase from St. griseoflavus (35% sequence identity over 90 residues), β-xylosidase from M. magnetotacticum (29% sequence identity over 94 residues) and a putative β-fructosidase from Sa. scabiei (39% sequence identity over 76 residues). In all of these sequences, the putative Ig-like domain immediately precedes a putative GH43 catalytic domain, which is known to adopt a fivefold β-propeller fold. The role of the putative Ig-like domain in these GHs is still currently unknown.
A calcium ion is present in the N-terminal domain on the surface near the start of the domain (Fig. 3 ▶). This calcium is octahedrally coordinated by two waters, a cacodylate ion, the carbonyl O atom of Lys103, the carboxylate group of Glu28 and the carboxylate group of Asp162 from a crystallographically related molecule. The role of this calcium (if any) is unclear. CBM9 from Thermotoga maritima xylanase 10A (PDB code 1i82; Notenboom et al., 2001 ▶) and the PKD domain of CtCel9D-Cel44A (PDB code 2c26; Najmudin et al., 2006 ▶) also contain calcium(s); however, the location of the calcium-binding site is different from that in BT2081. It has been postulated that these calcium ions may play a structural role given the nature of the buried binding sites (Najmudin et al., 2006 ▶; Notenboom et al., 2001 ▶). Unlike those proteins, however, the calcium in the BT2081 N-terminal domain is quite solvent-exposed and, as three of the six ligands are solvent molecules and another is a crystallographically related molecule, this bound calcium ion is most likely to be an artifact of crystallization.
Figure 3.

Molecular surface of the BT2081 N-terminal domain colored according to residue conservation by ConSurf (http://consurf.tau.ac.il; Landau et al., 2005 ▶). The most conserved residues are shown in magenta, the least conserved residues are shown in cyan and those with insufficient data to determine the conservation level are shown in yellow. The molecular surface of the C-terminal domain is shown in gray. (a) and (b) are views of opposite surfaces of the N-terminal domain, showing that residue conservation is predominantly on one side. The calcium ion in (b) is represented as a green sphere.
Analysis of BT2081 and its homologs reveals a highly conserved region of the N-terminal domain that may be functionally important. These conserved residues include Glu23, Ala24, Asn26, Glu28 and Ile31 situated between the N-terminus and β-strand 1, as well as residues from two adjacent regions: Phe69, Gly74, Ala75 and Ile77 between β-strands 4 and 5, and Val95, Thr96, Asp99 and Trp102 between β-strands 6 and 7. Together, these residues form a highly conserved patch on one surface of the N-terminal domain, in contrast to the minimal conservation that is observed on the opposite surface (Fig. 3 ▶).
In light of the fact that Ig-like domains in GHs are sometimes CBMs, one possibility is that the N-terminal domain of BT2081 may also be a CBM and, as such, the highly conserved region may be a carbohydrate-binding site. Another possible role for the BT2081 N-terminal domain is as a linker region, similar to the Ig-like domain in NedA. BT2081 is predicted to contain a signal sequence at the N-terminus, which, based on its predominantly hydrophobic amino-acid composition, may correspond to a single transmembrane helix that anchors BT2081 to the bacterial outer membrane. If this is indeed the case, the N-terminal domain may act as a linker region between the membrane surface and the C-terminal domain.
3.4. BT2081 C-terminal domain
Analysis with FATCAT indicates that most of the top structural neighbors of the BT2081 C-terminal domain are either catalytic modules or CBMs of GHs. Representative examples of these structural neighbors are the catalytic module of a Bacillus 1,3–1,4-β-glucanase (PDB code 1byh; Keitel et al., 1993 ▶) and the CBMs from CBM families 6, 11, 16 and 29, which include C. thermocellum xylanase 11A (PDB code 1gmm; Czjzek et al., 2001 ▶), C. thermocellum Lic26A-Cel5E endoglucanase (PDB code 1v0a; Carvalho et al., 2004 ▶), Thermoanaerobacterium polysaccharolyticum mannanase (PDB code 2zew; Bae et al., 2008 ▶) and Piromyces equi cellulose/hemicellulase complex (PDB code 1gwk; Charnock et al., 2002 ▶), respectively (see Table 2 ▶ b).
