

Abstract
The HELP assay is a technique that allows genome-wide analysis of cytosine methylation. Here we describe the assay, its relative strengths and weaknesses, and the transition of the assay from a microarray to massively-parallel sequencing-based foundation.
Keywords: Cytosine methylation, CpG island, epigenetic
1. Introduction
Description of theoretical basis and framework for the technique
Ideally, all studies of cytosine methylation would be performed by shotgun bisulphite sequencing, as recently used for the Arabidopsis thaliana (1-3) and the human (4) genomes. The current problem is the sheer amount of sequencing needed to provide adequate coverage, over 1 billion sequence reads of 75 bp being required for the human genome (4), which remains daunting even with today's massively-parallel sequencing technologies. For example, if the Illumina GAIIx technology is used, generating 10 million alignable sequences per lane, over 100 lanes are required, which usually incurs a cost of at least tens of thousands of US dollars today, and for most centers will be a six figure cost.
The use of shotgun bisulphite sequencing (BS-seq, MethylC-seq) is finding a role in defining reference epigenome datasets (4), which will be very useful for comparison studies, but the requirement for most investigators is an assay that allows a more limited survey of the genome, prompting the development of a number of alternatives. Elsewhere in this volume there are descriptions of such assays, for example based on reagents that bind selectively to methylated DNA or restriction enzymes that cleave DNA when unmethylated. The HELP assay uses a methylation-sensitive restriction enzyme to cut genomic DNA, but differs from many other assays based on the same approach by using the methylation-insensitive isoschizomer MspI as a control. Whereas HpaII on its own would allow the patterns of methylation of two cell samples to be compared, it does not allow a quantitative assessment of the degree of methylation at a specific locus within the genome, whereas the use of MspI as a reference allows at least some degree of quantitation. To put it another way, HpaII on its own allows intergenomic comparisons while a HpaII/MspI strategy allows intragenomic quantification of the degree of methylation also. We originally described loci in terms of being hypomethylated or hypermethylated (5), reflecting the semi-quantitative nature of the assay, but recent extensive bisulphite validation of HELP data showed a strong linear correlation (r=0.88, (6)), indicating that the MspI-normalised data are reasonably quantitative.
Where HELP differs from many other commonly-used assays is the testing of not only CG dinucleotide-rich but also the CG-depleted majority of the genome. While we have previously shown that the representations generated by the high-resolution HELP assay represent over 98% of CpG islands in the human genome (7), in Table 1 we demonstrate that the majority (∼93%) of loci tested reside outside CpG islands. This confers advantages and disadvantages for the investigator. The striking disadvantage is that we really don't know how to interpret cytosine methylation in most genomic contexts, whereas the acquisition of methylation by a CG-dense promoter is reliably associated with silencing of the gene. In a recent study from our lab, we studied ∼1 million loci in the rat genome using a custom-designed HELP microarray, testing whether cytosine methylation was dysregulated by intrauterine growth restriction (IUGR). We found that there were significant changes in cytosine methylation, but that these changes were almost all in intergenic sequences, at loci that were frequently highly-conserved between species, whereas the gene promoters were not altered in any way. When we tested whether these methylation changes were associated with gene expression changes at the nearest genes, we found concordant changes, with hypomethylation of intergenic sequences associated with increased transcription and vice versa (8). Had we focused on promoters and/or CpG islands, we would have found few changes associated with the IUGR phenotype. We conclude that the exploration of the CG-depleted majority of the genome may capture more information about the epigenome but that this information is often strikingly difficult to interpret.
TABLE 1.
| HpaII-amplifiable fragments | Size/bp | Number | In CpG island1 | In CG Cluster2 | |||
|---|---|---|---|---|---|---|---|
| 50-200 | 514,387 | 22.52% | 109,826 | 4.81% | 178,574 | 7.82% | |
| Version 1 HELP (5) | 200-2,000 | 1,016,980 | 44.53% | 44,086 | 1.93% | 104,242 | 4.56% |
| Version 2 HELP (7) | 50-2,000 | 1,531,367 | 67.05% | 153,912 | 6.74% | 282,816 | 12.38% |
| 293,598 | 12.86% | 509,407 | 22.30% | ||||
CpG Island annotations from the hg18 build of the human genome at the UCSC Genome Browser
CG Cluster annotations from Glass JL et al. (27)
Percentages shown are the number of HpaII-amplifiable fragments as a function of the total number in the genome.
