

Abstract
The most direct method for synthesizing 2-arylindoles is the oxidative coupling of an arene with an indole. We have shown that both the activity and regioselectivity of these cross-coupling reactions are correlated to the acidity of the medium. This insight has been applied to predict the best conditions for the oxidative cross-coupling of N-alkylindoles, an important class of substrates that was heretofore incompatible with the harsh conditions required for oxidative cross-coupling. Both experimental and computational data indicate that the mechanism for C-H palladation of both the indoles and simple arenes is best described as concerted metalation-deprotonation (CMD), regardless of the substitution on the arenes.
Introduction
The palladium catalyzed oxidative cross-coupling of arenes is a desirable process because the prefunctionalization of either substrate is not required prior to the coupling reaction; thus allowing for a rapid and green synthesis of biaryls.1–9 A consortium of leading pharmaceutical companies have described such processes as some of the most “aspirational” reactions that remain undeveloped in the “key green chemistry research areas”.10 Arylated indoles represent a privileged class of heterocycles that have diverse applications in pharmaceuticals, fragrances, dyes, and agrochemicals.11,12 Ideally the biaryl bond in these compounds could be synthesized by oxidative cross-coupling, but such reactions, involving double C-H bond functionalization, are difficult because the oxidative conditions often decompose electron-rich N-alkylindole substrates. Herein, we disclose that tuning the acidity of the reaction medium allows for the synthesis of arylated indoles via oxidative cross-coupling and prevents the oxidative decomposition of the important, but sensitive, indole substrates and products, and that these reaction conditions allow for three distinct palladation mechanisms.
We have previously shown that benzofuran (1) can be oxidatively cross-coupled with benzene (used as a co-solvent) to regioselectively form 2-phenylbenzofuran (2) (Figure 1, reaction a).13 The reaction was notable because of its high yield, short reaction time and use of molecular oxygen as the terminal oxidant. However, our initial efforts to apply these optimized conditions to indoles were unfruitful. The acidic, oxidizing conditions that were ideal for benzofurans rapidly decomposed N-alkylindole substrates (reaction b).14
Figure 1.

Limits of current oxidative coupling technology (SEM = 2-(Trimethylsilyl)ethoxymethyl, MOM = methoxymethyl, Bn = benzyl)
Subsequently, both we and the Fagnou group achieved the intra- and intermolecular oxidative arylation of N-acetylindoles by replacing the acidic media with a neutral co-solvent15 or by buffering it with either AgOAc or Cu(OAc)2, which can act as both oxidants and weak bases.15–17 Additionally, both groups found that the selectivity for arylation at either the 2- or 3-position of the indole was influenced by the choice of oxidant: AgOAc favoring the 2-aryl product, and Cu(OAc)2 favoring arylation at the 3-position.15,16 However, none of these conditions were compatible with N-alkylindoles, providing negligible yields and a loss of regiochemical control, regardless of the oxidant.
In light of this precedent, we hypothesized that the reactivity of the oxidative cross-coupling could be modulated as a function of the medium's acidity. This was verified by studying the oxidative coupling of benzofuran (1) with benzene as the concentration of organic acid in the reaction was modulated (Figure 2). 1 was an ideal substrate for these studies because of its reactivity in all of the conditions, and because it was not prone to oxidative decomposition. PivOH was used as the acidic additive because it minimized the formation of acetoxylated by-products.18
Figure 2.

Oxidative coupling of benzofuran (1) and benzene as a function of acid concentration
The most active conditions for the oxidative arylation of 1 were the most acidic. As the concentration of organic acid was lowered with respect to the concentration of AgOAc, which served as both a stoichiometric oxidant and a weak base, the rate of the reaction was retarded. All reactions containing PivOH and AgOAc produced nearly a 1:1 mixture of the two regiomeric products (2 and 3), but when PivOH was removed from the reaction, the 3-aryl product was preferentially formed (4:1, 3:2). Consequently, we concluded that both the activity and the regioselectivity of the oxidative cross-coupling were correlated to the acidity of the reaction medium.
Results and Discussion
Reaction Discovery
In light of these trends, we hypothesized that sensitive indole substrates might be amenable to oxidative cross-coupling with arene solvents using the “buffered” conditions which contained near equal amounts of PivOH and AgOAc, but we were cognizant of the regioselectivity issues that could be observed. Gratifyingly, when the “buffered” conditions were applied to the oxidative cross-coupling of N-SEM-indole 4, the reaction proceeded with modest yield and regioselectivity (Table 1, entries 2–4). Reactions containing no PivOH failed to completely convert the starting material (entry 1). The optimal conditions contained a slight excess of AgOAc compared to PivOH, which likely prevented the acid promoted oxidative decomposition of the indole substrate and products.
