

Abstract
We previously identified that overexpression of the platelet-derived growth factor receptor (PDGFR) is associated with metastatic medulloblastoma (MB) and showed that PDGF treatment increases ERK activity and promotes MB cell migration. In this study, we investigated whether ERK regulates Rac1/Pak1 signaling and is critically linked to MB cell migration. Herein we demonstrate that PDGF-BB treatment of MB cells induces concomitant activation of PDGFRβ, MEK1/ERK, Rac1 and Pak1, but suppresses Rho activity, which together significantly promotes cell migration. Conversely, cells transfected with either PDGFRβ or Pak1 siRNA or treated with an inhibitor of Rac1 (NSC23766) or N-myristoyltransferase-1 (Tris-dipalladium) are unable to activate Rac1 or Pak1 in response to PDGF, and consequently, are unable to undergo PDGF-mediated cell migration. Furthermore, we also demonstrate that either chemical inhibition of MEK/ ERK (U0126) or stable downregulation of PDGFRβ by shRNA similarly results in the loss of PDGF-induced ERK phosphorylation and abolishes Rac1/Pak1 activation and cell migration in response to PDGF. However, specific depletion of Pak1 by siRNA has no effect on PDGF-induced ERK phosphorylation, indicating that in MB cells ERK signaling is Pak1-independent, but PDGF-induced migration is dependent on ERK-mediated activation of Pak1. Finally, using tissue microarrays, we detect phosphorylated Pak1 in 53% of medulloblastomas and show that immunopositivity is associated with unfavorable outcome. We conclude that Rac1/Pak1 signaling is critical to MB cell migration and is functionally dependent on PDGFRβ/ERK activity.
Keywords: ERK, Medullobastoma, Migration, Pak1, PDGF
Introduction
Medulloblastoma (MB), the most common malignant brain tumor of children, is characterized by a propensity to spread throughout the central nervous system (CNS), with approximately one-third of patients presenting with metastasis at diagnosis and up to two-thirds at the time of tumor relapse [1, 2]. The presence of metastasis is the single most important prognostic indicator of poor outcome [3-5]. Understanding the signaling mechanisms regulating MB migration and invasion should aid in the development of novel therapeutic strategies aimed at preventing MB metastasis.
We previously identified overexpression of the platelet-derived growth factor receptor (PDGFR) in metastatic MB and showed that PDGF treatment of MB cells induces extracellular signal-regulated kinase (ERK) phosphorylation and promotes migration [6, 7]. PDGFR, a membrane-bound receptor tyrosine kinase (RTK), was first identified as being essential for the regulation of neural cell proliferation, migration and survival in embryologic CNS development [8]. These findings implicate PDGFR/ERK signaling as an important effector of MB growth and metastasis.
Small GTPases of the Ras and Rho families play critical roles in coupling RTK signaling to the cytoskeleton and to other signaling molecules that are essential for cell motility [9-11]. Members of the Ras family generally regulate cell proliferation and differentiation, whereas Rho predominantly controls cytoskeleton rearrangement [12-14]. The most common members of the Rho family are RhoA, Rac1 and Cdc42. RhoA promotes focal adhesion and regulates contractility via actin stress fiber assembly and Rho-kinase inhibition affects cell morphology, motility and invasion through activation of Rac1 in some cell types [15, 16]. Rac1 regulates lamellipodia formation to mediate cell migration [17, 18]. In many cell types, RhoA-GTP level is negatively regulated by Rac1 activation via RTK-mediated signaling [16, 19]. Thus, the balance between Rac1 and RhoA is critical to cell motility, cell–cell adhesion and cell morphology [20, 21].
The Rac1 downstream effectors include a number of proteins, of which the best characterized is the p21 activated kinase (Pak) family [22, 23]. Pak undergoes auto-phosphorylation on multiple sites and is activated upon binding to Rac1- or Cdc42-GTP [24, 25]. Pak1 is a serine/threonine protein kinase that regulates cytoskeletal remodeling and cell motility through actin and microtu-bules [26-31]. Overexpression of constitutively active Pak1 enhanced cancer cell growth and invasion, whereas overexpression of dominant negative Pak1 suppressed invasion [27, 28]. Pak1 has been reported to modulate activation of the Raf/MEK/ERK pathway by either directly activating Raf or priming MEK for activation of Raf in some cell types [32-37]. In HEK293 cells, Pak1 was shown to phosphorylate Raf-1 on serine 338 and MEK1 on serine 298 resulting in cross-activation of ERK [35]. Because of these reported interactions, we investigated whether PDGFR/ERK regulates Rac1/Pak1 signaling and is critically linked to PDGF-mediated MB cell migration and explored whether there is functionally associated cross-talk between PDGFR, ERK and Pak1 in MB cells.
Materials and methods
Cell culture and reagents
Daoy and D556 human medulloblastoma cells were cultured in EMEM with 10% fetal bovine serum (FBS). PDGF-BB was purchased from Sigma (St. Louis, MO). Rac1 inhibitor NSC23766 was purchased from Calbiochem (La Jolla, CA). Tris-dipalladium (Tris-DBA) was generously provided by Dr. Jack L. Arbiser (Emory University, Atlanta, GA).
Western blotting and antibodies
Western blot of whole cell lysates harvested in lysis buffer (Cell Signaling Technology, Danvers, MA) was performed with the following primary antibodies: RhoA and PDGFRβ (Santa Cruz, CA); Rac1 (BD Biosciences, San Jose, CA); phospho-PDGFRβ (Tyr751), phospho-MEK, phospho-ERK, phospho-Pak1 (Thr423)/Pak2 (Thr402) and Pak1 (Cell Signaling Technology); Ras (Upstate, Billerica, MA). Goat or rabbit anti-mouse horseradish peroxidase secondary antibodies (Santa Cruz) were used and the immunore-active bands were detected by ECL. Densitometric analysis of the visualized bands was used to quantitate and compare the relative changes in levels of target proteins between PDGF-treated and untreated cells.
siRNA transfection
Pak1 siRNA (L-003521-00 and J-003521-09), PDGFRβ siRNA (L-003163-00) and negative control non-targeting siRNA (D-001810-01-05 and D-001810-02-05) were purchased from Dharmacon (Chicago, IL). For transfections, 1.5 × 105 cells were seeded in each well of a six-well plate and grown to 50–60% confluency prior to transfection. Cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen, Carisbad, CA) for 48 h according to the manufacturer’s instruction. The final concentration of siRNA was 100 nmol/l.
