

Abstract
Memory CD8+ T cell responses have been considered to be independent of CD80/CD86-CD28 costimulation. However, recall responses are often severely blunted in CD28−/− mice. Whether this impairment represents a requirement for CD28 costimulation for proper memory CD8+ T cell development or a requirement during the recall response is unknown. Furthermore, how CD28 costimulation affects the phenotype and function of memory CD8+ T cells has not been characterized in detail. In this study, we investigate these questions by studying the role of the CD28 costimulatory pathway in memory CD8+ T cell responses to acute and persistent DNA virus infections. Memory CD8+ T cells against vaccinia virus (VV) infection which develop without CD28 costimulation exhibit lower expression of differentiation markers CD27 and CD122 (IL-15Rβ). These memory CD8+ T cells also fail to produce IL-2. Our data indicate that for an optimal recall response, CD28 costimulation is required both for T cell priming and also during the recall response. Similar requirements were observed for memory CD8+ T cell responses during persistent infection with murine gammaherpesvirus 68 (MHV-68) infection, indicating CD28 may play the same role in both acute and persistent infections. Finally, we show deficits in the recall response are restored by IL-2 signaling during recall, but not during priming. The data presented show that CD28 costimulation not only controls the magnitude of the primary response but also affects development of memory CD8+ T cells and is required during the recall response in addition to initial T cell priming.
Memory CD8+ T cells represent a crucial arm of immunological memory. When a host is infected by a pathogen, naive CD8+ T cells specific for the pathogen clonally expand to significantly higher frequencies and differentiate into effector cells to counter and clear the pathogen. Once the pathogen is cleared, the majority of effector CD8+ T cells die and a small proportion (5–10%) of them survive and differentiate into memory CD8+ T cells which provide protection against secondary challenge by the same pathogen (1). Upon secondary encounter, memory CD8+ T cells rapidly expand, secrete key effector cytokines (such as IFN-γ, TNF-α, and IL-2), and exhibit cytotoxicity. Efforts in understanding signals required for proper differentiation of memory cells and rapid secondary expansion are key in optimizing vaccine design against a wide variety of infections (2).
In addition to the recognition of Ag through the TCR (signal 1), costimulation (signal 2), and inflammatory signals such as IL-12 and type I/II IFNs (signal 3) are required for the clonal expansion of naive CD8+ T cells (3, 4). The CD80/CD86-CD28 costimulatory pathway is the major costimulatory pathway and is critical for primary CD8+ T cell responses against DNA, peptide, or dendritic cell (DC) vaccination and antitumor CD8+ T cell responses (5-7). The pathway is also important for controlling the magnitude of primary CD8+ T cell responses to various pathogens (8-22) and is critical for the generation of humoral immunity (23). CD80 and CD86 are up-regulated on APCs upon maturation and they bind to CD28 on the T cell. CD28 signaling is mediated through the PI3K-protein kinase B (PKB/Akt) pathway and growth factor receptor-bound protein 2, and results in up-regulation of CD25 (the IL-2R α-chain), entry of the T cell into the cell cycle, enhanced T cell survival through the up-regulation of the antiapoptotic molecule Bcl-xL, and the production of IL-2 (24, 25). T cell activation leads to up-regulation of CTLA-4, which binds to CD80 and CD86 at a higher affinity than CD28 and negatively regulates the T cell response (26). The CD80/CD86-CD28 pathway has also been shown to be bidirectional, signaling into CD80 and CD86 on DCs resulting in the production of IFN-γ and IL-6 (27).
In contrast to the well-defined nature of signals required for initiating a primary CD8+ T cell response, signals required for memory CD8+ T cell differentiation and secondary responses are less well characterized. Studies have shown that early IL-2 signals (28) and CD4+ T cell help early and/or late (29-31) are crucial for memory cell differentiation, though variations exist between different infection models (32). Duration of the TCR signal has been shown to have little effect on the recall response (33), and the roles of inflammatory signals are not clear. Costimulation, however, is considered to be dispensable for memory T cell responses. This well-established concept stems mainly from studies of memory CD4+ T cell responses in vitro (34, 35), as well as infection of CD28−/− mice with lymphocytic choriomeningitis virus (LCMV)3 (14). However, in contrast to these studies, recall CD8+ T cell responses to influenza virus, Listeria monocytogenes, Salmonella typhimurium, Toxoplasma gondii, and murine gammaherpesvirus 68 (MHV-68) are impaired in the absence of CD28 costimulation (12, 13, 19, 20, 22, 36, 37). Whether these observations indicate requirements for CD28 costimulation during the primary response or during the recall response is not clear and whether CD28 costimulation affects the phenotype and function of memory CD8+ T cells has not been investigated in detail. Recently, studies have shown that CD28 costimulation controls trafficking of memory CD8+ T cells (38), cytokine production and recall responses of memory CD4+ T cells (39), and generation and survival of memory CD4+ T cells (40). Furthermore, our previous work revealed that memory CD8+ T cells during low-level persistent viral infection with MHV-68 produce lower levels of IFN-γ and exhibit an altered phenotype (22). Because memory CD8+ T cell differentiation during MHV-68 infection differs from that during a normal acute viral infection (41), we asked whether CD28 costimulation plays a role in memory CD8+ T cell differentiation and recall responses in an acute viral infection model.
Vaccinia virus (VV) is a dsDNA virus which belongs to the Poxviridae family and the genus Orthopoxvirus (42). It shares high sequence homology to other orthopoxviruses such as variola virus (which causes smallpox infections in humans), ectromelia virus (mousepox), and monkeypox. The natural reservoir of VV is not known, but it is a very potent vaccine against smallpox in humans and replicates to high titers in mice. Despite the frequent contact of humans with smallpox throughout history and recent fears of smallpox as a bioterrorism agent and documented contact of humans with monkeypox (42, 43), the immune responses against this family of viruses is poorly characterized. The major natural route of human to human spread is caused by inhalation of droplets released by the oral, nasal, or pharyngeal mucosa of the infected patient, though some cases of infection are caused by transmission of virus through broken skin, and in the context of vaccination the virus is inoculated intradermally (44). Thus, in these studies, we used the intranasal (i.n.) infection model with the Western Reserve strain (VV-WR). In contrast, MHV-68 is also a dsDNA virus with significantly different characteristics. It belongs to the gammaherpesviridae family and is a natural pathogen in rodents, with similar characteristics to human EBV and Kaposi’s sarcoma-associated herpesvirus (KSHV) infections (45). After initial replication in the lungs after i.n. infection, it establishes a lifelong low-level (latent) persistent infection in the host.
Using the VV-WR model, we show that memory CD8+ T cells generated in the absence of CD28 costimulation exhibit an altered phenotype and do not produce IL-2. Recall responses are significantly impaired in these mice upon peptide and viral challenge, and this is due to a requirement of CD28 costimulation during both the primary and recall response. Similar requirements were observed in memory CD8+ T cell responses against MHV-68. Furthermore, our results indicate that the effect of CD28 costimulation on the recall response is not due to a lack of IL-2 during T cell priming. However, administration of IL-2 during the recall response restored the secondary response in CD28-deficient T cells. Our findings indicate that memory CD8+ T cell responses are not costimulation independent and that several key characteristics of CD8 T cell memory are controlled by the CD80/CD86-CD28 costimulatory pathway.