Despite the low sequence identity between the C-terminal domain of BT2081 and its top structural neighbors, they share the same core β-jelly-roll topology (Fig. 4 ▶). Another shared feature is a conserved calcium-binding site on the convex surface of the β-jelly roll (Fig. 5 ▶). The role of this conserved calcium is most likely to be structural, as it has previously been shown in the case of CBM4 to contribute greater stability to the protein fold (Abou-Hachem et al., 2002 ▶). In BT2081, this calcium is octahedrally coordinated by a water molecule, the carbonyl O atoms of Asn120, Thr174 and Lys176 and the carboxylate O atoms of Glu122 and Asp351. Of this cluster, Glu122 and Asp351 are strictly conserved, Lys176 is highly conserved and Asn120 and Thr174 are poorly conserved among BT2081 sequence homologs, although the latter three would not necessarily be expected to be conserved in amino-acid type as they only make main-chain contacts to the calcium. Several of the immediate neighboring residues (e.g. Phe133, Trp145, Lys237, Trp300 and Phe331) are also highly conserved. Comparison of this site in BT2081 and its structural neighbors reveals that it is most similar, in terms of residue conservation, to the corresponding sites in members from the CBM11 (PDB code 1v0a) and CBM16 (PDB code 2zew) families.
Figure 4.

Superposition of the BT2081 C-terminal domain (yellow) with two representative top structural neighbors as assessed by FATCAT: the catalytic module of a Bacillus 1,3–1,4-β-glucanase (red; PDB code 1byh) and CBM29 of a mannanase from T. polysaccharolyticum (green; PDB code 2zew). (a) View from above the convex surface of the β-jelly-roll core, highlighting the conserved calcium ion represented as spheres and circled. (b) View from above the concave surface of the β-jelly-roll core, highlighting the extended loop regions of BT2081 (circled) which help to form a pocket on the concave surface of the core.
Figure 5.

Coordination of the conserved C-terminal calcium ion of BT2081. Electron density from 2F o − F c (contoured at 2.5σ level) and F o − F c (contoured at 3.0σ level) maps is represented as green and blue mesh, respectively. A 13σ level anomalous signal obtained from data collected below the nickel edge was seen for the calcium and is shown here contoured at the 6.0σ level as magenta mesh. Distances between the calcium ion and its ligands are indicated in Å.
3.5. Putative carbohydrate-binding pocket in the C-terminal domain
The C-terminal domain of BT2081 contains extended solvent-exposed loop regions (Glu144–Pro165, Thr182–Gly199, Ile203–Ile228, Ala240–Cys258, Thr267–Asn284, Phe307–Lys324 and Ser333–Ser346) that are not found in its closest structural neighbors. Several of these loops also contain additional secondary-structural elements including β-strands, α-helices and 310-helices. As a consequence of these extended loop regions, a significant distinction between BT2081 and its structural counterparts is the presence of a deep solvent-accessible pocket in BT2081 formed by highly conserved residues from the concave surface of the β-jelly-roll core and five of the loop regions extending from this core. The concave surface of the β-jelly-roll core forms the base of this pocket, with loop 1 (residues 144–165) forming the pocket terminus, loop 2 (residues 182–199) and loop 3 (residues 203–228) comprising the middle section of the pocket, and loop 4 (residues 267–284) and loop 5 (residues 333–346) forming the side walls near the pocket entrance.
The pocket is lined with a combination of both hydrophobic and hydrophilic residues (aliphatic, Gly148, Gly151, Pro165, Gly189, Met194, Ile196, Ala197, Ala198, Ala211, Met212, Pro215, Leu216, Ala218, Gly276, Ala279, Val330, Leu335 and Gly337; aromatic, Tyr152, Phe202, Phe206, Phe221, Tyr261, Phe280 and Phe340; hydrophilic, Asn149, Thr155, Thr166, Thr185, Asn200, Thr219, Asp259, Thr281, Arg290, Thr332, Ser334, Glu336, Asp338 and His339; Fig. 6 ▶), most of which are highly conserved among BT2081 and its top 20 PSI-BLAST-derived sequence homologs, lending support to this pocket being functionally significant.