HpaII-amplifiable fragments are defined as those that can be (a) amplified using our LM-PCR approach and (b) into which we can place an oligonucleotide for microarray interrogation, giving us a lower limit of 50 bp in size. The 200-2,000 bp representation of our original HELP assay (5) is compared with the increased representation from the more recently-published version (7), demonstrating the substantially increased representation in CG-dense regions like CpG islands. It should also be noted that even with this increased coverage of CG-dense regions, the majority of loci interrogated reside outside these regions, with over 93% of HpaII-amplifiable fragments in the later version of the HELP assay in non-CpG island genomic contexts.
Another disadvantage of a restriction enzyme-based approach is that it only tests the minority of CG dinucleotides located within that restriction site. These assays are therefore innately not comprehensive, and to test for methylation at restriction sites in the genome is akin to the paradigm of looking at night for a lost wallet underneath lampposts. What these assays rely on is the existence of concordant methylation states in cis in the genome, so one CG's methylation is predictive of those nearby. There are empirical data from Eckhardt et al. supporting this long-held impression of methylation blocks, with their data indicating these blocks to be as long as several hundred basepairs in the human genome (9), consistent with our own repeated observations (example shown in Figure 1). Such a concordance of methylation in cis also facilitates affinity-based assays, as the effect of multiple CGs with concordant methylation on a segment of DNA should be strongly influential of binding in these assays.
Figure 1.
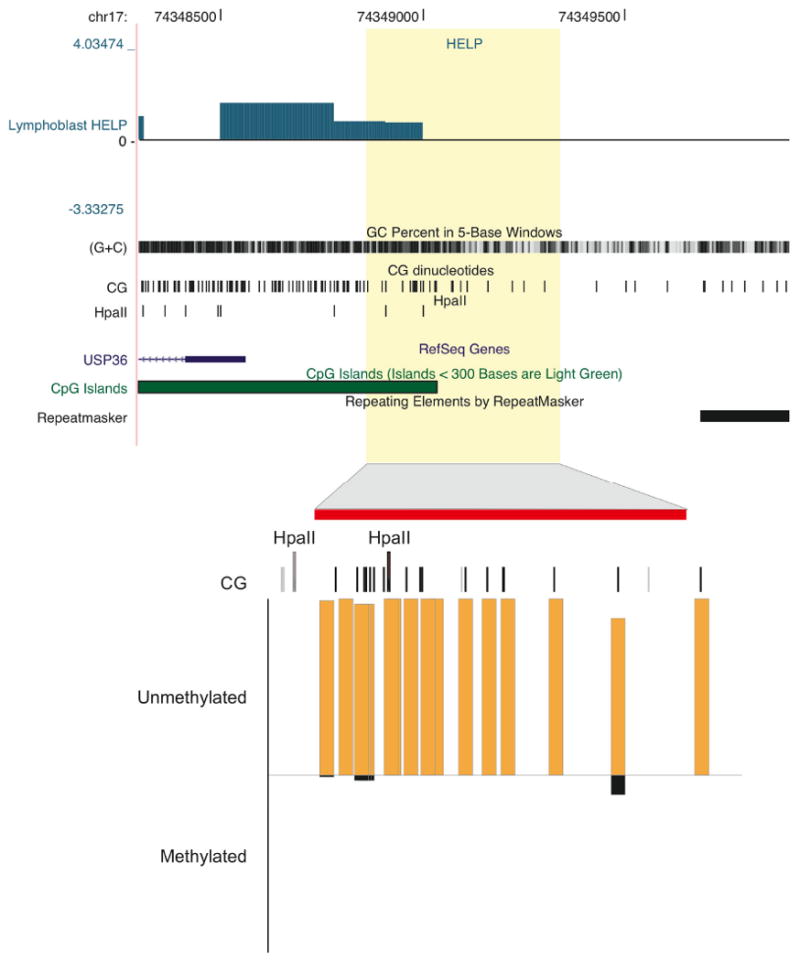
An example of HELP data from human lymphoblastoid cells. We observe hypomethylation (positive values) in a CpG island at the USP36 locus. Quantitative validation using bisulphite MassArray in the lower part of the figure shows near-complete hypomethylation not only at HpaII sites reported by the HELP assay but also at adjacent CGs that are not part of HpaII sites. This represents an example of how methylation at HpaII sites tends to be concordant with that of adjacent CG dinucleotides.
It is important to validate any genome-wide assay with an orthogonal, quantitative, locus-specific assay, not only to test whether the data generated with the genome-wide assay are accurate (technical validation) but also to determine whether differences observed between the cell samples being compared are genuine (biological validation). The gold standard for measurement of cytosine methylation is bisulphite sequencing in some form. Our preference is the use of the MassArray EpiTyper platform from Sequenom, but using software that we have developed to support the platform (10). Technical validation of restriction enzyme-based assays is simplified by the ability to focus on the CG dinucleotide in the informative restriction enzyme digestion site, whereas the adjacent CGs in hundreds of basepairs can influence binding in an affinity system, making technical validation more onerous.