Table 1.
Discovery and optimization of regioselective oxidative arylation with electron-rich indoles
| Entry | R | PivOH | Conv. [a] | Yield[a] | Ratio[b] |
|---|---|---|---|---|---|
| 1 | H | 0 equiv | 62% | 43% | 2.1:1 |
| 2 | H | 1 equiv | 81% | 58% | 2.2:1 |
| 3 | H | 2.5 equiv | 96% | 69% | 2.6:1 |
| 4 | H | 5 equiv | 98% | 65% | 2.5:1 |
| 5 | CO2Me | 2.5 equiv | 100% | 97% | 8.7:1 |
| 6 | OMe | 2.5 equiv | 100% | 56% | 2.7:1 |
Unreacted starting material and products were isolated by flash chromatography.
5:6, 8:9 and 11:12 determined by GC.
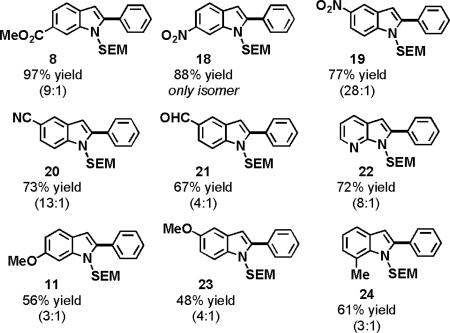
The regioselectivities of the indole reactions were not dramatically affected by the acidity of the reaction medium; rather, they were modulated by substitution on the indole substrate. The electron-withdrawing ester functionality (7) tripled the regioselectivity of the oxidative arylation, favoring the 2-phenylindole (8) and significantly increased the overall yield (entry 5). Substitution with an electron-donating methyl ether (10) had minimal effect on the regioselectivity and a slight deleterious effect on the yield (entry 6).
Scope of the Oxidative Cross-Coupling Reaction
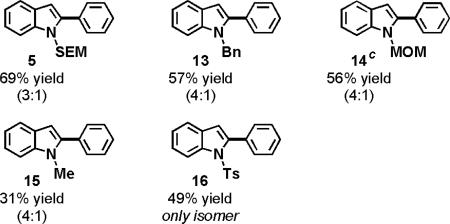
The optimized oxidative coupling conditions were amenable to N-alkylindoles decorated with a wide variety of functional groups (Table 2). Indoles substituted with N-protecting groups such as benzyl, MOM and SEM were all amenable to the oxidative coupling reaction (5, 13, 14). Protection of the indole with the electron withdrawing tosyl group did not prevent the oxidative coupling (16). N-Methylindole provided a diminished yield, despite rapid conversion, which implies that oxidative decomposition of the starting material, the product, or both may be a competitive process for this substrate.
Table 2.
Scope of oxidative arylation reactiona
| Variation of N-Substituentb |
|---|
|
| Variation of Indole Substrateb |
|---|
|
| Variation of Benzene Substrateb |
|---|
|
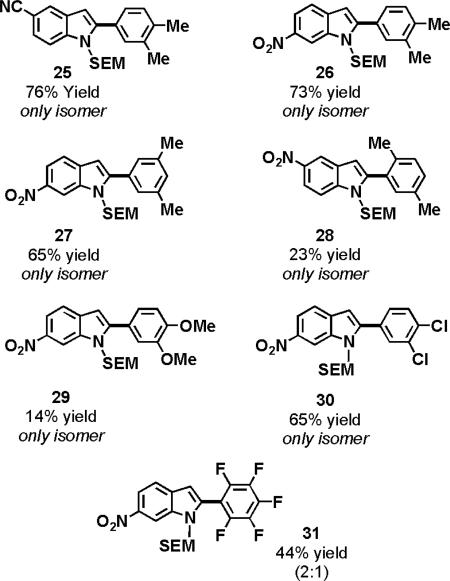
0.15 mmol indole, 4 mL arene, 10 mol % Pd(OAc)2, 3 equiv AgOAc, 2.5 equiv PivOH, 120 °C (MW).
Only the major isomer is shown. The minor isomer is the 3-arylindole. Yields and regioselectivities were obtained by isolation of both isomers following flash chromatography.