GTPase pull-down assays
Rac and Rho GTPase activity was analyzed using respective GTPase assay kits (Millipore, Temecula, CA). Briefly, for each assay 200 μg of freshly prepared cell extracts were reconstituted in 500 μl of lysate buffer and added to 10–20 μg of the respective GTPase assay reagent; Pak-1 RBD for Rac activity and Rhotekin RBD for Rho activity. The GTPases were detected by Western blot with anti-Rac1 for Rac-GTP and anti-RhoA for Rho-GTP. The significance of the experiments was determined by the Student’s t-test.
Boyden chamber migration assay
Fibronectin-mediated cell adhesion and migration was assessed using the Chemicon QCM-FN Quantitative Cell Migration Assay (Chemicon International, Temecula, CA) as previously described [6]. Briefly, cells were cultured to 80% confluency and either treated with 5 μM Tris-DBA in serum-free EMEM for 16 h or serum-starved for 24 h, harvested with trypsin/EDTA and resuspended in serum-free EMEM and treated with 100 μM NSC23766 or 10 μM U0126 for 1 h at 37°C. Following incubation, 1.5 × 105 cells in 200 μl serum-free EMEM were seeded into the pre-coated (fibronectin or BSA) upper chambers. To determine whether Pak1 contributes to MB migration, aliquots of 4 × 104 cells (transfected with Pak1 or control siRNA) were placed into the upper chambers. The lower well contained 500 μl serum-free EMEM, with or without 20 ng/ml PDGF-BB. The cells were incubated for 5 h at 37°C (3 h for Tris-DBA), stained with cell staining solution and then washed in distilled water. Cells were eluted with extraction solution and 100 μl transfered to a 96-well microtiter plate for absorbance reading (570 nm). The commercial cell migration assay kits were used in accordance with the manufacturer’s instructions for the accurate measurement of migrating cell number, including performing a pre-test cellular dye absorption nomogram calibration that correlates the OD reading to a known quantity of serially diluted stained tumor cells and subtraction of background membrane absorbance of dye, performed with and without cells in the presence of control BSA. In all cases background membrane absorbance of dye was negligible. P value was calculated using Student’s t-test.
Establishment of stable cell lines
To make stable medulloblastoma cell lines with continuous knockdown of PDGFRβ expression, 2 × 104 Daoy and D556 cells were seeded in 24-well plates, 0.4 μg PDGFRβ shRNA (from SA Biosciences, Frederick, MD. Cat# KH00477 N) gently mixed with 4 μl SureFECT transfection reagent (SA Biosciences, Cat# SA-01) in 50 μl OPTI-MEM solution and then added into each well. 48 h later, cells were split into 60 mm dishes with G418 for further culture. Western Blots were used to verify whether expression of PDGFRβ was downregulated by shRNA in the transfected cells, comparing to the cells transfected with negative control shRNA. After preliminary selection, 200 cells with knockdown of PDGFRβ expression was seeded on each 100 mm dish with G418 for colonies formation. Single colony was picked up and Western Blots were performed at least three times to verify the stable knockdown cells. In this study, we defined A4 and A11 as Daoy PDGFRβ-knockdown cells, B7 and B9 as D556 PDGFRβ-knockdown cells, and NC1 and NC2 as negative control cells.
Tissue microarray
Construction of the tissue microarray (TMA) was performed at the National Insitutes of Health (NIH) consisting of 144 previously untreated paraffin-embedded childhood medulloblastoma tumor tissue specimens, obtained from the pathology libraries of Children’s National Medical Center (CNMC) and the Armed Forces Institute of Pathology (AFIP). IRB approval was obtained from each institution for this TMA construction and analysis. In brief, paraffin-embedded tissue samples were extracted using 0.6 μm diameter needles and mechanically embedded in a paraffin block into an array. Each sample was spotted into the block twice, thus providing two identical spots for each tissue sample. A microtome was used to cut the paraffinized TMA blocks and 5 micron-thick sections were mounted onto positively charged slides for immunohistochemical analysis. A database relating each tumor specimen to its clinical and histologic characteristics was constructed and used for all correlations to the immunostainings. Of the 144 specimens, 94 were deemed evaluable for analysis.
Immunohistochemistry for phospho-Pak1
Standard immunohistochemistry (IHC) for phospho-Pak1Thr212 (#P3237, Sigma Chemical, St Louis, MO) was performed using the TMA described above. Immunostaining for phospho-Pak1 was first performed using normal brain and classic MB tissue samples to serve as negative control and for optimization of the staining procedure, respectively, prior to using the TMA for IHC. TMAs were treated with DAKO antigen retrieval solution and blocked with horse serum prior to application of the primary antibody. Incubation with anti-phospho-Pak1 (1:150 dilution) was performed overnight at 4°C. Immunodetection was performed using the Elite Vectastain ABC system (Vector Laboratories, Burlingame, CA). Color visualization was performed using 3,3′-diaminobenzide as the chromagen substrate (Innovex Biosciences, Pinole, CA). Haematoxylin was used as the counterstain for nuclear detail.
Each tissue sample in the array was independently scored for positivity by two neuropathologists (M.S. and E.J.R.). Scoring was performed blinded and the immunostaining results for each tumor specimen were defined as either negative or positive for phospho-Pak1. The positive or negative grading definitions used were established by the neurophathologists and were based on the relative diffuse cellular homogeneity observed with the specific target staining for each specimen tested. Of the 144 MB specimens, 50 were designated “not-amenable“ for analysis to indicate that in the opinion of the grading neuropathologist, that either the IHC staining or the tissue integrity on the TMA was not of suitable quality to confidently grade the staining. The significance was determined using two sample test of proportion (z test) in STATA software by statistician.