Figure 6.

Putative carbohydrate-binding pocket as seen from above the concave surface of the jelly-roll fold. An arrow indicates the pocket entrance. The residues that line the pocket are highlighted in stick representation and are color-coded according to type as follows: aromatic, green; hydrophobic, light green; polar, lilac; acidic, red; basic, blue.
The pocket, which has a volume of ∼1160 Å3 as calculated by PDBsum (Laskowski, 2009 ▶), has a slight kink near its entrance and measures ∼4 Å from the entrance to this bend and ∼19 Å from there to the bottom of the pocket. The width varies along the length of the pocket, ranging from ∼10 Å at the entrance to ∼7.5 Å at the narrowest portion at the bend and widening to ∼13.5 Å near the pocket terminus (Fig. 7 ▶).
Figure 7.

Cutaway side view of the putative carbohydrate-binding pocket highlighting the nature of the residues lining the pocket, including several that may play a role in catalysis. The color scheme of the residues is the same as in Fig. 6 ▶.
Several features of this pocket suggest that it may be a carbohydrate-binding site. First, it is located in a similar location to the carbohydrate-binding sites of structurally similar GH catalytic modules and CBMs. In CBMs that can be classified as ‘glycan-chain-binding’ or type B CBMs (Boraston et al., 2004 ▶), the carbohydrate-binding site is a cleft that extends along the concave surface of the β-jelly-roll fold. These clefts consist of several subsites to which individual sugar moieties of a polysaccharide chain can bind. The topography of the cleft is defined in a large part by several aromatic residues, which are key determinants of polysaccharide binding specificity. Second, hydrogen bonding between the polysaccharide and polar residues in the binding site is also important for binding affinity in these CBMs (Boraston et al., 2004 ▶).
Similar to the carbohydrate-binding sites found in type B CBMs, the putative binding pocket of BT2081 contains several highly conserved aromatic residues arranged in such a way that they form a twisted platform to which a potential polysaccharide could bind. These aromatic residues include Phe202, Phe206, Phe221, Tyr261, Phe280 and Phe340, of which Phe202, Phe206, Tyr261 and Phe340 are strictly conserved among sequence homologs. Moreover, the BT2081 pocket contains a number of polar residues (e.g. Asn149, Thr155, Thr166, Thr185, Asn200, Thr219 and Thr332) whose side chains are positioned for potential hydrogen bonding with a bound polysaccharide, in another characteristic feature which helps to define carbohydrate-binding specificity in type B CBMs.
The evidence from genomic context analysis and the similarity of the fold and putative binding pocket of BT2081 to those of CBMs and catalytic modules of GHs strongly suggest that the pocket in BT2081 may be a carbohydrate-binding site. If so, the next question is whether the C-terminal domain of BT2081 displays GH enzymatic activity or whether it acts instead as a noncatalytic CBM.
3.6. BT2081 as a potential GH
The hydrolysis of glycosidic bonds by GHs usually occurs via general acid catalysis involving two GH carboxylate residues that act as a proton donor and a nucleophile/base (Koshland, 1953 ▶; Sinnott, 1990 ▶). Hydrolysis can occur via two canonical mechanisms, which result in either the inversion or retention of the configuration of the anomeric carbon that undergoes nucleophilic attack.
In Bacillus 1,3–1,4-β-glucanase (PDB code 1byh), a member of GH family 16 (GHF16) and one of the top structural neighbors of the BT2081 C-terminal domain, Glu105 and Glu109 act as the nucleophile and acid/base in a retaining mechanism for the glycosidic bond cleavage (Keitel et al., 1993 ▶). A comparison with the BT2081 pocket reveals that BT2081 lacks these two catalytic glutamate residues at the corresponding locations. However, several other charged residues line the pocket, including Asp259, Arg290, Glu336 and Asp338, all of which are near each another at the pocket entrance (Figs. 5 ▶, 6 ▶ and 7 ▶). Of these residues, Asp259 and Asp338 at the bend of the pocket are particularly interesting candidates for performing catalytic roles because they are positioned opposite each other in the pocket, with their carboxylate O atoms being sufficiently far apart (∼7.5 Å) to accommodate a carbohydrate substrate. The feasibility of Asp259 as a potential catalytic residue in a hydrolysis reaction may further be supported by its interaction with the neighboring Arg290, which would be expected to reduce the pK a of Asp259 and thereby prime it for a role as a nucleophile in a retaining mechanism (Fig. 7 ▶).