Indoles bearing electron-withdrawing groups (8, 18–21) provided higher yields and regioselectivities than those containing electron-donating groups (10, 23, 24). Notably, N-SEM-7-azaindole, which could potentially deactivate the catalyst via ligation, coupled with good regioselectivity and yield (22).
A variety of functional groups were also tolerated on the benzene substrate. Ortho, meta and para-disubstituted benzenes provided only one regiomeric product (25–28). However, the steric interaction associated with the coupling of para-xylene diminished its yield (28). Additionally, there appears to be a limit to the electron density of the arene that can be coupled to the indole, as the yield for coupling ortho-xylene (26) was five times greater than that of veratrole (29). Interestingly, benzene substrates bearing halogens, which are generally considered to be deactivated for electrophilic aromatic substitutions, could also be oxidatively cross-coupled with indoles, albeit in slightly diminished yields (30, 31).
Mechanism of Indole Palladation
In general, two C-H bond activation mechanisms are commonly used to describe the substitution of arenes by electrophilic metals such as Pd(II). The first involves the electrophilic palladation via a SEAr process. For indoles, this proposed mechanism involves the initial palladation at their nucleophilic 3-position, after which a 1,2-migration of the metal to the substrate's 2-position can follow.19 Alternatively, palladation via a concerted metalation-deprotonation (CMD) process with a 6-member transition state has been described as a possible mechanism in the metalation of many heterocyclic arenes, and the regioselectivity of such a process for N-methylindole substrates has been reported recently.20
To probe these two mechanistic possibilities we conducted a series of experiments (Figure 3). We first confirmed that N-SEM-indoles, which contain both electron-donating and electron-withdrawing groups, are nucleophilic at their 3-position using a simple deuteration study (reaction a). This showed that, regardless of the substitution on the indole's benzenoid ring, the palladation of the indole substrate does not occur via direct electrophilic substitution at the 2-position, a pyrrole-like mechanism. However, the longer reaction times required for deuterium incorporation into 33 confirmed the well-known fact that the electrophilic aromatic substitution of electron poor indoles at their 3-position (e.g. 33-d) was less favourable than that of electron-rich substrates (e.g. 32-d).
Figure 3.

Probing the mechanism of indole palladation
A competition experiment between N-SEM-6-methoxyindole (10) and N-SEM-indole (4) found that the indole bearing an electron donating group (10 → 11) reacted 1.34 times faster than its unsubstituted counterpart (Figure 3, reaction b). Additionally, N-SEM-6-nitroindole (34) reacted three times slower than its unsubstituted counterpart. These data seem to indicate that, during the course of the oxidative coupling reaction, the palladation of the indole substrate proceeds via an electrophilic substitution process. As previously described, the electrophilic palladium should attack at the aromatic heterocycle's nucleophilic 3-position, followed by a 1,2-migration of the metal to the indole's 2-position (Figure 4, mechanism b). However, such a mechanism is not in line with the observations shown in Table 2, where the attachment of electron-withdrawing groups to the indole's 6-position, which should destabilize the product of such a migration (37), actually enhanced the formation of 2-arylated indole products (compare 11 with 18, Table 2). Consequently, we hypothesized that indole substrates bearing both electron-donating and electron-withdrawing groups were palladated by the aforementioned CMD mechanism (Figure 4, mechanism a).
Figure 4.

Potential mechanisms for palladating indole
Computational analysis of the palladation of substituted N-methylindoles corroborated the CMD mechanism. The reaction barriers (Gibbs free energy of activation ΔG‡) were evaluated with indoles bearing electron donating and electron withdrawing groups (Table 3). In accord with experimental outcomes, the ΔG‡ for the CMD pathway was found to be 2.0 kcal mol−1 lower for the indole bearing the 6-methoxy group (entry 1) and 2.5 kcal mol−1 higher for the indole bearing the 6-nitro group (entry 3), relative to the non-substituted indole (entry 2).
Table 3.
Distortion/interaction analysis (kcal mol−1) for the lowest free energy CMD transition states at the C-2 sites of N-methylindoles bearing electron donating and withdrawing groups on their 6-positions
| Entry | R | δG‡ | δE‡ | Edist (ArH) | Edist (Pd)a | −Eint | BPd-C2b |
|---|---|---|---|---|---|---|---|
| 1 | OMe | 15.7 | 5.5 | 39.1 | 25.7 | 59.2 | 0.813 |
| 2 | H | 17.7 | 7.6 | 36.1 | 24.8 | 53.3 | 0.794 |
| 3 | NO2 | 20.2 | 10.1 | 31.4 | 23.7 | 45.0 | 0.742 |
Pd(OAc)2.