Materials and Methods
Mice, virus, and reagents
VV-WR strain was obtained from Dr. W. R. Green (Dartmouth Medical School, Lebanon, NH) and was propagated and titered on the 143B cell line. MHV-68 virus (clone G2.4) was originally obtained from Prof. A. A. Nash (University of Edinburgh, Edinburgh, U.K.) and was propagated and titered as previously described (46). A recombinant vaccinia virus expressing the ORF6487–495/Db epitope of MHV-68 (rVV-ORF6) was obtained from Dr. P. Doherty (St. Jude Children’s Research Hospital, Nashville, TN) (47). Intranasal infections with VV-WR (1000 PFU) or MHV-68 (400 PFU) mice were performed under anesthesia with ketamine/xylazine. For i.p. infections with VV-WR or rVV-ORF6, 2 × 106 PFU were given unless stated otherwise.
C57BL/6 and congenic B6-Ly5.2-Cr (which are Ly5.1/CD45.1+) mice were purchased from The National Cancer Institute (Bethesda, MD). CD80/CD86−/− mice on the C57BL/6 background were obtained from Dr. L. Kasper (Dartmouth Medical School) and CD28−/− mice were obtained from The Jackson Laboratory and bred in the Dartmouth-Hitchcock Medical Center mouse facility. The Animal Care and Use Program of Dartmouth College approved all animal experiments.
Tissue preparation
Single-cell suspensions of spleen and lung lymphocytes were prepared as described previously (22). For the preparation of PBL, peripheral blood was collected in medium containing 10 U/ml heparin and RBC were lysed.
Plaque assay and body weight measurements
Infectious virus titers in the lungs and spleens were determined by standard plaque assays similar to the MHV-68 plaque assay previously described (46), except that the assay was performed on 143B cells for 2 days and were fixed with a 1:1 mixture of acetone and methanol. For measuring weight loss, infected mice were weighed on the day of infection and every other day after infection. Data are represented as percentage of initial body weight (at the day of infection).
MHC/peptide tetramer, Ab staining, and flow cytometric analysis
MHC/peptide tetramers for the VV-WR epitope B8R20–27 (TSYKFESV)/Kb or the MHV-68 epitopes ORF6487–495 (AGPHNDMEI)/Db and ORF61524–531 (TSINFVKI)/Kb, which were conjugated to allophycocyanin, were obtained from the National Institutes of Health Tetramer Core Facility (Emory University, Atlanta, GA). Cells were stained for 1 h at room temperature in the dark as previously described (48). Cells were further stained with PerCP-conjugated anti-CD8α (clone 53-6.7) and Abs against surface markers as described previously (49).
In vivo cytotoxicity assay
Naive C57BL/6 splenocytes were pulsed with 1 μg/ml B8R20–27 (B8R) peptide or no peptide and were labeled with 2.5 or 0.25 μM CFSE (Molecular Probes), respectively. Cells were mixed at a 1:1 ratio, and ~2 × 107 total cells were injected i.v. One day later, mice were sacrificed and collected spleen cell suspensions were incubated with 20 μg/ml 7-aminoactinomycin D (Sigma-Aldrich) for 15 min at room temperature in the dark to label dead cells. Cells were analyzed by flow cytometry and specific lysis was calculated using the formulas: ratio = (number CFSElow:number CFSEhigh) and percentage of specific lysis = (1 − (ratio of naive:ratio of infected) × 100).
Intracellular cytokine staining
Splenocytes were incubated with 1 μg/ml B8R peptide plus 10 U/ml IL-2 and 10 μg/ml brefeldin A in complete medium at 37°C for 5 h. Cells were stained with anti-CD8 and anti-CD44 Ab, then fixed, and rendered permeable before staining with allophycocyanin-conjugated anti-IFN-γ (XMG1.2) and PE-conjugated anti-TNF-α (MP6-XT22), anti-IL-2 (JES6-5H4), PE-conjugated anti-granzyme B (clone 100), or isotype control (50). The percentage of cells producing cytokines were calculated by subtracting the background observed with the no peptide control.
Adoptive transfer and challenge experiments
For direct challenge experiments in the VV-WR system, naive or infected B6 or CD28−/− mice were injected i.v. with B8R20–27 peptide (50 μg) and LPS (30 μg). In the MHV-68 system, naive or infected B6 or CD28−/− mice were challenged with 2 × 106 PFU of rVV-ORF6 i.p., and expansion of virus-specific CD8+ T cells in the spleen was enumerated by MHC/peptide tetramer staining at the indicated days postchallenge.
For adoptive transfer experiments, CD8+ T cells were purified (typically <95% pure) from spleens of VV-WR- or MHV-68-infected mice (2+ months postinfection (p.i.)), and equal numbers of CD8+ T cells (1.5–5 × 106 cells) were injected into congenic B6-CD45.1 (B6-Ly5.2-Cr) mice i.v. Cells from each group were stained to determine the percentage of CD8+ T cells and tetramer+ cells and, from that data, the exact number of virus-specific memory CD8+ T cells that were injected into each mouse was calculated. One day after transfer, mice were challenged with 2 × 106 PFU of VV-WR or VV-ORF6 i.p. Spleens were harvested 5 or 6 days later as indicated and stained with the appropriate MHC-peptide tetramer complexes, anti-CD8, and anti-CD45.2 or CD45.1 Abs (which marks the donor population). To calculate fold expansion of transferred virus-specific memory CD8+ T cells, the total number of virus-specific memory CD8+ T cells in the spleen of each mouse (5 or 6 days postchallenge) was calculated and the cells were divided by the number of virus-specific CD8+ T cells initially transferred into the recipient (before challenge). Four to six recipients per group were used in each experiment.
IL-2 immune complex injection
For restoration of IL-2 signaling during priming, C57BL/6 or CD28−/− mice were infected with VV-WR i.n., and mice in the CD28−/− group were injected with either 50 μg of anti-IL-2 mAb (clone S4B6) mixed with 1.5 μg of murine IL-2 (mIL-2; eBioscience) or 50 μg of control rat IgG (R-IgG; Jackson ImmunoResearch Laboratories) i.p. daily, from days 0 to 6 p.i., as described previously by Boyman et al. (51). Control C57BL/6 mice were treated with R-IgG. On day 7, mice were bled and PBL were stained for CD8, CD44, and CD122 expression to confirm the efficacy of the IL-2 immune complex.
For restoration of IL-2 signaling during recall, CD8+ T cells from VV-WR-infected C57BL/6 or CD28−/− mice were purified and transferred into naive congenic B6-CD45.1 mice, and 1 day later mice were challenged with 2 × 106 PFU of VV-WR i.p. (as described above). The IL-2 immune complex (anti-IL-2 mAb plus mIL-2) was given daily from days 0 to 5 postchallenge.
Statistical analysis
Values of p were calculated using Student’s t test. A value of p < 0.05 was considered significant.
Results
Virulence and viral replication of VV-WR in CD28−/− mice after i.n. infection
To study the role of CD28 costimulation in the development of memory CD8+ T cells during an acute infection, we chose the VV-WR i.n. infection model, which represents a natural route of infection for poxvirus infections. VV-WR elicits severe virulence upon i.n. infection (52). To determine the sublethal dose which elicits a potent T cell response, we titered the viral dose. Female C57BL/6 mice were infected i.n. with either 105, 104, or 103 PFU of VV-WR and were monitored daily for survival. None of the mice (0 of 8) survived infection with 105 PFU (median survival, 7.3 ± 0.5 days), whereas 50% survived (4 of 8) when infected with 104 PFU, and all mice survived when infected with 103 PFU (7 of 7). Therefore, in experiments described below, all mice were infected with 103 PFU.