Results
PDGF concomitantly induces activation of PDGFRβ/MEK1/ERK and Rac1, but suppresses RhoA activity in medulloblastoma (MB) cells
We hypothesize that in MB cells PDGFR signaling functions to increase ERK/Rac1 basal activity, which then promotes migration via activation of the downstream effector Pak1. Daoy cells, which we have previously shown have an mRNA expression profile that is similar to metastatic MB [6] and are metastatic in mouse xenografts [38], would thus be expected to express abundant Rac1 protein and to have significantly increased Rac1 and Pak1 activity over basal level in response to PDGF. To determine the effects of PDGF-BB on the temporal activation of PDGFRβ/ERK/Rac1/RhoA in Daoy cells, we examined phosphorylation (activation) of PDGFRβ and MEK1/ERK and the level of Rac1- or RhoA-GTP activity at different time points following PDGF-BB treatment. As shown in Fig. 1a, PDGF increases phosphorylation of PDGFRβ at 8 and 15 min (P = 0.006 and P = 0.028, respectively, as measured by densitometry comparing PDGF treated and untreated cells). An increase of Ras-GTP also was detected at 8 and 15 min (data not shown). Similarly, PDGF induced increased phosphorylation of MEK1 and ERK at 8 and 15 min. As shown in Fig. 1b, Rac-GTP (indicated Rac1 activity) is marginally increased (P = 0.027, comparing the Rac-GTP/total Rac1 ratios at 0 and 15 min), while Rho-GTP is marginally decreased following PDGF treatment (P = 0.003, comparing the Rho-GTP/total RhoA ratios at 0 and 15 min). Together, these results demonstrate that PDGF activates the PDGFR/MEK1/ERK pathway and in turn concomitantly increases Rac1 activity, but suppresses RhoA activity.
Fig. 1.

PDGF concomitantly induces activity of PDGFRβ/MEK1/ERK and Rac1, but suppresses RhoA activity, in medulloblastoma (MB) cells, and Rac activity is essential for MB cell migration. a Daoy MB cells were serum-deprived in medium containing 0.1% FBS for 24 h and stimulated with 10 ng/ml PDGF-BB for 8 and 15 min (0 min represents no PDGF treatment), and then Western blot was performed to detect changes in phosphorylation of PDGFRβ or MEK1/ERK, respectively, at the time points indicated. b Rac-GTP and Rho-GTP levels were assessed by pull down assays using PAK-1 RBD and Rhotekin RBD, respectively. Data are representative of at least three separate experiments. c, d Daoy cells were pretreated with or without 100 μM of the Rac1 inhibitor NSC23766 for 1 h or 5 μM of Tris-DBA for 16 h, stimulated with or without PDGF-BB (10 ng/ml for 15 min), and then Rac- and Rho-GTP pull down assays were performed. Aliquots of 1.5 × 105 cells in serum-free EMEM were placed in the upper well of Boyden chambers and allowed to migrate toward a PDGF-BB (20 ng/ml) gradient in the lower well. NSC23766 and Tris-DBA treatments blocked the PDGF-induced increase in Rac-GTP and concomitant decrease in Rho-GTP (c, d, upper panels) and abolished PDGF-mediated cell migration, without affecting basal migration (c, d, lower panels). Each bar represents the S.E. of triplicate wells
Rac1 activity is essential for MB cell migration
Based on the finding that PDGF induces Rac1 activity in MB, we next sought to determine whether Rac1 activation is critical to PDGF-mediated MB cell migration. To address this question, we first determined whether the Rac1 inhibitor, NSC23766, specifically suppresses PDGF-mediated Rac1 activation in Daoy. As shown in Fig. 1c (upper panel), in comparison to untreated control cells, cells treated with NSC23766 showed marginally decreased levels of Rac-GTP, but maintained levels of Rho-GTP, in response to PDGF (P = 0.03). Importantly, no significant difference was observed on basal levels of Rac-GTP or Rho-GTP without PDGF treatment between NSC 23766 treated and untreated cells, indicating that the action of the inhibition is specifically growth factor-dependent.
Subsequently, we used fibronectin-coated Boyden chamber assays to determine whether Rac1 inhibition alters cell migration. As shown in Fig. 1c (lower panel), PDGF stimulation increased migration by 69% in comparison to unstimulated control cells (P < 0.05), while pretreatment with NSC23766 completely abolished PDGF-mediated migration without effecting basal level migration. We also performed scratch assays in the presence or absence of NSC23766 and found that PDGF-mediated cell motility is markedly blocked by treatment with the Rac1 inhibitor (data not shown), even in the presence of 10% serum (data not shown), indicating that Rac1 plays an important role overall in growth factor-mediated Daoy migration. To confirm that Rac1 activation is required for PDGF-mediated cell migration, we repeated the Rac/Rho-GTP pull down and Boyden chamber migration assays in the presence of Tris-DBA, an inhibitor of the enzyme N-myri-stoyltransferase-1 (NMT-1), which has been reported to act as an anti-neoplastic agent by inhibiting several Ras/Rac downstream signaling pathways, including Akt and ERK, which were correlated to Rac activation [39, 40]. As shown in Fig. 1d, Tris-DBA prevented an increase in the level of Rac1-GTP and a decrease of Rho-GTP in response to PDGF and completely abolished PDGF-mediated cell migration, but had no effect on the basal level growth factor-independent migration.