Mayer bond order for Pd-CAr interaction in the TS.
To explain the impact of electron donating and withdrawing groups on direct arylation reactivity of indoles, the contributions to free energy of activation ΔG‡ were calculated using the distortion/interaction analysis of the corresponding transition states. In this approach, the distortion energy Edist, associated with distortion of both the arene (ArH) and the catalyst (Pd(OAc)2) from their ground state geometries to the geometries in the transitions state, and the electronic interaction energy Eint, between Pd(OAc)2 and ArH in the CMD transition state, were evaluated (Table 3). The OMe group in the indole increases Edist(ArH) by 3.0 kcal mol−1 (entries 1 vs 2). On the other hand, the NO2 group in the indole decreases Edist(ArH) by 4.7 kcal mol−1 (entries 3 vs 2). The Edist(Pd) (between 23.7 and 25.7 kcal mol−1) showed little variation for reactions of different indoles. The presence of an electron donating and withdrawing substituent was also found to influence the interaction energy Eint (Table 3). Eint becomes more negative for the indole bearing an OMe substituent and less negative for the indole bearing an NO2 substituent. The analysis of the covalent interactions in the CMD transition states also indicates that the Pd−C bond order decreases in order OMe > H > NO2. Since the influence of the substituents on Edist(ArH) is less than their influence on Eint, the observed reactivity trend (OMe > H > NO2) for these indole substrates is caused by a change in electronic interaction energy (Eint) between the indole and the Pd(II) catalyst. The changes in Eint can be traced to how these electron donating and withdrawing groups tune the energy and electron distribution of the two highest occupied molecular orbitals of the indole (the HOMO and HOMO-1) upon the substitution (Figure 5).
Figure 5.

The occupied frontier orbitals for the indoles that contribute to the interaction with the Pd(II) catalyst in the CMD transition state. The C-2 atomic contribution (%) and the energy (eV) for each orbital are shown
According to the analysis of fragment orbital interactions in the CMD transition state, significant contributions to indole-to-metal charge transfer interactions only involve two donor orbitals (the HOMO and HOMO-1) of the indole. When the CMD transition state is formed, these two orbitals change their electron population by 13.0% and 10.0%, respectively. The OMe substituent increases the energy of the HOMO to −5.35 eV and increases the HOMO density at the C-2 site (20%). As a result, for OMe-indole, the HOMO becomes the only donor orbital that participates in indole-to-metal charge transfer and, upon the formation of the CMD transition state, this orbital changes its population by 21.1%. On the other hand, NO2 substituent lowers the energies of the occupied orbitals of indole. As a result, the electronic interaction between NO2-indole and Pd(II) is weakened and the Eint is only −45.0 kcal mol−1 (Table 3, entry 3), compared to Eint of −59.2 kcal mol−1 in OMe-indole.
Mechanism of Palladation of the Simple Arene Substrate
The ability to couple both electron-rich and electron-poor arenes to indole substrates is incongruent with our previous work (Table 2, 25–31), in which acidic conditions could not couple heteroaromatic substrates with pentafluorobenzene,13 and basic conditions could not couple N-acetylindoles with anisole.15 To explain these phenomena, we hypothesized that the optimized, buffered reaction conditions allowed for mechanistic promiscuity, whereby the simple arene, like the indole substrate, could be palladated by either electrophilic substitution or proton abstraction processes. Alternatively, the palladation of the simple arene could proceed via concerted metalation-deprotonation mechanism.20
To test these two hypothetical reaction pathways we performed a competition experiment in which the indole 34 was mixed with equimolar amounts of ortho-dichlorobenzene (40) and ortho-xylene (41) (Figure 6, reaction a). Our optimal conditions for oxidative cross-coupling resulted in a near equal mixture of products (1.1:1, 26:30). This product ratio did not change significantly as the acidity of the medium was modulated.
Figure 6.

Probing the mechanism of palladation of the simple arene substrate
This unpredicted lack of chemoselectivity was further exemplified by the regioselective competition reaction that occurs in the oxidative coupling of ortho-trifluoromethyltoluene (42), (Figure 6, reaction b). Interestingly, a 1.4:1 ratio of products (43:44) was observed, with the major product arising from palladation para to the methyl group. The results of these competition studies, in which there is no marked chemoselectivity, are neither consistent with an electrophilic palladation mechanism nor a proton abstraction mechanism involving the build-up of charged intermediates.