Mice display signs of disease upon i.n. infection with VV-WR, which is accompanied by severe weight loss (53). We did not observe any significant differences in degree of weight loss in wild-type C57BL/6 and CD28−/− mice after infection, although CD28−/− mice displayed a slightly enhanced degree of weight loss (Fig. 1A). We next measured the kinetics of viral replication in various organs during the acute infection. In both wild-type and CD28−/− mice, the viral titers in the lungs peaked at day 5 and were mostly cleared by 16 days p.i., and no significant differences were observed (Fig. 1B). The virus also replicated to high titers in the ovaries similarly in both strains and minimal replication was observed in the spleens (data not shown). Therefore, CD28 costimulation does not affect the course of disease or kinetics of viral replication upon i.n. infection with VV-WR.
FIGURE 1.
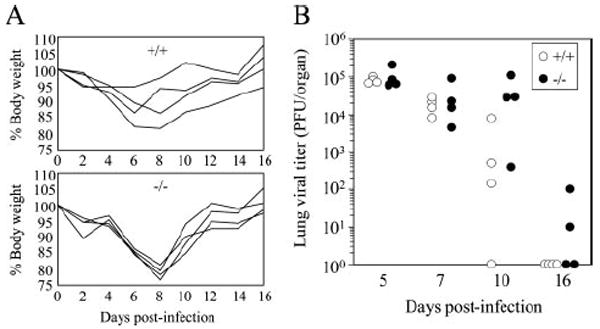
Viral replication and associated weight loss are not affected by CD28. A, Weight loss was measured at the indicated days p.i. as described in Materials and Methods. Each line represents an individual mouse. B, Virus titers in the lung and spleen were measured at the indicated time points. Representative data from two independent experiments consisting of four to eight mice per group are shown.
Kinetics of the CD8+ T cell responses to VV-WR in the absence of CD28
Next, the kinetics of the antiviral CD8+ T cell response was measured. First, the virus-specific CD8+ T cell response to the immunodominant B8R epitope (54) was quantified at the peak of the response by tetramer staining. At day 10 p.i., there was a significant reduction in both the frequency (Fig. 2A) and total number (Fig. 2B) of B8R-specific CD8+ T cells in the lung and spleen in CD28−/− mice. Next, we followed the kinetics of the CD8+ T cell response in the peripheral blood over time. There was a sharp increase in the percentage of CD8+ T cells (Fig. 2C), CD8+ T cells that were CD62Llow (Fig. 2D), and the frequency of B8R-specific CD8+ T cells (Fig. 2E), which peaked at day 10 p.i. In CD28−/− mice, the increases in these parameters were significantly blunted. However, the CD8+ T cell response was comparable to that of wild-type C57BL/6 mice later in the response (Fig. 2, C–E), and the frequency of virus-specific memory CD8+ T cells was similar during the memory phase (>30 days; Fig. 2E). The frequency (Fig. 2F) and total number (Fig. 2G) of memory CD8+ T cells in the lung and spleen at 66 days p.i. were also comparable in C57BL/6 and CD28−/− mice. Therefore, as has been previously reported in various models, the primary CD8+ T cell response is significantly affected by CD28 costimulation, but the numbers of virus-specific memory CD8+ T cells are comparable.
FIGURE 2.
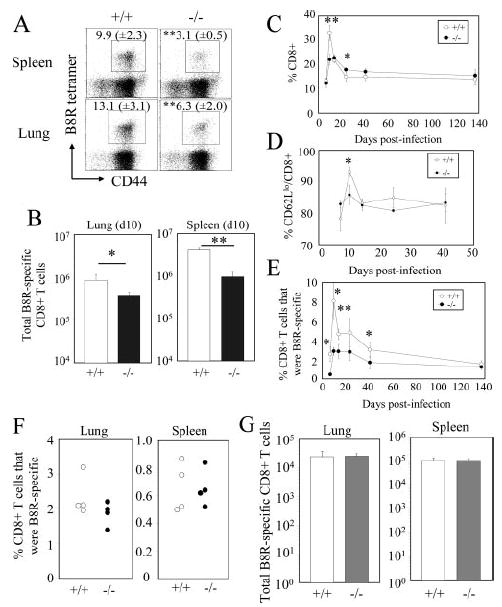
Kinetics of the CD8+ T cell response against i.n. VV-WR infection in CD28−/− mice. A and B, The B8R-specific primary CD8+ T cell response in the lung and spleen was quantified by tetramer staining at 10 days p.i. A, Representative plots gated on CD8+ cells are shown. Numbers indicate percentage of B8R tetramer+ CD8+ T cells. B, Total numbers of B8R-specific CD8+ T cells in each organ are graphed. C–E, Kinetics of the CD8+ T cell responses in the peripheral blood upon VV-WR infection. The percentages of CD8+ (C), CD62LlowCD8+ (D), and B8R tetramer+CD8+ (E) were measured at the indicated time points. F and G, B8R-specific memory CD8+ T cells in the lung and spleen were measured by tetramer staining at 66 days p.i. F, Percentages of B8R tetramer+CD8+ T cells are graphed. Each dot represents an individual mouse. G, Total number of B8R-specific CD8+ T cells in each organ. Representative data from two independent experiments consisting of four mice per group are shown. Error bars and numbers in parentheses indicate SD. *, p < 0.05; **, p < 0.01.
Memory CD8+ T cells in CD28−/− mice exhibit an altered phenotype and do not produce IL-2
We investigated the impact of CD28 deficiency on CD8+ T cell memory by first examining the phenotype of VV-WR-specific memory CD8+ T cells. The expression of activation/differentiation markers, CD44, CD62L, CD69, CD25, CD127, and CD122, were analyzed on CD8+ B8R tetramer+ cells in the spleen, the lung, and the bone marrow (BM, which is known to be a major reservoir of the central memory T cell subset) (55). The B8R-specific memory CD8+ T cells in all organs of CD28−/− mice expressed lower levels of CD122 (IL-15Rβ), measured by mean fluorescence intensity and percentage of cells positive for its expression (Fig. 3A). Furthermore, the B8R-specific memory CD8+ T cells expressed lower levels of CD27 in both the spleen and the BM. Memory CD8+ T cells in the lung expressed low levels of CD27 in both wild-type and CD28−/− mice, which is expected because memory CD8+ T cells in the peripheral organs are known to be mainly CD27low (56). Expression of CD44, CD62L, CD69, CD25, and CD127 was not affected (data not shown). From these phenotyping studies, we suspected that the memory CD8+ T cells that develop in the absence of CD28 costimulation may be functionally impaired.
FIGURE 3.