PDGF induces phosphorylation of Pak1 through PDGFR/Rac1 signaling and siRNA depletion of Pak1 suppresses PDGF-mediated MB cell migration
Our findings show that Rac1 is necessary for growth factor-induced MB cell motility. Pak1, a major common downstream effector for Rac1 in the brain [41], undergoes autophosphorylation on multiple sites and is activated upon binding to Rac-GTP [26, 42, 43]. Phosphorylation of Pak1 at the site of Thr212 is associated with neurite growth and motility of neurons [29, 42] and phosphorylation of Thr423 has been directly associated with Pak1 activity in many cell types. To determine the role of Pak1 in PDGF-induced MB cell migration, we first examined Pak1 phosphorylation of Thr212 and Thr423 in Daoy cells at different time points following PDGF-BB treatment and then tested whether inhibition of PDGFR or Rac1 alters Pak1 phosphorylation in response to PDGF. As shown in Fig. 2a, there is an increase of Pak1 phosphorylation at both sites (Thr212 and Thr423) at various times following PDGF treatment, indicating that PDGF induces phosphorylation of Pak1 in Daoy. This is consistent with the temporal increase in Rac1 activity in response to PDGF. Conversely, as shown in Fig. 2b, depletion of PDGFRβ by transient siRNA transfection abolished PDGF-induced phosphorylation of PDGFRβ and activation of Rac1 and Pak1. Moreover, the expression level of a similar RTK-family protein, EphB1, was not altered by siRNA depletion of PDGFRβ, indicating that the PDGFRβ siRNA specifically targets PDGFRβ expression. Likewise, inhibition of downstream Rac1 activity by NSC23766 or Ras/Rac activity by Tris-DBA similarly blocked PDGF-induced Pak1 activation (Fig. 2b), as well as blocked the activation of Pak1 in response to 10% serum (data not shown); thereby supporting a direct linkage between the PDGFR signaling pathway and Rac1 and Pak1 activation in MB cells.
Fig. 2.

PDGF induces phosphorylation of Pak1 through PDGFR/Rac1 signaling and siRNA depletion of Pak1 suppresses PDGF-mediated MB cell migration. a Daoy cells were starved in serum-free medium overnight, stimulated with 10 ng/ml PDGF-BB, and Western blot for detection of changes in Pak1 phosphorylation using anti-phospho-Pak1Thr212 and anti-phospho-Pak1Thr423 was performed at the indicated time points following PDGF stimulation. b Left panel, Daoy cells were transfected with control or PDGFRβ siRNA (lane 1 and 2, control siRNA; lane 3 and 4, PDGFRβ siRNA), treated with or without PDGF-BB (10 ng/ml for 15 min), and assayed for changes in Rac-GTP and phosphorylation of PDGFRβ and Pak1Thr212. PDGFRβ depletion results in loss of PDGF-induced activation of Rac1 and phosphorylation of Pak1. Right panel, treatment of Daoy cells with NSC23766 (100 μM for 1 h) or Tris-DBA (5 μ M for 16 h), with or without 10 ng/ml PDGF-BB stimulation for 15 min, inhibits PDGF-induced Pak1 phosphorylation. Data are representative of at least two separate experiments. c Daoy cells transfected with control or Pak1 siRNA, stimulated with or without PDGF-BB (10 ng/ml for 15 min), and assayed for changes in Pak1 phosphorylation by Western blot. Pak1 siRNA knockdown cells show absence of PDGF-mediated phosphorylation of Pak1 at Thr212 and Thr423(upper panel). Lower panel showed that control and Pak1 siRNA transfected Daoy cells were assessed for migration in the presence or absence of a PDGF-BB (20 ng/ml) chemoattractant gradient using the Boyden chamber migration assay. Pak1-depleted cells maintain basal migration, but demonstrate ablation of PDGF-induced migration. Each bar represents the S.E. of six transwells. Experiments were performed separately for three times
We then tested whether Pak1 activity correlates with MB migration. To address this question we examined whether depletion of Pak1 by siRNA transient transfection alters PDGF-induced MB cell migration. As shown in Fig. 2c, depletion of Daoy Pak1 levels abrogated PDGF-induced phosphorylation of Pak1 at both sites (Thr212 and Thr423). In the Pak1-deficient cells, PDGF-induced cell migration was significantly attenuated compared to Daoy cells transfected with control siRNA (Fig. 2c). These results were validated using another MB cell type, D556, by again showing that PDGF treatment induces Pak1 phosphorylation and that Pak1 depletion and loss of PDGF-induced Pak1 phosphorylation concurrently and significantly attenuates MB cell migration (Supplementary Fig. 1).
PDGF-mediated activation of Rac1/Pak1 is ERK signaling dependent, but ERK activation is Pak1 independent in MB cells
Studies have shown that inhibition of ERK activation in colon carcinoma cells by treatment with the MEK inhibitor, U0126, results in an increase in cytoskeletal stress fibers and decreased cell motility, while the repression of stress fiber formation by ERK-MAP kinase signaling in Ras-transformed fibroblasts promotes cell motility [40, 44]. One study also reported that PDGF-mediated Pak1 phosphorylation on Thr212 in smooth muscle cells was dependent in part on ERK signaling [36]. To test whether ERK is similarly involved in the regulation of PDGF-mediated activation of Rac1/Pak1 in MB cells, we examined the effect of pretreatment with the MEK/ERK specific inhibitor U0126 in MB cells stimulated with PDGF. We demonstrate that pretreatment of Daoy cells with U0126 completely inhibits PDGF-mediated activation of Rac1, without affecting the basal level of Rac-GTP, and abolishes PDGF-mediated Pak1 phosphorylation at Thr212 and Thr423 (Fig. 3a, P = 0.016 for Thr212, P = 0.04 for Thr423), comparing PDGF stimulated cells with or without U0126 pretreatment). Complete inhibition of PDGF-mediated Pak1 activation by U0126 was confirmed in D556 cells (data not shown), indicating that PDGF-mediated activation of Rac1/Pak1 in MB cells is dependent on the ERK signaling pathway. However, other studies have shown that in a number of cell types specific downstream inhibition of Pak1 can result in the loss of ERK activation and diminished MEK/ERK pathway signaling [32-34, 36]. To determine whether the cross-talk regulation between ERK and Pak1 in MB cells is bi-directional, we depleted Pak1 by siRNA and examined PDGF-mediated phosphorylation of ERK in Daoy and D556. Interestingly, in contrast to these reports in other cell types, downregulation of Pak1 does not affect the basal or PDGF-induced levels of phospho-ERK in MB cells (Fig. 3b), confirming that depletion of Pak1 alone is insufficient to alter activation of ERK and suggesting that cross-talk regulation between ERK/Pak1 is unidirectional in MB cells.
Fig. 3.