Among the top 20 BT2081 homologs derived from PSI-BLAST Asp259 is almost absolutely conserved (19/20 = 95%), while Asp338 (12/20 = 60%) is less so (Fig. 8 ▶). In the eight sequences in which the position equivalent to Asp338 is not an aspartate, seven have an alanine and one has a glycine. The lack of strict conservation at these sites, particularly Asp338, may raise doubts as to the functional role of these residues; however, such an apparent lack of conservation among active-site residues is not unprecedented. In GH family 97 (GH97), for example, its members have evolutionarily diverged into two main subfamilies which differ in their catalytic mechanism, so that the members of one subfamily are retaining GHs while the members of the other are inverting GHs. In addition to these two main subfamilies, six outlier sequences in the GH97 family were found which lacked key catalytic residues, indicating that these members may be inactive or have evolved a different catalytic mechanism (Gloster et al., 2008 ▶). It is feasible that a similar evolutionary divergence may also have occurred in the BT2081 protein family.
Figure 8.

Sequence alignment between BT2081 and its top 20 PSI-BLAST homologs (the alignment is only shown for residues 255–361 of BT2081 for clarity). Residues in white characters on a red background are strictly conserved, while those in red characters with a white background are highly conserved. Red arrows indicate the two residues of BT2081 (Asp259 and Asp338) which may play a role in catalysis. Sequences with aspartates at these positions are highlighted in yellow.
The fact that the putative carbohydrate-binding site in BT2081 is a pocket rather than a tunnel or a cleft, as seen in other carbohydrate-active enzymes, suggests that BT2081, if it were indeed a GH, would be likely to be classified as an ‘exo’ GH (i.e. it would cleave the polysaccharide chain at its ends rather than in the middle) and in this regard would be similar to other exopolysaccharidases, such as glucoamylase and β-amylase (Davies & Henrissat, 1995 ▶).
Manual docking of various polysaccharides (e.g. cellopentaose, mannopentaose and cellohexaose) into the BT2081 pocket reveals that the pocket can accommodate a polysaccharide as large as a pentaose, with three of the sugar units fitting into the section of the pocket extending from the bend to the pocket terminus and the other two sugar units fitting into the portion that extends from the bend to the pocket entrance. Consequently, if BT2081 were indeed proven to be a GH with Asp259 and Asp338 being the catalytic residues, it would be feasible that up to three terminal sugar units of a bound polysaccharide chain could be cleaved off at one time.
4. Conclusions
BT2081 is the first structural representative of a new protein family which is likely to play a role in carbohydrate metabolism in the distal human gut based on structural and genomic context analyses. BT2081 contains Ig-like and jelly-roll domains, which have been observed to be modules in carbohydrate-active enzymes, such as GHs. The N-terminal Ig-like domain may act as a CBM and/or a linker region between the outer membrane and the C-terminal domain. The C-terminal domain contains a pocket that is similar in many respects to carbohydrate-binding sites in CBMs and catalytic modules of GHs. This domain may be catalytic, with Asp259 and Asp338 acting as the catalytic residues in glycosidic bond cleavage. Further biochemical investigation of BT2081 is needed to ascertain whether this is indeed the case.
Additional information about BT2081 is available from TOPSAN (Krishna et al., 2010 ▶) at http://www.topsan.org/explore?PDBid=3hbz.