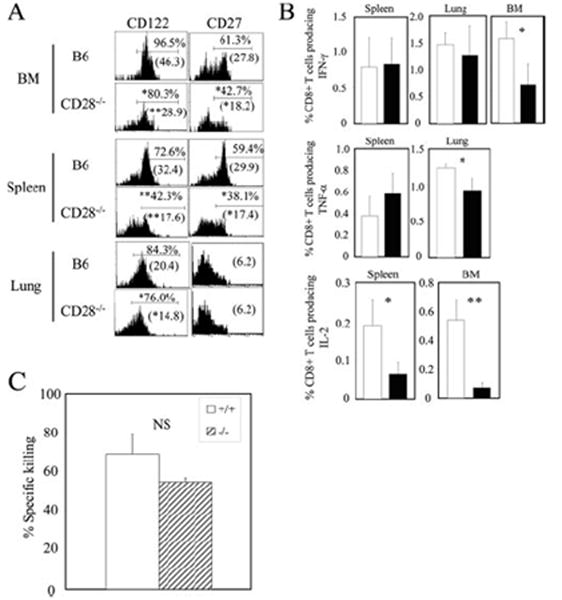
VV-specific memory CD8+ T cells exhibit an altered phenotype and do not produce IL-2 in the absence of CD28 costimulation. A, CD122 and CD27 expression on B8R-specific memory CD8+ T cells in various organs was analyzed by tetramer staining at day 66 (lung) and day 90 (spleen and BM) p.i. Representative plots gated on CD8+ B8R tetramer+ cells are shown. Numbers above indicate percentage of cells staining positive for the respective markers, and numbers below in parentheses represent the mean fluorescence intensity. B, Cytokine production by B8Rspecific memory CD8+ T cells was measured on day 66 (lung) and day 131 (spleen and BM) p.i. by intracellular cytokine staining following stimulation by B8R peptide. C, Cytotoxicity was assessed by an in vivo cytotoxicity assay as described in Materials and Methods. Representative data from two independent experiments consisting of three or four mice per group are shown. *, p < 0.05; **, p < 0.01.
To assess the functions of the memory cells, we measured cytokine production by the memory CD8+ T cells using intracellular cytokine staining. The absence of CD28 costimulation did not affect the percentage of CD8+ T cells producing IFN-γ in the spleen and lung, but the percentage in the BM was reduced 2-fold (Fig. 3B). TNF-α production was not affected in the spleen and a slight reduction was observed in memory CD8+ T cells in the lungs. The most striking result was that IL-2-producing memory CD8+ T cells were virtually absent in both the spleen and the BM of CD28−/− mice (Fig. 3B). Memory cells in the lungs produced minimal amounts of IL-2 in wild-type and CD28−/− mice (data not shown), which was expected because the cells residing in this tissue are mainly effector memory cells which do not produce IL-2 (56). Therefore, CD28 costimulation is crucial for IL-2 production by memory CD8+ T cells. Next, we assessed the cytotoxicity of memory CD8+ T cells that developed in the absence of CD28 costimulation using an in vivo cytotoxicity assay. We observed no significant differences in killing of viral peptide-pulsed targets (Fig. 3C), indicating that CD28 costimulation plays a minimal role in the development of cytotoxicity of memory CD8+ T cells.
Impaired recall responses by memory CD8+ T cells in the absence of CD28 costimulation
The ability to mount a rapid and robust recall response is a hallmark of immunological memory. We next tested the ability of memory CD8+ T cells in CD28−/− mice to mount a recall response. We were unable to reinfect these mice with the same virus because CD28 costimulation is critical for the humoral response; therefore, wild-type and CD28−/− mice would differ in their ability to neutralize the virus during secondary challenge. We circumvented this problem by taking two distinct approaches. First, we directly challenged VV-WR-infected C57BL/6 or CD28−/− mice with B8R peptide plus LPS i.v. and analyzed the expansion of B8R-specific CD8+ T cells in the spleen 6 days later. Using this immunization scheme, minimal expansion was observed in uninfected C57BL/6 mice (0.7 ± 0.1% B8R specific). When infected C57BL/6 mice were challenged, a robust recall response was observed (Fig. 4, A and B). However, the secondary response was significantly impaired in CD28−/− mice.
FIGURE 4.
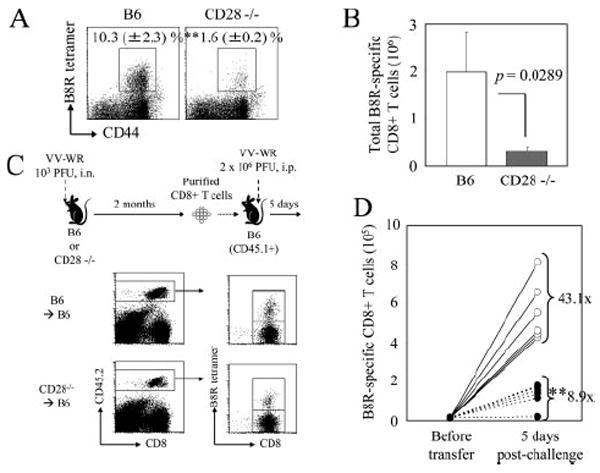
CD28 costimulation is required for secondary expansion of VV-specific memory CD8+ T cells. A and B, VV-WR-infected C57BL/6 or CD28−/− mice were challenged on day 125 p.i. with B8R peptide plus LPS i.v. and expansion of B8R-specific CD8+ T cells in the spleen was measured by tetramer staining 6 days later. A, Representative plots gated on CD8+ cells are shown and numbers indicate percentages of B8R tetramer+CD8+ T cells (±SD). B, Total number of B8R-specific CD8+ T cells in the spleen 6 days later. Error bars indicate SD. C and D, 2.5 × 106 purified CD8+ T cells from spleens of VV-WR-infected B6 or CD28−/− mice (number of B8R-specific memory CD8+ T cells determined by tetramer staining) were transferred into naive B6-CD45.1+ mice. One day posttransfer, mice were challenged with 2 × 106 PFU of VV-WR i.p., and expansion of B8R-specific CD8+ T cells in the spleen was enumerated by tetramer staining 5 days later. Experimental scheme (C, above), gating (C, below), and total number of memory cells before and after challenge (D) are shown. D,○, B6→B6; ●, CD28−/− →B6. Each set of connected dots represent individual mice, and numbers indicate average fold expansion of transferred memory cells after 5 days in each group, calculated as written in Materials and Methods. **, p < 0.01.
The second approach we took was an adoptive transfer approach. As shown in Fig. 4C, equal numbers of purified CD8+ T cells (numbers of B8R-specific memory CD8+ T cells in each population were quantified by tetramer staining) from VV-WR-infected C57BL/6 or CD28−/− mice were transferred into naive congenic recipients and challenged the next day with VV-WR i.p. Five days postchallenge, fold expansion of transferred B8R-specific CD8+ T cells was calculated by dividing the total number of recovered B8R-specific CD8+ T cells by the number of B8R-specific CD8+ T cells transferred into individual naive recipients before challenge. Compared with the B8R-specific memory CD8+ T cells derived from the wild-type host, which expanded on average 43.1-fold, cells derived from CD28−/− hosts expanded on average 8.9-fold, a significant decrease in expansion (Fig. 4D). A similar defect in expansion was also observed when the secondary challenge was administered by the i.n. route (data not shown). Results from these two experiments clearly demonstrate that CD28 costimulation is required for robust secondary expansion of VV-specific memory CD8+ T cells.
CD28 costimulation is required both during priming and recall to maximize the secondary response of VV-specific memory CD8+ T cells
The results clearly show the requirement of CD28 costimulation for a robust secondary response of VV-specific memory CD8+ T cells. However, it was important to determine whether CD28 costimulation is required during priming, during the recall response, or both. In experiments directly challenging CD28−/− mice (Fig. 4, A and B), as well as adoptive transfer of CD28−/− CD8+ T cells (Fig. 4, C and D), the memory cells receive no CD28 signal throughout the experiment. Therefore, we designed the following adoptive transfer experiments to distinguish whether the signal is required during the primary response (for priming and for the development of “competent” memory cells) or during the recall response.