PDGF-mediated activation of Rac1/Pak1 is ERK signaling dependent, but ERK activation is Pak1 independent in MB cells. a Daoy cells grown in serum-free medium were pretreated with or without 10 μM of the ERK inhibitor U0126 for 1 h, stimulated with or without 10 ng/ml PDGF-BB for 15 min, and assayed for changes in phosphorylation of ERK and Pak1Thr212 and Pak1Thr423 by Western blot and Rac-GTP by pull down assay. U0126 treatment significantly suppresses PDGF-induced ERK phosphorylation and completely abolishes PDGF-mediated increased activity of Rac1 or Pak1, without affecting the Rac1-GTP basal level (unable to assess the effect on background Pak1 activity since basal level of phospho-Pak1 is undetectable). U0126 (–) represents vehicle control (DMSO) alone. b Daoy and D556 cells were transfected with Pak1 siRNA, stimulated with or without PDGF-BB (10 ng/ml for 15 min), and Western blot performed to detect resultant changes in ERK phosphorylation. Both MB cell types with downregulation of Pak1 by siRNA maintain basal and PDGF-mediated phosphorylation of ERK compared to control cells. Data in Fig. 3 are representative of at least three separate experiments
PDGF-mediated ERK activation is PDGFRβ-dependent and is necessary for migration of MB cells
To determine the extent to which PDGFR signaling regulates ERK activity in MB cells, we used human PDGFRβ shRNA to establish two MB cell lines with stable knockdown of PDGFRβ expression and examined if sustained downregulation of PDGFRβ affects the level of ERK phosphorylation. As shown in Fig. 4a, shRNA-induced knockdown of PDGFRβ leads to significant loss of PDGF-mediated PDGFRβ phosphorylation, and markedly decreases the level of PDGF-induced ERK phosphorylation in both MB cell types (t-tests of P < 0.05, as measured by densitometry comparing PDGF treated A4 and NC1 or B9 and NC1 for Daoy or D556, respectively). PDGF stimulation of control shRNA cells with intact PDGFR induces PDGFR phosphorylation, which can result in a concomitant decrease in PDGFR expression following PDGFR activation, This is a well described growth-factor mediated event in receptor-tyrosine kinases secondary to proteosomal degradation of the receptor following its activation as a regulatory mechanism to extinguish receptor activation. shRNA-induced knockdown of PDGFRβ resulting in the loss of PDGF-mediated PDGFRβ and ERK phosphorylation was validated in a separate set of shRNA PDGFRβ positive and negative stable cell clones in both MB cell types (Supplementary Fig. 2). Loss of phosphorylation of ERK was also observed in Daoy transiently transfected with PDGFRβ siRNA (data not shown), confirming that PDGFRβ acts as an important upstream modulator of ERK signaling and is required for PDGF-mediated ERK activation in MB cells. To confirm the functional role of ERK in the regulation of MB cell migration, we treated cells with U0126 and performed Boyden chamber migration assays. The results show that U0126 completely blocks PDGF-mediated cell migration (Fig. 4b), indicating that ERK activity is necessary for growth factor-induced cell migration of MB cells.
Fig. 4.
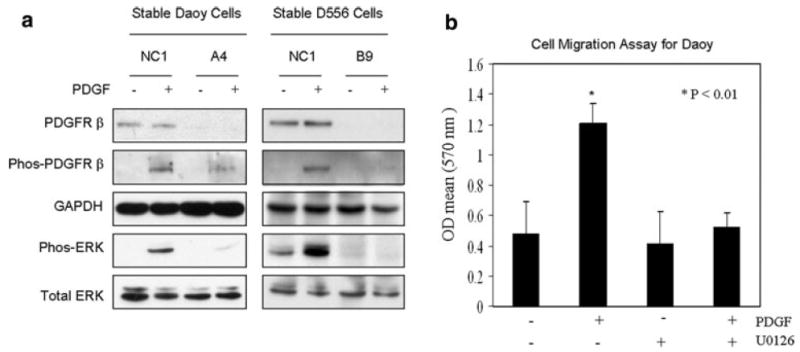
PDGF-mediated ERK activation is PDGFRβ-dependent and is necessary for migration of MB cells. a Daoy and D556 cells were transfected with control or shRNA PDGFRβ and stable clones for each were established (NC1, Daoy or D556 cells with negative control shRNA; A4, Daoy with shRNA knockdown of PDGFRβ; B9, D556 with shRNA knockdown of PDGFRβ), stimulated with or without PDGF-BB (10 ng/ml for 15 min), and Western blot was performed for the detection of changes in PDGFRβ and ERK phosphorylation. MB cells with stable downregulation of PDGFRβ show significant suppression of PDGF-mediated phosphorylation of PDGFRβ and ERK (P < 0.05 for each). Data are representative of at least three separate experiments. b Boyden chamber migration assay performed, as previously described, shows that U0126 (10 μM) pretreatment of Daoy cells for 1 h completely blocks PDGF-directed cell migration, without affecting basal migration. Each bar represents the S.E. of triplicate wells
Medulloblastoma expression levels of phosphorylated Pak1 correlate with poor clinical outcome
Aberrant Rac1 activity has recently been associated with the invasive and malignant phenotype in a variety of cancers [45, 46] and an increased level of tumor-associated phosphorylated Pak1 has been associated with shorter survival time in patients with glioblastoma [29]. To explore the clinical and prognostic relevance of activated Pak1 in MB, we performed immunohistochemistry for phospho-Pak1 on 94 evaluable MB specimens. Of these MB specimens, we detected positive phosphorylated Pak1 immunostaining in 53% of all MB (Table 1). Positive immunostaining for phospho-Pak1, when present, was generally diffusely positive throughout the tumor sample (Fig. 5a). Furthermore, immunohistochemical analysis using the same TMA revealed that Rac1 is diffusely and ubiquitously expressed by nearly all 94 MB evaluated (data not shown). There was no association of immunopositivity with either a specific tumor histologic subtype or with clinical evidence of metastasis (Table 1). However, a highly significant positive rate for phospho-Pak1 was observed in the confirmed deceased MB patients (82%), compared to those who are known to have survived and are confirmed alive without disease recurrence or progression more than 5 years after the completion of the initial treatment for MB (22%; P < 0.001) (Table 1). In addition, Kaplan–Meier survival curve showed a significant decrease of 5-year survival rate in the MB patients with positive phospho-Pak1 (36%, 5 out of 14), compared to the MB patients with negative phospho-Pak1 (90%, 18 out of 20, P < 0.01) (Fig. 5b). These results suggest that the presence of activated Pak1 is a poor prognostic indicator in MB.