Computational analysis of the arene 42 provides insight into the mechanism by which it is palladated (see supporting information for details). The carbon atom para to the methyl group contains the largest partial negative charge, so it should be most amenable to a palladation via an SEAr mechanism. The anion corresponding to abstraction of the position para to the trifluoromethyl group is more stable than that of the anion para to the methyl group, indicating that the former proton would be more easily abstracted by a traditional acid-base reaction. Interestingly, the lengths of these two C-H bonds are computed to be identical (1.075 Å). Previous computational and experimental work has shown the CMD mechanism is favoured by longer C-H bonds.20 Thus the CMD mechanism predicts the minimal selectivity that was observed in the oxidative coupling of 42. Importantly, these computations can be performed using readily available, user-friendly software (such as Spartan21) and a desktop/laptop computer, indicating that predictions about the reactivity of a given substrate in palladation reactions involving CMD mechanisms can be easily obtained by a conventional synthetic organic chemist.
Finally, to differentiate between the possibility of two competing mechanisms and the action of a single, unique mechanism, we performed kinetic isotope studies on the oxidative coupling reaction and varied the concentration of the PivOH (Figure 6, reaction c). It has been previously suggested that that electrophilic palladation processes, in which minimal C-H bond breaking is involved the transition state (KIE < 3), should be favoured by higher concentrations of acid (usually AcOH; in our case PivOH). Conversely, reactions containing less acid, should be more basic, and should favor a proton abstraction process, where considerable C-H bond breaking can be observed in the transition state (KIE > 3).22,23 As predicted, large KIE values were observed in a competition reaction between equimolar amounts of benzene and benzene-d6, indicating that the palladation proceeded through a CMD mechanism and that the cleavage of the solvent arene's C-H bond was rate-determining. Furthermore, the data shows that the KIE for the oxidative coupling did not vary dramatically as a function of the acidity of the medium, which indicates that the palladation of the simple arene occurs via a single CMD mechanism, as opposed to competing electrophilic palladation and proton abstraction mechanisms.
Conclusions
The data presented in this article is one of a small number of studies involving complementary experimental and computational evidence supporting a C-H palladation mechanism involving a concerted metalation-deprotonation mechanism.20 Other researchers have described similar metalation processes, which have been called proton abstraction24–26 or σ-bond metathesis,27 and numerous palladium catalyzed C-H functionalizing reactions have been characterized as having both the ability to react with both electron-rich and electron-poor heterocycles and KIEs in the 3–5 range. Consequently, we hypothesize that the CMD mechanism demonstrated herein, may not be limited to a select set of reactions, but may be operating in a variety of C-H functionalizations. Our work further validates the idea that the CMD mechanism, though often called proton abstraction, is not traditional acid-base chemistry, as it is not affected by the acidity of the reaction media. Rather, a given C-H bond's propensity towards participation in a CMD process is better predicted by the length of the bond, as opposed to its relative acidity.
In summary, we have discovered a mild and efficient method for oxidatively cross-coupling indoles with benzene substrates via double C-H functionalization. Both the indole and the simple arene substrates are palladated by concerted metalation-deprotonation processes. This work represents the first example of the CMD mechanism operating in acidic reaction media, as is common for oxidative cross coupling reactions. However, we have shown that the acidity of the media does not significantly affect the CMD process for the palladation of simple, benzene-derived arenes, but does affect the regioselectivity of the palldation of indole-derived arenes in oxidative cross-coupling reactions. Future work in our laboratory will seek to develop oxidative cross-coupling reactions for new classes of high-value substrates and will be guided by the mechanistic investigations that were disclosed in this article.
Methods
Representative Procedure
A magnetically stirred solution of 1-[2-(trimethylsilyl)-ethoxymethyl]-indole-6-carboxylate (64 mg, 0.21 mmol), palladium acetate (4.7 mg, 0.021 mmol), silver acetate (104 mg, 0.63 mmol), 2,2-dimethylpropanoic acid (53 mg, 0.52 mmol) in 5 mL of benzene was microwave heated at 120°C for 3 h. After evaporation of the solvent, the mixture was then diluted with 40 mL of water and extracted with three 50 mL portions of ethyl acetate. The combined organic extracts were washed with two portions of 40 mL of brine followed by two portions of 40 mL of water and dried with anhydrous magnesium sulfate. After filtration, the solvent is removed at reduced pressure and crude product was purified by column chromatography to give pure 8 (70 mg, 87% yield) and the corresponding 3-phenyl product (8 mg, 10% yield). Both compounds were analytically pure, as determined by 1H-NMR, 13C-NMR and mass spectrometry (see supporting information for complete characterization).