We first sought to determine whether CD28 costimulation is required during priming. In this experiment, CD28 costimulation was not delivered during the primary response due to the absence of CD80/CD86, but upon transfer into naive wild-type recipients the memory cells received the CD28 signal during the recall response. Although CD80/CD86−/− mice differ from CD28−/− mice due to their inability to signal through CTLA-4 in addition to CD28, we observed no differences in the memory CD8+ T cells recovered from CD80/CD86−/− mice including the frequency of virus-specific CD8 T cells (which were not significantly different from wild-type mice). Equal numbers of CD8+ T cells from VV-WR-infected wild-type or CD80/CD86−/− mice were purified and transferred into naive wild-type congenic recipients, challenged, and analyzed 5 days later for their expansion by dividing the total number of recovered memory CD8+ T cells by the input number (Fig. 5A). Memory CD8+ T cells derived from CD80/CD86−/− mice showed reduced expansion compared with memory cells derived from wild-type mice suggesting a role of CD28 costimulation in programming of memory CD8+ T cell development. However, the reduction was not as dramatic as that observed with memory cells from CD28−/− mice (Fig. 4C).
FIGURE 5.
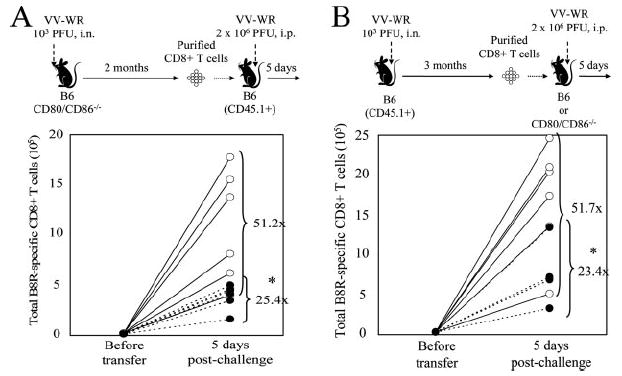
CD28 costimulation is required both during the priming and recall phase of VV-specific memory CD8+ T cells to maximize the recall response. A, CD28 costimulation during the primary response is required for an optimal VV-specific recall response. In brief, 1.5 × 106 purified CD8+ T cells from spleens of VV-WR-infected B6 or CD80/CD86−/− mice (number of B8R-specific memory CD8+ T cells determined by tetramer staining) were transferred into naive B6-CD45.1+ mice. One day posttransfer, mice were challenged with 2 × 106 PFU of VV-WR i.p. and expansion of B8R-specific CD8+ T cells in the spleen was enumerated by tetramer staining 5 days later. B, CD28 costimulation during challenge maximizes the recall response. In brief, 4 × 106 purified CD8+ T cells from spleens of VV-WR-infected B6-CD45.1+ mice (number of B8R-specific memory CD8+ T cells determined by tetramer staining) were transferred into naive B6 or CD80/CD86−/− mice. One day posttransfer, mice were challenged with 2 × 106 PFU of VV-WR i.p. and expansion of B8R-specific CD8+ T cells in the spleen was enumerated by tetramer staining 5 days later. Experimental schemes are shown above, and representative data from two individual experiments are shown. ○, B6→B6; ●, CD80/CD86−/− →B6 (A) or B6→CD80/CD86−/− (B). Each set of connected circles represent individual mice, and numbers indicate average fold expansion of transferred memory cells after 5 days in each group, calculated as written in Materials and Methods. *, p < 0.05.
Next, to explore the requirement of CD28 costimulation during the recall response, we designed an experiment in which memory CD8+ T cells were primed in the presence of CD28 costimulation, but did not receive the signal during the recall response when transferred into CD80/CD86−/− recipients. Equal numbers of CD8+ T cells from VV-WR-infected wild-type congenic mice were purified and transferred into wild-type or CD80/CD86−/− recipients, challenged, and analyzed 5 days later for their expansion (Fig. 5B). B8R-specific memory CD8+ T cells recalled in the absence of CD28 costimulation showed a reduced recall response; but again, the reduction was modest compared to that of CD28−/− mice (Fig. 4C). From these experiments, we conclude that CD28 costimulation maximizes the secondary response by acting both during the primary and recall response.
CD28 costimulation is required for secondary responses of memory CD8+ T cells during persistent infection
MHV-68 establishes a low-level persistent infection and memory CD8+ T cells specific for the virus exhibit unique characteristics distinct from that of memory cells in acute viral infections (41). We have previously reported that MHV-68 ORF61-specific memory CD8+ T cells require CD28 costimulation to elicit a robust recall response (22). We confirmed similar requirements for ORF6-specific memory CD8+ T cells by challenging MHV-68-infected wild-type or CD28−/− mice with rVV-ORF6 at 5 mo p.i. Expansion of ORF6-specific memory CD8+ T cells was significantly blunted in CD28−/− mice (Fig. 6, A and B). Next, we sought to determine whether the stage in which CD28 costimulation is required during persistent infection (MHV-68) differs from that of an acute infection (VV-WR). To test this hypothesis, adoptive transfer experiments similar to those performed in Fig. 5 were performed using the MHV-68 system. First, to test the requirement of CD28 costimulation during the primary response, equal numbers of CD8+ T cells from MHV-68-infected wild-type or CD80/CD86−/− mice were transferred into naive congenic wild-type recipients and challenged with rVV-ORF6, and the fold expansion of transferred ORF6-specific CD8+ T cells was calculated as described above (Fig. 6C). Memory CD8+ T cells from CD80/CD86−/− mice expanded significantly less compared with those from wild-type mice, showing a requirement of CD28 costimulation during the primary response. Next, to determine costimulatory requirements during recall, equal numbers of CD8+ T cells from MHV-68-infected wild-type congenic mice were transferred into naive wild-type or CD80/CD86−/− recipients and challenged with rVV-ORF6, and fold expansion of transferred ORF6-specific CD8+ T cells were analyzed (Fig. 6D). When transferred into CD80/CD86−/− recipients, MHV-68-specific CD8+ T cells were unable to mount a recall response, suggesting that CD28 costimulation is also required during the recall response. In summary, CD28 costimulation is required both during the primary and the recall response for MHV-68-specific memory CD8+ T cells to mount a secondary response. Therefore, we conclude that costimulatory requirements and the timing in which it is required by memory CD8+ T cells are similar during acute infections and low-level persistent infections.
FIGURE 6.