Supplementary Material
PDB reference: putative glycoside hydrolase, 3hbz
Acknowledgments
This work was supported by the NIH, National Institute of General Medical Sciences, Protein Structure Initiative grant No. U54 GM074898. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL). The SSRL is a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health (National Center for Research Resources, Biomedical Technology Program and the National Institute of General Medical Sciences). Genomic DNA from B. thetaiotaomicron VPI-5482 (ATCC No. 29148D-5) was obtained from the American Type Culture Collection (ATCC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
- Abou-Hachem, M., Karlsson, E. N., Simpson, P. J., Linse, S., Sellers, P., Williamson, M. P., Jamieson, S. J., Gilbert, H. J., Bolam, D. N. & Holst, O. (2002). Biochemistry, 41, 5720–5729. [DOI] [PubMed]
- Andreeva, A., Howorth, D., Brenner, S. E., Hubbard, T. J. P., Chothia, C. & Murzin, A. G. (2004). Nucleic Acids Res.32, D226–D229. [DOI] [PMC free article] [PubMed]
- Bae, B., Ohene-Adjei, S., Kocherginskaya, S., Mackie, R. I., Spies, M. A., Cann, I. K. O. & Nair, S. K. (2008). J. Biol. Chem.283, 12415–12425. [DOI] [PubMed]
- Bolam, D. N., Ciruela, A., McQueen-Mason, S., Simpson, P., Williamson, M. P., Rixon, J. E., Boraston, A., Hazlewood, G. P. & Gilbert, H. J. (1998). Biochem. J.331, 775–781. [DOI] [PMC free article] [PubMed]
- Boraston, A. B., Bolam, D. N., Gilbert, H. J. & Davies, G. J. (2004). Biochem. J.382, 769–781. [DOI] [PMC free article] [PubMed]
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res.37, D233–D238. [DOI] [PMC free article] [PubMed]
- Carvalho, A. L., Goyal, A., Prates, J. A. M., Bolam, D. N., Gilbert, H. J., Pires, V. M. R., Ferreira, L. M. A., Planas, A., Romao, M. J. & Fontes, C. (2004). J. Biol. Chem.279, 34785–34793. [DOI] [PubMed]
- Charnock, S. J., Bolam, D. N., Nurizzo, D., Szabo, L., McKie, V. A., Gilbert, H. J. & Davies, G. J. (2002). Proc. Natl Acad. Sci. USA, 99, 14077–14082. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cohen, A. E., Ellis, P. J., Miller, M. D., Deacon, A. M. & Phizackerley, R. P. (2002). J. Appl. Cryst.35, 720–726. [DOI] [PMC free article] [PubMed]
- Cohen, S. X., Morris, R. J., Fernandez, F. J., Ben Jelloul, M., Kakaris, M., Parthasarathy, V., Lamzin, V. S., Kleywegt, G. J. & Perrakis, A. (2004). Acta Cryst. D60, 2222–2229. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Comstock, L. E. & Coyne, M. J. (2003). Bioessays, 25, 926–929. [DOI] [PubMed]
- Cruickshank, D. W. J. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed]
- Czjzek, M., Bolam, D. N., Mosbah, A., Allouch, J., Fontes, C., Ferreira, L. M. A., Bornet, O., Zamboni, V., Darbon, H., Smith, N. L., Black, G. W., Henrissat, B. & Gilbert, H. J. (2001). J. Biol. Chem.276, 48580–48587. [DOI] [PubMed]
- Davies, G. & Henrissat, B. (1995). Structure, 3, 853–859. [DOI] [PubMed]
- Davies, G. J., Gloster, T. M. & Henrissat, B. (2005). Curr. Opin. Struct. Biol.15, 637–645. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Gaskell, A., Crennell, S. & Taylor, G. (1995). Structure, 3, 1197–1205. [DOI] [PubMed]
- Gloster, T. M., Turkenburg, J. P., Potts, J. R., Henrissat, B. & Davies, G. J. (2008). Chem. Biol.15, 1058–1067. [DOI] [PMC free article] [PubMed]
- Hashimoto, H. (2006). Cell. Mol. Life Sci.63, 2954–2967. [DOI] [PMC free article] [PubMed]
- Henrissat, B. & Davies, G. (1997). Curr. Opin. Struct. Biol.7, 637–644. [DOI] [PubMed]
- Keitel, T., Simon, O., Borriss, R. & Heinemann, U. (1993). Proc. Natl Acad. Sci. USA, 90, 5287–5291. [DOI] [PMC free article] [PubMed]
- Kleywegt, G. J. (2000). Acta Cryst. D56, 249–265. [DOI] [PubMed]
- Klock, H. E., Koesema, E. J., Knuth, M. W. & Lesley, S. A. (2008). Proteins, 71, 982–994. [DOI] [PubMed]
- Koshland, D. E. (1953). Biol. Rev. Camb. Philos. Soc.28, 416–436.