Table 1.
Phospho-Pak1 immunostaining results in medulloblastoma (MB)
| Negative | Positive | Positive (%) | |
|---|---|---|---|
| Histologic subtype | |||
| Classic | 11 | 14 | 56 |
| Anaplastic | 10 | 14 | 58 |
| Desmoplastic | 10 | 11 | 52 |
| N/Aa | 13 | 11 | 46 |
| Total | 44 | 50 | 53 |
| Non-metastatic | 41 | 45 | 52 |
| Metastatic | 3 | 5 | 62 |
| Deceased | 2 | 9 | 82 |
| Survivedb | 18 | 5 | 22* |
Histologic subtype of the tumors of deceased = classic (3), anaplastic (1), desmoplastic (2), N/A (5) and subtype of tumors of survived = classic (4), anaplastic (3), desmoplastic (6), N/A (10)
Not able to confirm subtype of MB
Alive without disease recurrence or progression >5 years from the end of initial treatment
P < 0.001, comparing positive % phospho-Pak1 tumors between deceased and survived
Fig. 5.

Immunohistochemistry of MB tissue microarrays for phosphorylated Pak1 and Kaplan–Meier survival curve. a Representative immunostaining of tissue microarray containing 144 MB tumors, of which include 94 were evaluable for phospho-Pak1 analysis. (i) Negative control image for immunostaining for phosphorylated Pak1Thr212 performed on separate MB tumor tissue in order to validate the specificity of the antibody. (ii) Positive control image for immunostaining for phosphorylated Pak1Thr212 performed on separate MB tumor tissue in order for optimization for immunostaining of tissue microarray. Representative images of (iii) negative and (iv) positive phosphorylated Pak1Thr212 immunostaining of MB tumors on the tissue microarrays. b Kaplan–Meier survival curve showed a decrease of 5 year survival rate in the MB patients with positive phospho-Pak1 (36%), comparing to the MB patients with negative phospho-Pak1 (90%) (P < 0.01)
Discussion
Aberrant PDGFR signaling disrupts neuronal migration and normal cerebellar development [47], while expression of PDGFR-related genes is associated with metastasis and somatic mutations of the Shh and Wnt developmental pathways in medulloblastoma [6, 48]. Because Rho family GTPases have been implicated in the invasiveness of various cancer cells [49-52], we hypothesized that PDGFR promotes medulloblastoma (MB) cell migration via regulation of Rho GTPases. Here, we demonstrate that Pak1, a major effector of Rac1 RhoGTPase, is essential for PDGFR-mediated MB cell migration, based on our results that siRNA depletion of Pak1 completely suppresses PDGF-mediated cell migration. Moreover, ERK-dependent phosphorylation of Pak1 is regulated by PDGF-mediated ERK activation in MB cells while inhibition of ERK activation abolishes PDGF-mediated Pak1 activation and MB cell migration. Importantly, we also show that phosphorylation of Thr212 on Pak1 is associated with poor clinical outcome in medulloblastoma. Thus, taken together, we demonstrate for the first time a functional and clinical linkage between medulloblastoma and PDGFR-ERK-Pak1 signaling, which could have important therapeutic relevance.
We previously demonstrated that PDGF promotes ERK phosphorylation in MB cells [6] and here we show that PDGF-mediated activation of ERK is specifically PDGFRβ-dependent. In keeping with reports in other cell types, PDGF-mediated activation of PDGFRβ/Rac1/Pak1 signaling in medulloblastoma cells is rapid, as evidenced by significantly increased downstream effector phosphorylation as early as 8 min after PDGF treatment [16]. Early changes in receptor signaling activity has been linked to cell migration occurring at much later time points since RTK-mediated events responsible for cell migration, such as secretion of proteases and microtubule or intermediate filament dynamics, have been shown to be prolonged following the initiating RTK signal propagation [53]. For example, recent studies have similarly reported PDGF-mediated regulation of cell migration, as measured by Boyden chamber assay 4 h after PDGF stimulation, via transiently increased Rac-GTP and cellular cGMP levels at 5 min after PDGF stimulation [54].
In some cell types, Pak1 activity has been shown to be required for maximal activation of the canonical Raf/MEK/ERK signaling cascade by PDGF, likely because of Pak co-activation of Raf and MEK [35, 36]. However, the convergence of RTK signaling and Pak modulation of MEK/ERK activation remains controversial, and in our study, specific downregulation of Pak1 in MB cells did not alter PDGF-mediated activation of ERK. Our result compares favorably to a study in osteoclast cells similarly showing that Pak1 does not modulate Raf-mediated MEK activation by M-CSF [37]. Thus, RTK-mediated interactions between Rac1/Pak1 and MEK/ERK is likely growth factor- and cell type-dependent, and possibly due to the underlying status of oncogenic Ras.
Although ERK dependency of Pak1 Thr212 phosphorylation by PDGF-BB has been demonstrated previously [36], a functional significance of Pak Thr212 phosphorylation by ERK was not described. Moreover, this earlier report primarily identified Pak1 as a facilitator of ERK signaling. In contrast, our results show that in MB cells Pak1 does not impact ERK activation and that ERK regulation of Pak1 is unidirectional rather than bi-directional. Furthermore, our results demonstrate phosphorylation of both Thr212 and Thr423 on Pak1 by ERK in response to PDGF. Phosphorylation of both Thr sites on Pak1 appears to correlate with Pak1 functional activity in MB cells since decreased phosphorylation at both sites was observed with inhibition of ERK and Rac1, which correlated with ablation of cell migration, but the dependency of function on either specific phosphorylation site remains to be determined. This is significant in light of the finding that Pak1 Thr423 phosphorylation is required to maintain Pak1 in the open active conformation [55]. The absence of functional changes following ERK activation may thus suggest the inability of ERK to phosphorylate Pak1 Thr423 in certain cell types.