A biochemical variation of cytosine methylation that was discovered to exist in mammalian cells recently is 5-hydroxymethylation (11). 5-hydroxymethylcytosine can be generated from 5-methylcytosine by TET enzymes working cooperatively with the MLL histone methyltransferase (12). At present, it is not known how the hydroxymethylation modification may influence either restriction enzyme or affinity-based assays.
Whereas in the early days of cytosine methylation assays it was sufficient to identify loci with substantial differences in methylation between samples, we now appreciate that in non-cancer human diseases the degree of difference in cytosine methylation may be relatively modest (13). This requires that assays used to test human specimens be sufficiently quantitative that they can discriminate such moderate changes in methylation between samples. Probably the best characterized example of a quantitative, genome-wide assay for cytosine methylation is the reduced representation bisulphite sequencing (RRBS) assay by Meissner et al. (14), which uses bisulphite conversion of a CG-rich subset of the genome for a quantitative, nucleotide resolution study targeting promoters. More recently, the methylation-sensitive cut counting (MSCC) technique was described by Ball et al (15). In their assay, they used a methylation-sensitive restriction enzyme, creating digested loci onto which adapters were ligated, allowing a restriction endonuclease to cleave the adjacent sequence which could then be sequenced using massively-parallel sequencing. The quantitative resolution of their assay appears to be of the order of 20%, and tests over 1.3 million sites in the human genome. As platforms for epigenome-wide association studies in human disease both RRBS and MSCC appear to be extremely powerful potential ways of using massively-parallel sequencing for quantitative studies.
2. Material and Methods
2.1 Sample preparation issues
The HELP protocol has been published a number of times at this point (5, 7, 16, 17) so we will restrict our discussion here to some of the critical issues involved. The first step in HELP assays is to create the genomic representations. We found that the use of dual adapters rather than a single adapter allowed us to avoid the ‘panhandle effect’ in which self-complementary adapter sequences located at both ends of the digested DNA undergo preferential intramolecular annealing below a certain fragment size, preventing us from amplifying the smaller HpaII fragments (7). The dual adapter approach was essential in allowing greater representation of the more CG-dense regions of the genome, where HpaII fragments are correspondingly shorter.
The HELP-seq approach described to use Illumina sequencing (7) can be adapted to any massively-parallel sequencing platform. Our experience is that reads of >30 bp allow unambiguous mapping of a high proportion of sequences flanking HpaII sites, so it is not necessary to extend the sequencing to 70 bp or longer. We described previously the modification of the microarray-based HELP assay as the basis for the sequencing approach (7), but the library preparation could more simply involve the dual adapter approach inherent to Illumina library preparation or a Y-adapter design, each of which would avoid the panhandle effect in creating the genomic representations.
2.2 Microarray design
Microarrays are designed by identifying in a reference genome all of the HpaII/MspI sites located 50-2,000 bp apart as the first step, then designing within each of these fragments an oligonucleotide that reports that locus uniquely. We have published exclusively to date the use of Roche-NimbleGen microrrays, but other customizable platforms could be used in the same manner, or the subset of oligos from a tiling design that is located within the informative HpaII/MspI fragments could be defined and used. When multiple oligos represent a fragment, their information has to be summarized in some way. We have explored various ways of calculating an average value for these oligonucleotides, the most important criterion appears to be to remove oligos that have failed for any reason, usually inadequate signal strength. We define inadequate signal strength as less than 2.5 median average deviations above the median of the control, random probes on the microarray.
2.3 Data analysis issues
The key to the successful adoption of a genome-wide assay is the availability of software resources allowing analysis of the data generated. We have published the software pipeline that supports HELP analysis (18) and have made it available as an opensource Bioconductor package (available online at link provided below). The pipeline includes data quality assessment tools, testing how homogeneous the hybridization was across the microarray, and representations of the signal strength by HpaII/MspI fragment size, which should give a characteristic pattern of low at the extremes and high in the middle of the range. Familiarity with these outputs allows the user to get a sense of whether the data are reliable or the root cause for experimental failure (poor PCR, inadequate labeling, hybridization artefacts etc).