- Krishna, S. S., Weekes, D., Bakolitsa, C., Elsliger, M.-A., Wilson, I. A., Godzik, A. & Wooley, J. (2010). Acta Cryst. F66, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- Landau, M., Mayrose, I., Rosenberg, Y., Glaser, F., Martz, E., Pupko, T. & Ben-Tal, N. (2005). Nucleic Acids Res.33, W299–W302. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A. (2009). Nucleic Acids Res.37, D355–D359. [DOI] [PMC free article] [PubMed]
- Lesley, S. A. et al. (2002). Proc. Natl Acad. Sci. USA, 99, 11664–11669.
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- McPhillips, T. M., McPhillips, S. E., Chiu, H.-J., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M. & Kuhn, P. (2002). J. Synchrotron Rad.9, 401–406. [DOI] [PubMed]
- Najmudin, S., Guerreiro, C., Carvalho, A. L., Prates, J. A. M., Correia, M. A. S., Alves, V. D., Ferreira, L. M. A., Romao, M. J., Gilbert, H. J., Bolam, D. N. & Fontes, C. (2006). J. Biol. Chem.281, 8815–8828. [DOI] [PubMed]
- Notenboom, V., Boraston, A. B., Kilburn, D. G. & Rose, D. R. (2001). Biochemistry, 40, 6248–6256. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Santarsiero, B. D., Yegian, D. T., Lee, C. C., Spraggon, G., Gu, J., Scheibe, D., Uber, D. C., Cornell, E. W., Nordmeyer, R. A., Kolbe, W. F., Jin, J., Jones, A. L., Jaklevic, J. M., Schultz, P. G. & Stevens, R. C. (2002). J. Appl. Cryst.35, 278–281.
- Sauve, V., Bruno, S., Berks, B. C. & Hemmings, A. M. (2007). J. Biol. Chem.282, 23194–23204. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sinnott, M. L. (1990). Chem. Rev.90, 1171–1202.
- Terwilliger, T. (2004). J. Synchrotron Rad.11, 49–52. [DOI] [PubMed]
- Tomme, P., Warren, R. A. & Gilkes, N. R. (1995). Adv. Microb. Physiol.37, 1–81. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2007). Methods Mol. Biol.364, 215–230. [DOI] [PubMed]
- Vriend, G. (1990). J. Mol. Graph.8, 52–56. [DOI] [PubMed]
- Winn, M. D., Murshudov, G. N. & Papiz, M. Z. (2003). Methods Enzymol.374, 300–321. [DOI] [PubMed]
- Xu, J., Bjursell, M. K., Himrod, J., Deng, S., Carmichael, L. K., Chiang, H. C., Hooper, L. V. & Gordon, J. I. (2003). Science, 299, 2074–2076. [DOI] [PubMed]
- Yang, H., Guranovic, V., Dutta, S., Feng, Z., Berman, H. M. & Westbrook, J. D. (2004). Acta Cryst. D60, 1833–1839. [DOI] [PubMed]
- Ye, Y. & Godzik, A. (2004). Protein Sci.13, 1841–1850. [DOI] [PMC free article] [PubMed]
- Yu, X., Visweswaran, G. R., Duck, Z., Marupakula, S., MacIntyre, S., Knight, S. D. & Zavialov, A. V. (2009). Biochem. J.418, 541–551. [DOI] [PubMed]
- Zocco, M. A., Ainora, M. E., Gasbarrini, G. & Gasbarrini, A. (2007). Dig. Liver Dis.39, 707–712. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: putative glycoside hydrolase, 3hbz