Overexpression of Pak1 has been shown in several cancers, and an association between phosphorylated Pak1Thr212 and decreased progression-free survival of patients with glioblastoma has been reported [29, 56-58]. We show a highly significant association between poor clinical outcome and positive phosphorylated Pak1Thr212. Since the number of metastatic tumors in our cohort is small, it is impossible to make a conclusion about the relation of Pak1 phosphorylation to metastasis. Confirmation of clinical correlations in this study was also limited by the lack of an available IHC antibody to reliably detect phosphorylation of tumor-assocaiated Pak1Thr423. Because Pak1 can also regulate survival and transcription, it is possible that the increased frequency of Pak1 phosphorylation observed in poor outcome tumors is related to these cellular functions. Thus, together these results provide crucial rationale for future expanded testing in both human MB and mouse MB models to validate that this pathway is mechanistically linked to in vivo metastasis and/or other oncogenic processes, and suggest that one novel therapeutic strategy for MB may be multi-targeted inactivation of PDGFR/ERK/Pak1.
Supplementary Material
Acknowledgments
Liangping Yuan, Mariarita Santi and Tobey J. MacDonald are supported by NIH R01 grant CA111835.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10585-010-9337-9) contains supplementary material, which is available to authorized users.
Contributor Information
Liangping Yuan, Emory Children’s Center, Aflac Center for Cancer and Blood Disorders, Emory University School of Medicine, 2015 Uppergate Drive NE, Room 442, Atlanta, GA 30322, USA.
Mariarita Santi, Department of Pathology, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA.
Elisabeth J. Rushing, Department of Neuropathology, Armed Forces Institute of Pathology, Washington, DC 20306, USA
Robert Cornelison, Cancer Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Tobey J. MacDonald, Emory Children’s Center, Aflac Center for Cancer and Blood Disorders, Emory University School of Medicine, 2015 Uppergate Drive NE, Room 442, Atlanta, GA 30322, USA tobey.macdonald@emory.edu
References
- 1.Packer RJ. Brain tumors in children. Arch Neurol. 1999;56:421–425. doi: 10.1001/archneur.56.4.421. [DOI] [PubMed] [Google Scholar]
- 2.Packer RJ, Cogen P, Vezina G, et al. Medulloblastoma: clinical and biologic aspects. Neuro Oncol. 1999;1:232–250. doi: 10.1215/15228517-1-3-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725–1731. doi: 10.1056/NEJM199306173282401. [DOI] [PubMed] [Google Scholar]
- 4.Geyer JR, Sposto R, Jennings M, et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. J Clin Oncol. 2005;23:7621–7631. doi: 10.1200/JCO.2005.09.095. [DOI] [PubMed] [Google Scholar]
- 5.Duffner PK, Horowitz ME, Krischer JP, et al. The treatment of malignant brain tumors in infants and very young children: an update of the Pediatric Oncology Group experience. Neuro Oncol. 1999;1:152–161. doi: 10.1093/neuonc/1.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacDonald TJ, Brown KM, LaFleur B, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29:143–152. doi: 10.1038/ng731. [DOI] [PubMed] [Google Scholar]
- 7.Chopra A, Brown KM, Rood BR, et al. The use of gene expression analysis to gain insights into signaling mechanisms of metastatic medulloblastoma. Pediatr Neurosurg. 2003;39:68–74. doi: 10.1159/000071317. [DOI] [PubMed] [Google Scholar]
- 8.Ferns GA, Sprugel KH, Seifert RA, et al. Relative platelet-derived growth factor receptor subunit expression determines cell migration to different dimeric forms of PDGF. Growth Factors. 1990;3:315–324. doi: 10.3109/08977199009003674. [DOI] [PubMed] [Google Scholar]
- 9.Berrier AL, Mastrangelo AM, Downward J, et al. Activated R-ras, Rac1, PI 3-kinase and PKCepsilon can each restore cell spreading inhibited by isolated integrin beta1 cytoplasmic domains. J Cell Biol. 2000;151:1549–1560. doi: 10.1083/jcb.151.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox D, Chang P, Zhang Q, et al. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J Exp Med. 1997;186:1487–1494. doi: 10.1084/jem.186.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bashour AM, Fullerton AT, Hart MJ, et al. IQGAP1, a Rac-and Cdc42-binding protein, directly binds and cross-links microfilaments. J Cell Biol. 1997;137:1555–1566. doi: 10.1083/jcb.137.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 13.Amano M, Chihara K, Kimura K, et al. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science. 1997;275:1308–1311. doi: 10.1126/science.275.5304.1308. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe N, Kato T, Fujita A, et al. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- 15.Salhia B, Rutten F, Nakada M, et al. Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res. 2005;65:8792–8800. doi: 10.1158/0008-5472.CAN-05-0160. [DOI] [PubMed] [Google Scholar]
- 16.Wildenberg GA, Dohn MR, Carnahan RH, et al. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 17.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 18.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 19.Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 20.Sander EE, ten Klooster JP, van Delft S, et al. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leeuwen FN, Kain HE, Kammen RA, et al. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J Cell Biol. 1997;139:797–807. doi: 10.1083/jcb.139.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim L, Manser E, Leung T, et al. Regulation of phosphorylation pathways by p21 GTPases. The p21 Ras-related Rho subfamily and its role in phosphorylation signalling pathways. Eur J Biochem. 1996;242:171–185. doi: 10.1111/j.1432-1033.1996.0171r.x. [DOI] [PubMed] [Google Scholar]
- 23.Sells MA, Knaus UG, Bagrodia S, et al. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- 24.Manser E, Huang HY, Loo TH, et al. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol Cell Biol. 1997;17:1129–1143. doi: 10.1128/mcb.17.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin GA, Bollag G, McCormick F, et al. A novel serine kinase activated by Rac1/CDC42Hs-dependent autophosphorylation is related to PAK65 and STE20. EMBO J. 1995;14:4385. doi: 10.1002/j.1460-2075.1995.tb00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 27.Vadlamudi RK, Adam L, Wang RA, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–36244. doi: 10.1074/jbc.M002138200. [DOI] [PubMed] [Google Scholar]