A critical normalization step recognizes the inherent heterogeneity in the ability to amplify different fragment sizes in PCR generation of libraries. We developed a quantile normalization strategy to take the signals from each fragment size range and adjust the range of signals to be similar for all. This was an essential component in the improved performance of the assay compared with validation data that we described (18). Adjustment of the range of signal intensities (mean-centering) between samples is also a valuable step, but care needs to be taken when dealing with an extremely hypomethylated sample, such as those from some tumours, as the signal distribution may be very skewed towards the higher range of values because of underlying biological, not technical artefactual reasons. Readjusting the range of signal intensities on the basis of bisulphite validation assays is a reasonable means of addressing this potential problem.
The final part of the pipeline involves visualizing the data as tracks in the UCSC genome browser. While the data are reasonably quantitative, as described earlier, the HpaII/MspI distributions form a bimodal distribution (5) indicating the greatest discriminatory capacity of the assay for highly-methylated from relatively hypomethylated loci. We therefore distinguish these categories by separating the peaks of the bimodal distribution as negative and positive values, charting them as histogram values using the UCSC genome browser ‘wiggle’ track format, thus highlighting the hypomethylated regions as being distinct from the methylated majority of the genome.
2.4 Validation approach
Bisulphite validation studies target the HpaII sites generating the signals obtained from the microarray. Each microarray signal is the result of digestion at both HpaII/MspI sites flanking the informative fragment. The total amount of digestion occurring to create this fragment is dependent on the flanking HpaII site with the greater amount of methylation, therefore it is this value that we use when correlating microarray with these bisulphite data.
If the flanking area contains more than one HpaII site, it is possible that they drive the representation and consequently the microarray signal. In those infrequent situations in which validation is not correlating with microarray data, such sites should be tested. The more common reason for discordance between microarray and bisulphite data is sequence polymorphism, as described in the next section.
3. Troubleshooting
Hints for troubleshooting
The HELP assay requires intact DNA of a reasonable molecular weight that is clean enough to digest readily to completion. A gel image of HpaII digestion will never be able to distinguish between poor quality DNA and incomplete digestion because of methylation of the majority of HpaII sites in the genome, another reason why the concurrent use of MspI is valuable. When analyzing the data, we find it useful to use the mitochondrial DNA sequences included on the microarray design as a control. Mitochondrial DNA is unmethylated and in high copy number relative to nuclear DNA loci, so a high signal intensity should be seen for all mtDNA loci in both HpaII and MspI channels. Anything otherwise is an indication of poor sample quality or poor digestion of the DNA.
As mentioned earlier, a problem that can occur is due to polymorphic HpaII sites in the genome. The microarrays are designed based on a consensus genomic DNA sequence, anticipating that a given oligonucleotide on the microarray will be associated with a HpaII/MspI fragment of a defined size. The acquisition of new HpaII/MspI sites or the loss of annotated sites will change whether the oligos are still located at an informative locus (the fragment size range may no longer be in the 50-2,000 bp size range of the genomic representation) or may change the size of the fragment generated, which will cause the quantile normalization approach based on predicted fragment size to distort the data. A sequencing-based approach bypasses these concerns, and in fact reveals the striking polymorphism of these CG dinucleotide-containing sites (at least 3% between individuals (7)). When loci identified in microarray studies fail to validate by bisulphite approaches, such polymorphisms should be tested for.
Acknowledgments
The personnel of Einstein's Center for Epigenomics are acknowledged for their contributions.
Abbreviations
- HELP
HpaII tiny fragment Enrichment by Ligation-mediated PCR
- CG/CpG
cytosine-guanine dinucleotide
- RRBS
reduced representation bisulphite sequencing
- MSCC
methyl-sensitive cut counting
- IUGR
intrauterine growth restriction
- mtDNA
mitochondrial DNA
4. Appendices
NimbleGen custom microarrays for human, mouse, rat
These design identifiers need to be specified when ordering these microarrays from Roche-NimbleGen.
| Species | Roche-NimbleGen microarray design ID |
| Human | HG18_HELP |
| Mouse | AE_Mouse_HD2_HELP |
| Rat | 080402_RN4_Greally_HELP_HX1 |
| Cow | 080829_Btau4_Help_Tiling_HX1 |
Software publications and online sources
Thompson RF, Reimers M, Khulan B, Gissot M, Richmond TA, Chen Q, Zheng X, Kim K, Greally JM. An analytical pipeline for genomic representations used for cytosine methylation studies. Bioinformatics. 2008 May 1;24(9):1161-7. Epub 2008 Mar 18. PubMed PMID: 18353789.
Bioconductor HELP package: http://www.bioconductor.org/packages/2.3/bioc/html/HELP.html
Other publications using HELP
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Masako Suzuki, Email: masako.suzuki@einstein.yu.edu.
John M. Greally, Email: john.greally@einstein.yu.edu.