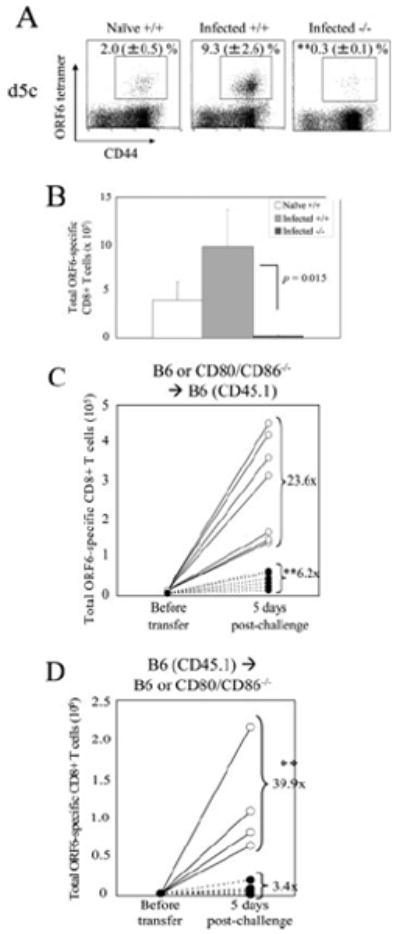
Costimulatory requirements for recall responses of memory CD8+ T cells during a low-level persistent infection. Naive C57BL/6, MHV-68-infected C57BL/6, or CD28−/− mice were challenged with 2 × 106 PFU of VV-ORF6 i.p. and were analyzed for ORF6-specific CD8+ T cell expansion in the spleens by tetramer staining 5 days postchallenge. Representative plots gated on CD8+ cells (A) and total number of ORF6-specific CD8+ T cells (B) are shown. Numbers indicate percentage of ORF6–tetramer+CD8+ T cells (±SD). Representative data from two independent experiments consisting of three or four mice per group are shown. C and D, CD28 costimulation is required both during the priming and recall phase for maximal recall responses of MHV-68-specific CD8+ T cells. C, In brief, 2 × 106 purified CD8+ T cells from spleens of MHV-68-infected B6 or CD80/CD86−/− mice (number of ORF6-specific memory CD8+ T cells determined by tetramer staining) were transferred into naive B6-CD45.1+ mice. One day posttransfer, mice were challenged with 2 × 106 PFU of VV-ORF6 i.p. and expansion of B8R-specific CD8+ T cells in the spleen were enumerated by tetramer staining 5 days later. D, In brief, 8 × 106 purified CD8+ T cells from spleens of MHV-68-infected B6-CD45.1+ mice (number of ORF6-specific memory CD8+ T cells determined by tetramer staining) were transferred into naive B6 or CD80/CD86−/− mice. One day posttransfer, mice were challenged with 2 × 106 PFU of VV-ORF6 i.p. and expansion of ORF6-specific CD8+ T cells in the spleen were enumerated by tetramer staining 5 days later. Representative data from two individual experiments are shown. ○, B6→B6; ●, CD80/CD86−/−→B6 (C) or B6→CD80/CD86−/− (D). Each set of connected circles represent individual mice, and numbers indicate average fold expansion of transferred memory cells after 5 days in each group, calculated as written in Materials and Methods. *, p < 0.05; **, p < 0.01.
Recall responses in CD28-deficient mice are restored by IL-2 signaling during recall but not priming
One major consequence of CD28 costimulation is the production of IL-2 by the T cell (57). Recent reports have shown that direct IL-2 signaling during priming is required for programming of memory CD8+ T cell differentiation to elicit robust secondary expansion (28, 58). The inability of memory CD8+ T cells to efficiently expand upon secondary Ag encounter in the absence of CD28 costimulation lead us to hypothesize that the defect is due to insufficient production of IL-2 and signaling into the CD8+ T cell in the absence of costimulation during priming. Boyman et al. (51) previously reported that coadministration of anti-IL-2 (clone S4B6) mAb with mIL-2 efficiently delivers IL-2 signals to T cells, which has been used in the context of virus-specific CD8+ T cells (28). We tested our hypothesis by using the IL-2 immune complex to replenish IL-2 signaling during the primary response in CD28−/− mice and examined whether this intervention rescued the recall response of VV-specific CD8+ T cells. Wild-type or CD28−/− mice were infected with VV-WR i.n. and were treated with R-IgG or the IL-2 immune complex (wild-type mice were treated with R-IgG only) daily from days 0 to 6. We confirmed the efficacy of the IL-2 immune complex by its ability to expand memory-phenotype CD8+ T cells as previously reported (51). Treatment with IL-2 immune complex lead to a dramatic increase in the percentage of CD44highCD122highCD8+ T cells in the peripheral blood at day 7 (C57BL/6 R-IgG treated: 44.8 ± 5.1%; CD28−/− R-IgG treated: 24.8 ± 1.8%; CD28−/− IL-2 immune complex treated: 62.2 ± 12.1% of CD8+ T cells; CD28−/− R-IgG vs IL-2: p > 0.001). Two months p.i., CD8+ T cells from each group were transferred into naive congenic wild-type recipients and were challenged with VV-WR, and fold expansion of transferred B8R-specific memory CD8+ T cells was analyzed by tetramer staining, as shown in Fig. 7A. Memory CD8+ T cells from wild-type mice treated with R-IgG expanded on average 38.3-fold, whereas memory cells from CD28−/− mice treated with R-IgG showed a significantly reduced expansion (average, 6.9-fold), confirming earlier results (Fig. 7B). When memory cells from CD28−/− mice treated with the IL-2 immune complex were transferred and challenged, the cells were still impaired in their ability to respond (average, 4.8-fold expansion), thus showing the inability of increased IL-2 signaling during priming to rescue secondary responses in CD28−/− mice.
FIGURE 7.
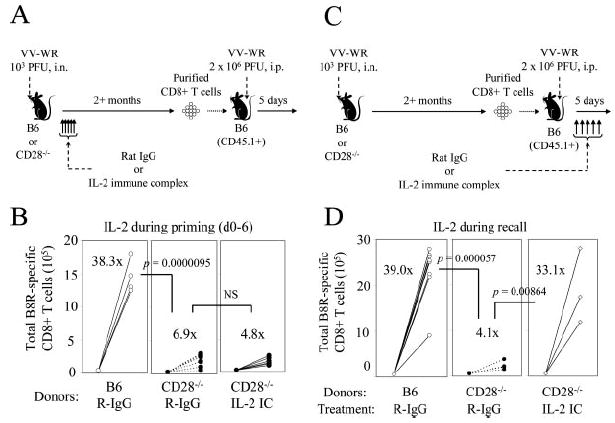
Recall responses in CD28-deficient mice are restored by IL-2 signaling during recall but not priming. A and B, C57BL/6 or CD28−/− mice were infected i.n. with VV-WR and were treated daily with R-IgG or 50 μg of anti-IL-2 plus 1.5 μg of mIL-2 (IL-2 immune complex (IC), see Materials and Methods) during priming from days 0 to 6. After 2 mo, 2.8 × 106 purified CD8+ T cells from each group (number of B8R-specific memory CD8+ T cells determined by tetramer staining) were transferred into naive B6-CD45.1+ mice. One day posttransfer, mice were challenged with VV-WR i.p., and expansion of B8R-specific CD8+ T cells in the spleen were enumerated by tetramer staining 5 days postchallenge. A, Experimental scheme. B, Total numbers of B8R-specific CD8+ memory cells before and after challenge are shown. Donor groups are indicated below. C and D, Experiment similar to A and B was conducted, except R-IgG or IL-2 immune complex was given daily during the recall response. C, Experimental scheme. D, Total numbers of B8R-specific CD8+ memory cells before and after challenge are shown. Each set of connected circles represent individual mice, and numbers indicate average fold expansion of transferred memory cells after 5 days in each group, calculated as indicated in Materials and Methods. **, p < 0.01.