- 28.Adam L, Vadlamudi R, Mandal M, et al. Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J Biol Chem. 2000;275:12041–12050. doi: 10.1074/jbc.275.16.12041. [DOI] [PubMed] [Google Scholar]
- 29.Aoki H, Yokoyama T, Fujiwara K, et al. Phosphorylated Pak1 level in the cytoplasm correlates with shorter survival time in patients with glioblastoma. Clin Cancer Res. 2007;13:6603–6609. doi: 10.1158/1078-0432.CCR-07-0145. [DOI] [PubMed] [Google Scholar]
- 30.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 31.Lee SH, Kunz J, Lin SH, et al. 16-kDa prolactin inhibits endothelial cell migration by down-regulating the Ras-Tiam1-Rac1-Pak1 signaling pathway. Cancer Res. 2007;67:11045–11053. doi: 10.1158/0008-5472.CAN-07-0986. [DOI] [PubMed] [Google Scholar]
- 32.Eblen ST, Slack JK, Weber MJ, et al. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol. 2002;22:6023–6033. doi: 10.1128/MCB.22.17.6023-6033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith SD, Jaffer ZM, Chernoff J, et al. PAK1-mediated activation of ERK1/2 regulates lamellipodial dynamics. J Cell Sci. 2008;121:3729–3736. doi: 10.1242/jcs.027680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park ER, Eblen ST, Catling AD. MEK1 activation by PAK: a novel mechanism. Cell Signal. 2007;19:1488–1496. doi: 10.1016/j.cellsig.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coles LC, Shaw PE. PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene. 2002;21:2236–2244. doi: 10.1038/sj.onc.1205302. [DOI] [PubMed] [Google Scholar]
- 36.Sundberg-Smith LJ, Doherty JT, Mack CP, et al. Adhesion stimulates direct PAK1/ERK2 association and leads to ERK-dependent PAK1 Thr212 phosphorylation. J Biol Chem. 2005;280:2055–2064. doi: 10.1074/jbc.M406013200. [DOI] [PubMed] [Google Scholar]
- 37.Bradley EW, Ruan MM, Oursler MJ. PAK1 is a novel MEK-independent raf target controlling expression of the IAP survivin in M-CSF-mediated osteoclast survival. J Cell Physiol. 2008;217:752–758. doi: 10.1002/jcp.21550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacDonald TJ, Tabrizi P, Shimada H, et al. Detection of brain tumor invasion and micrometastasis in vivo by expression of enhanced green fluorescent protein. Neurosurgery. 1998;43:1437–1442. doi: 10.1097/00006123-199812000-00101. [DOI] [PubMed] [Google Scholar]
- 39.Bhandarkar SS, Bromberg J, Carrillo C, et al. Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma growth in vitro and in vivo. Clin Cancer Res. 2008;14:5743–5748. doi: 10.1158/1078-0432.CCR-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4:67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 41.Manser E, Leung T, Salihuddin H, et al. A brain serine/ threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- 42.Rashid T, Banerjee M, Nikolic M. Phosphorylation of Pak1 by the p35/Cdk5 kinase affects neuronal morphology. J Biol Chem. 2001;276:49043–49052. doi: 10.1074/jbc.M105599200. [DOI] [PubMed] [Google Scholar]
- 43.Sells MA, Pfaff A, Chernoff J. Temporal and spatial distribution of activated Pak1 in fibroblasts. J Cell Biol. 2000;151:1449–1458. doi: 10.1083/jcb.151.7.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahai E, Olson MF, Marshall CJ. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 2001;20:755–766. doi: 10.1093/emboj/20.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engers R, Ziegler S, Mueller M, et al. Prognostic relevance of increased Rac GTPase expression in prostate carcinomas. Endocr Relat Cancer. 2007;14:245–256. doi: 10.1677/ERC-06-0036. [DOI] [PubMed] [Google Scholar]
- 46.Baugher PJ, Krishnamoorthy L, Price JE, et al. Rac1 and Rac3 isoform activation is involved in the invasive and metastatic phenotype of human breast cancer cells. Breast Cancer Res. 2005;7:R965–974. doi: 10.1186/bcr1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andrae J, Afink G, Zhang XQ, et al. Forced expression of platelet-derived growth factor B in the mouse cerebellar primordium changes cell migration during midline fusion and causes cerebellar ectopia. Mol Cell Neurosci. 2004;26:308–321. doi: 10.1016/j.mcn.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 48.Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaffe AB, Hall A. Rho GTPases in transformation and metastasis. Adv Cancer Res. 2002;84:57–80. doi: 10.1016/s0065-230x(02)84003-9. [DOI] [PubMed] [Google Scholar]
- 50.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 51.Allen WE, Jones GE, Pollard JW, et al. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J Cell Sci. 1997;110:707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- 52.Allen WE, Zicha D, Ridley AJ, et al. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141:1147–1157. doi: 10.1083/jcb.141.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- 54.Guo D, Tan YC, Wang D, et al. A Rac-cGMP signaling pathway. Cell. 2007;128:341–355. doi: 10.1016/j.cell.2006.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chong C, Tan L, Lim L, et al. The mechanism of PAK activation Autophosphorylation events in both regulatory and kinase domains control activity. J Biol Chem. 2001;276:17347–17353. doi: 10.1074/jbc.M009316200. [DOI] [PubMed] [Google Scholar]
- 56.Schraml P, Schwerdtfeger G, Burkhalter F, et al. Combined array comparative genomic hybridization and tissue microarray analysis suggest PAK1 at 11q13.5-q14 as a critical oncogene target in ovarian carcinoma. Am J Pathol. 2003;163:985–992. doi: 10.1016/S0002-9440(10)63458-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balasenthil S, Sahin AA, Barnes CJ, et al. p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J Biol Chem. 2004;279:1422–1428. doi: 10.1074/jbc.M309937200. [DOI] [PubMed] [Google Scholar]
- 58.Carter JH, Douglass LE, Deddens JA, et al. Pak-1 expression increases with progression of colorectal carcinomas to metastasis. Clin Cancer Res. 2004;10:3448–3456. doi: 10.1158/1078-0432.CCR-03-0210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.