Supplementary Material
Acknowledgements
In memory of Keith Fagnou. The authors thank CEM Corp. for use of a Discover® microwave synthesizer; William Euler, Ben Reukberg and Melanie Sanford for helpful discussions; and Christopher Latendresse for computational assistance. This work was supported by the National Science Foundation (CAREER 0847222), the Petroleum Research Fund (47019-G1) and a Pilot/Feasibility Grant from the Rhode Island INBRE Program (NIH-NCRR P20RR016457). S.G. thanks the Centre for Catalysis Research and Innovation (CCRI), University of Ottawa for funding.
Footnotes
Supplemental Information Complete experimental details, compound characterization and analysis of product purity. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009;38:2447. doi: 10.1039/b805701j. [DOI] [PubMed] [Google Scholar]
- (2).Chen X, Engle K, Wang D, Yu J-Q. Angew. Chem. Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Joucla L, Djakovitch L. Adv. Synth. Cat. 2009;351:673. [Google Scholar]
- (4).Li C-J. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]
- (5).Li B, Yang S, Shi Z. Synlett. 2008;2008:949. [Google Scholar]
- (6).Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]
- (7).Rong Y, Li R, Lu W. Organometallics. 2007;26:4376. [Google Scholar]
- (8).Brasche G, García-Fortanet, Buchwald SL. Org. Lett. 2008;10:2207. doi: 10.1021/ol800619c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhao X, Yeung CS, Dong VM. J. Am. Chem. Soc. 2010;132:5837. doi: 10.1021/ja100783c. [DOI] [PubMed] [Google Scholar]
- (10).Constable DJC, Dunn PJ, Hayler JD, Humphrey GR, Leazer JL, Jr., Linderman RJ, Lorenz K, Manley J, Pearlman BA, Wells A, Zaks A, Zhang TY. Green Chem. 2007;9:411. [Google Scholar]
- (11).Bandini M, Eichholzer A. Angew. Chem. Int. Ed. 2009;48:9608. doi: 10.1002/anie.200901843. [DOI] [PubMed] [Google Scholar]
- (12).Sundberg RJ. The Chemistry of Indoles. Academic Press; 1970. [Google Scholar]
- (13).Dwight TA, Rue NR, Charyk D, Josselyn R, DeBoef B. Organic Letters. 2007;9:3137. doi: 10.1021/ol071308z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Witkop B, Patrick JB, Rosenblum M. Journal of the American Chemical Society. 1951;73:2641. [Google Scholar]
- (15).Potavathri S, Dumas AS, Dwight TA, Naumiec GR, Hammann JM, DeBoef B. Tetrahedron Lett. 2008;49:4050. doi: 10.1016/j.tetlet.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Stuart DR, Villemure E, Fagnou K. J. Am. Chem. Soc. 2007;129:12072. doi: 10.1021/ja0745862. [DOI] [PubMed] [Google Scholar]
- (17).Stuart DR, Fagnou K. Science. 2007;316:1172. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]
- (18).Liégault B, Lee D, Huestis MP, Stuart DR, Fagnou K. J. Org. Chem. 2008;73:5022. doi: 10.1021/jo800596m. [DOI] [PubMed] [Google Scholar]
- (19).Lane BS, Brown MA, Sames D. J. Am. Chem. Soc. 2005;127:8050. doi: 10.1021/ja043273t. [DOI] [PubMed] [Google Scholar]
- (20).Gorelsky SI, Lapointe D, Fagnou K. J. Am. Chem. Soc. 2008;130:10848. doi: 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]
- (21).Spartan '08. Wavefunction, Inc.; [Google Scholar]
- (22).Campeau L, Parisien M, Jean A, Fagnou K. J. Am. Chem. Soc. 2006;128:581. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- (23).Pascual S, de Mendoza P, Echavarren AM. Org. Biomol. Chem. 2007;5:2727. doi: 10.1039/b707940k. [DOI] [PubMed] [Google Scholar]
- (24).García-Cuadrado D, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2006;128:1066. doi: 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]
- (25).García-Cuadrado D, de Mendoza P, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2007;129:6880. doi: 10.1021/ja071034a. [DOI] [PubMed] [Google Scholar]
- (26).Lafrance M, Rowley CN, Woo TK, Fagnou K. J. Am. Chem. Soc. 2006;128:8754. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]
- (27).Hull KL, Sanford MS. J. Am. Chem. Soc. 2009;131:9651. doi: 10.1021/ja901952h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.