References
- 1.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Nat Genet. 2007;39:61–9. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 2.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Nature. 2008;452:215–9. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Cell. 2008;133:523–36. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khulan B, Thompson RF, Ye K, Fazzari MJ, Suzuki M, Stasiek E, Figueroa ME, Glass JL, Chen Q, Montagna C, Hatchwell E, Selzer RR, Richmond TA, Green RD, Melnick A, Greally JM. Genome Res. 2006;16:1046–55. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, Valk PJ, Lowenberg B, Delwel R, Melnick A. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oda M, Glass JL, Thompson RF, Mo Y, Olivier EN, Figueroa ME, Selzer RR, Richmond TA, Zhang X, Dannenberg L, Green RD, Melnick A, Hatchwell E, Bouhassira EE, Verma A, Suzuki M, Greally JM. Nucleic Acids Res. 2009;37:3829–39. doi: 10.1093/nar/gkp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM. J Biol Chem. 2010;285:15111–8. doi: 10.1074/jbc.M109.095133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson RF, Suzuki M, Lau KW, Greally JM. Bioinformatics. 2009;25:2164–70. doi: 10.1093/bioinformatics/btp382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kriaucionis S, Heintz N. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heijmans BT, Kremer D, Tobi EW, Boomsma DI, Slagboom PE. Hum Mol Genet. 2007;16:547–54. doi: 10.1093/hmg/ddm010. [DOI] [PubMed] [Google Scholar]
- 14.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES. Nature. 2008;454:766–70. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Nat Biotechnol. 2009;27:361–8. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Figueroa ME, Melnick A, Greally JM. Methods Mol Biol. 2009;538:395–407. doi: 10.1007/978-1-59745-418-6_20. [DOI] [PubMed] [Google Scholar]
- 17.Oda M, Greally JM. Methods Mol Biol. 2009;507:77–87. doi: 10.1007/978-1-59745-522-0_7. [DOI] [PubMed] [Google Scholar]
- 18.Thompson RF, Reimers M, Khulan B, Gissot M, Richmond TA, Chen Q, Zheng X, Kim K, Greally JM. Bioinformatics. 2008;24:1161–7. doi: 10.1093/bioinformatics/btn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Figueroa ME, Reimers M, Thompson RF, Ye K, Li Y, Selzer RR, Fridriksson J, Paietta E, Wiernik P, Green RD, Greally JM, Melnick A. PLoS ONE. 2008;3:e1882. doi: 10.1371/journal.pone.0001882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, Fernandez H, Tallman MS, Greally JM, Carraway H, Licht JD, Gore SD, Melnick A. Blood. 2009;114:3448–58. doi: 10.1182/blood-2009-01-200519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Figueroa ME, Wouters BJ, Skrabanek L, Glass J, Li Y, Erpelinck-Verschueren CA, Langerak AW, Lowenberg B, Fazzari M, Greally JM, Valk PJ, Melnick A, Delwel R. Blood. 2009;113:2795–804. doi: 10.1182/blood-2008-08-172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gissot M, Choi SW, Thompson RF, Greally JM, Kim K. Eukaryot Cell. 2008 Mar;7(3):537–40. doi: 10.1128/EC.00448-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopes EC, Valls E, Figueroa ME, Mazur A, Meng FG, Chiosis G, Laird PW, Schreiber-Agus N, Greally JM, Prokhortchouk E, Melnick A. Cancer Res. 2008;68:7258–63. doi: 10.1158/0008-5472.CAN-08-0344. [DOI] [PubMed] [Google Scholar]
- 24.Morey Kinney SR, Zhang W, Pascual M, Greally JM, Gillard BM, Karasik E, Foster BA, Karpf AR. Cancer Prev Res (Phila Pa) 2009;2:1065–75. doi: 10.1158/1940-6207.CAPR-09-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sohal D, Yeatts A, Ye K, Pellagatti A, Zhou L, Pahanish P, Mo Y, Bhagat T, Mariadason J, Boultwood J, Melnick A, Greally J, Verma A. PLoS One. 2008;3:e2965. doi: 10.1371/journal.pone.0002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Einstein F, Thompson RF, Bhagat TD, Fazzari MJ, Verma A, Barzilai N, Greally JM. PLoS One. 5:e8887. doi: 10.1371/journal.pone.0008887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glass JL, Thompson RF, Khulan B, Figueroa ME, Olivier EN, Oakley EJ, Van Zant G, Bouhassira EE, Melnick A, Golden A, Fazzari MJ, Greally JM. Nucleic Acids Res. 2007;35:6798–807. doi: 10.1093/nar/gkm489. [DOI] [PMC free article] [PubMed] [Google Scholar]