Next, we investigated whether restoring IL-2 signaling during the recall phase could rescue recall responses in CD28−/− mice. Similar experiments as explained above were conducted; with the exception of the IL-2 immune complex or control R-IgG being delivered daily to recipients on days 0–5 postchallenge (Fig. 7C). B8R-specific memory CD8+ T cells from wild-type mice expanded on average 39.0-fold upon challenge when treated with R-IgG during the recall response, while memory cells from CD28−/− mice treated with R-IgG expanded on average 4.1-fold (Fig. 7D). However, when memory CD8+ T cells from CD28−/− mice were challenged and treated with the IL-2 immune complex during the recall response, the cells expanded on average 33.1-fold. The data altogether indicate that the impaired recall response by memory CD8+ T cells in the absence of CD28 costimulation can be restored by IL-2 signaling during the recall phase but not during priming.
Discussion
Activation of memory T cells has generally been accepted to be independent of the CD80/CD86-CD28 costimulatory pathway, although differences exist in results obtained from various studies using different models. In polyclonal or in vitro priming models, memory CD4+ T cells have been shown to elicit robust recall responses to Ag in vitro in the absence or with low levels of CD80/CD86 on the APC (34, 35). However, the survival of memory CD4+ T cells is impaired significantly when primed in the absence of this costimulatory pathway (40). Ndejembi et al. (39) have recently reported that blocking this pathway during recall responses by CTLA4-Ig results in diminished IL-2 and TNF-α production and recall responses of transgenic and influenza-specific polyclonal memory CD4+ T cells in vivo. These results clearly show that certain aspects of CD4+ T cell memory are controlled by CD80/CD86-CD28 costimulation.
Conflicting data exist for the role of CD80/CD86-CD28 costimulation in CD8+ T cell memory. During acute LCMV (Armstrong) infection, virus-specific memory CD8+ T cell number and function are maintained in the absence of CD28 costimulation (14, 16). However, recall responses are impaired during influenza challenge in CD28−/− mice (12, 13), during L. monocytogenes challenge in CD28−/− mice or with CTLA4-Ig treatment during recall (20, 32), and to vesicular stomatitis virus challenge in vitro in the absence of CD80/86 (11). In studies using CD28−/− mice, it is unclear whether the signal is required during priming or during the recall response. CD28 costimulation also seems to impact memory CD8+ T cell responses to chronic viral infections. The virus-specific CD8+ T cell response is lost during LCMV clone 13 infection in CD28−/− mice (15), memory and recall IFN-γ responses are impaired during intravaginal HSV-2 infection (10), and MHV-68-specific memory CD8+ T cells in CD28−/− mice produce lower amounts of IFN-γ, express lower levels of CD122 and CD27, and are defective in secondary responses (22). These reports point to a role for the CD28 costimulatory pathway in CD8+ T cell memory. Therefore, we were prompted to examine its role in the development of virus-specific memory CD8+ T cells during acute i.n. VV-WR infection.
CD28 costimulation did not affect the kinetics of viral replication or the course of the disease (Fig. 1). As previously reported in various infection models (8-22), the primary CD8+ T cell response was severely impaired (Fig. 2, A–E). However, the frequencies and numbers of virus-specific CD8+ T cells were not affected at later time points (Fig. 2, E–G). We assessed whether the memory CD8+ T cells were altered in terms of function or phenotype. VV-specific memory CD8+ T cells in CD28−/− mice expressed lower levels of CD122 (IL-15Rβ) in all organs and lower CD27 in lymphoid organs (Fig. 3A). This was an interesting observation, since we had previously reported that MHV-68-specific memory CD8+ T cells in CD28−/− and CD80/CD86−/− mice have low expression of these two markers (22). IL-15 is an essential survival factor for memory CD8+ T cells (59), and the costimulatory molecule CD27 has been shown to play an important role in memory CD8+ T cell responses to influenza challenge (60). Thus, the low expression of these two molecules may account for the functional impairments of VV and MHV-68-specific memory CD8+ T cells (Figs. 3-6) and Ref. 22). In contrast, CD27 and CD122 have both been used as differentiation markers for CD8+ T cells and the altered phenotype may indicate suboptimal differentiation.
The memory CD8+ T cells in CD28−/− mice were capable of producing the cytokines IFN-γ and TNF-α, although IFN-γ production was reduced in the BM (Fig. 3B). However, in both the spleen and the BM, the memory cells were unable to produce IL-2 (Fig. 3B). It has been reported previously that memory CD4+ T cells fail to produce TNF-α and IL-2 when restimulated in the presence of CTLA4-Ig (39). Clearly, these results demonstrate that CD28 costimulation is required for IL-2 production by memory T cells. The studies on memory CD4+ T cells indicated that the CD28 signal is required during restimulation in vitro, and our current studies extend these studies to in vivo requirements for CD28 costimulation during recall in a nontransgenic infection model. The differences observed with TNF-α production is unclear, but this may indicate an intrinsic difference between memory CD4+ and CD8+ T cells in the requirement of costimulation for cytokine production.
The recall proliferative response of VV-specific memory CD8+ T cells was severely impaired in CD28−/− mice, both in response to peptide and viral challenge (Fig. 4). This observation has been made in other infection models, although the magnitude of reduction varies between the models (12, 13, 19, 20, 22, 36, 37), including a persistent viral infection MHV-68 (Fig. 6). Impaired IL-2 production and recall responses were reminiscent of CD4-helpless memory CD8+ T cells. However, transfer of naive wildtype CD4+ T cells into CD28−/− mice before the primary response did not restore the recall response in CD28−/− mice (data not shown). Furthermore, CD4-helpless memory cells during VV-WR infection elicit a different phenotype and further impairments in addition to IL-2 production and recall responses (S. Fuse, C. Y. Tsai, and E. J. Usherwood, manuscript in preparation). Although we cannot conclusively rule out indirect effects due to a lack of CD4 T cell help, these data suggest that CD28 costimulation directly into CD8+ T cells plays a significant role in the development of VV-specific memory CD8+ T cells.
Earlier studies did not differentiate whether CD28 costimulation was required during the priming phase (and thus the proper differentiation of “fit” memory cells) or during the recall phase. We assessed these two possibilities using adoptive transfer experiments. Our results indicated that CD28 costimulation during both priming and recall phases affect the magnitude of the recall response, and the overall defect in the recall response observed in CD28−/− mice was the sum of these two events (Fig. 5). The requirement for CD28 costimulation during priming may reflect its role in controlling TCR signal strength, which has been proposed to control T cell fitness (61). Furthermore, dendritic cells are required for memory CD8+ T cells to elicit a robust recall response (62), which will be fully armed with costimulatory molecules. Therefore, it is natural to assume that costimulatory signals play a role in maximizing the recall response. In line with our findings, CTLA4-Ig treatment during recall reduces the recall response during L. monocytogenes infection (32). Similar requirements in both the priming and recall phases were also observed with MHV-68-specific memory CD8+ T cells (Fig. 6, C and D). Because during persistent MHV-68 infection CD8+ T cells encounter Ag more frequently, a stronger requirement for costimulation may be necessary to maintain their fitness, thus resulting in the significant reduction in the recall response when CD28 costimulation was absent during priming of the CD8+ T cells (Fig. 6C). However, a strong requirement for CD28 costimulation during the recall response was also observed (Fig. 6D). This was somewhat surprising since MHV-68-specific memory CD8+ T cells have more frequent encounters with Ag and exhibit a more effector memory cell phenotype (41), and it has been shown in vitro that effector cells have lower requirements for costimulation (63).
A major consequence of CD28 costimulation on T cells is the production of IL-2, which plays a major role in the programming of memory CD8+ T cell differentiation (28). Therefore, we hypothesized that the reduced recall response in the absence of CD28 costimulation during priming or recall is due to the lack of IL-2 signaling. Interestingly, restoration of IL-2 signaling during recall, but not priming, completely restored the recall response in the absence of costimulation (Fig. 7, B and D). These results may indicate that memory CD8+ T cells are in a partly anergic state in the absence of CD28 costimulation, which can be restored by IL-2. It has been reported previously that the majority of responding CD8+ T cells during a recall response express IL-2 (64) and that IL-2 signals can significantly enhance recall responses (65, 66). We hypothesize that the inability of the memory cells to produce IL-2 (Fig. 3B) plays a role in the defect in the recall response; however, we cannot rule out IL-2 provided from other sources. The results also indicate that the effect of CD28 costimulation during priming is not due to insufficient IL-2 production (Fig. 7B). In this regard, it is interesting that supplementation of IL-2 during priming cannot rescue T cell proliferation and survival in the absence of CD28 costimulation or the reduction in the generation/ survival of memory CD4+ T cells (5, 40, 67, 68). One possibility may be that CD28 costimulation is regulating the strength of the initial TCR signal, thus controlling its fitness to elicit an optimal recall response (61, 69).
Various parameters such as route of infection, infectious dose, tissue tropism, and/or cytopathic characteristics affect costimulatory requirements. Viral infections administered locally in low doses or with localized tropisms tend to have a stronger requirement for costimulation (20), as observed in our experiments (103 PFU for VV-WR, 400 PFU for MHV-68, i.n.; see Fig. 1B and Refs. 70-73) and others (HSV-2, influenza) (10, 12). In contrast, CD28−/− VV-specific memory CD8+ T cells after systemic VV infection proliferate normally in response to secondary ectromelia virus challenge (74). The differences between this study and our data may be due to a systemic vs i.n. challenge or possibly a difference in the virus used for secondary challenge (VV vs ectromelia). In addition to dose and tropism, other costimulatory molecules such as 4-1BB and CD27 can also impact memory CD8+ T cell responses (37, 49, 60). These molecules likely also partly compensate for the lack of CD28 signaling to facilitate a primary T cell response to viruses. Overall, our work and the work of others clearly demonstrate that CD80/CD86-CD28 costimulation controls certain key aspects of CD4+ and CD8+ T cell memory differentiation, such as IL-2 production and recall responses (39). Information provided by our study should be considered in efforts to maximize memory responses and optimize vaccine design.
Acknowledgments
We thank Dr. William R. Green for the VV-WR strain stock and Michael J. Molloy for critical reading of this manuscript.
Footnotes
This work was supported by National Institutes of Health Grant CA103642.
Abbreviations used in this paper: LCMV, lymphocytic choriomeningitis virus; MHV-68, murine gammaherpesvirus 68; VV-WR, vaccinia virus Western Reserve; i.n., intranasal; rVV, recombinant VV; p.i., postinfection; mIL-2, murine IL-2; BM, bone marrow; R-IgG, rat IgG.
Disclosures The authors have no financial conflict of interest.
References
- 1.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 2.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 3.Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, Popescu F, Xiao Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 4.Haring JS, Badovinac VP, Harty JT. Inflaming the CD8+ T cell response. Immunity. 2006;25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Kundig TM, Shahinian A, Kawai K, Mittrucker HW, Sebzda E, Bachmann MF, Mak TW, Ohashi PS. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity. 1996;5:41–52. doi: 10.1016/s1074-7613(00)80308-8. [DOI] [PubMed] [Google Scholar]
- 6.Horspool JH, Perrin PJ, Woodcock JB, Cox JH, King CL, June CH, Harlan DM, St Louis DC, Lee KP. Nucleic acid vaccine-induced immune responses require CD28 costimulation and are regulated by CTLA4. J Immunol. 1998;160:2706–2714. [PubMed] [Google Scholar]
- 7.Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, Kedl RM. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J Exp Med. 2004;199:775–784. doi: 10.1084/jem.20031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lumsden JM, Roberts JM, Harris NL, Peach RJ, Ronchese F. Differential requirement for CD80 and CD80/CD86-dependent costimulation in the lung immune response to an influenza virus infection. J Immunol. 2000;164:79–85. doi: 10.4049/jimmunol.164.1.79. [DOI] [PubMed] [Google Scholar]
- 9.Zimmermann C, Seiler P, Lane P, Zinkernagel RM. Antiviral immune responses in CTLA4 transgenic mice. J Virol. 1997;71:1802–1807. doi: 10.1128/jvi.71.3.1802-1807.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thebeau LG, Morrison LA. Mechanism of reduced T-cell effector functions and class-switched antibody responses to herpes simplex virus type 2 in the absence of B7 costimulation. J Virol. 2003;77:2426–2435. doi: 10.1128/JVI.77.4.2426-2435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McAdam AJ, Farkash EA, Gewurz BE, Sharpe AH. B7 costimulation is critical for antibody class switching and CD8+ cytotoxic T-lymphocyte generation in the host response to vesicular stomatitis virus. J Virol. 2000;74:203–208. doi: 10.1128/jvi.74.1.203-208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertram EM, Lau P, Watts TH. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol. 2002;168:3777–3785. doi: 10.4049/jimmunol.168.8.3777. [DOI] [PubMed] [Google Scholar]
- 13.Bertram EM, Tafuri A, Shahinian A, Chan VS, Hunziker L, Recher M, Ohashi PS, Mak TW, Watts TH. Role of ICOS versus CD28 in antiviral immunity. Eur J Immunol. 2002;32:3376–3385. doi: 10.1002/1521-4141(200212)32:12<3376::AID-IMMU3376>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 14.Suresh M, Whitmire JK, Harrington LE, Larsen CP, Pearson TC, Altman JD, Ahmed R. Role of CD28–B7 interactions in generation and maintenance of CD8 T cell memory. J Immunol. 2001;167:5565–5573. doi: 10.4049/jimmunol.167.10.5565. [DOI] [PubMed] [Google Scholar]
- 15.Christensen JE, Christensen JP, Kristensen NN, Hansen NJ, Stryhn A, Thomsen AR. Role of CD28 co-stimulation in generation and maintenance of virus-specific T cells. Int Immunol. 2002;14:701–711. doi: 10.1093/intimm/dxf037. [DOI] [PubMed] [Google Scholar]
- 16.Andreasen SO, Christensen JE, Marker O, Thomsen AR. Role of CD40 ligand and CD28 in induction and maintenance of antiviral CD8+ effector T cell responses. J Immunol. 2000;164:3689–3697. doi: 10.4049/jimmunol.164.7.3689. [DOI] [PubMed] [Google Scholar]
- 17.Edelmann KH, Wilson CB. Role of CD28/CD80–86 and CD40/CD154 costimulatory interactions in host defense to primary herpes simplex virus infection. J Virol. 2001;75:612–621. doi: 10.1128/JVI.75.2.612-621.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shedlock DJ, Whitmire JK, Tan J, MacDonald AS, Ahmed R, Shen H. Role of CD4 T cell help and costimulation in CD8 T cell responses during Listeria monocytogenes infection. J Immunol. 2003;170:2053–2063. doi: 10.4049/jimmunol.170.4.2053. [DOI] [PubMed] [Google Scholar]
- 19.Mittrucker HW, Kohler A, Mak TW, Kaufmann SH. Critical role of CD28 in protective immunity against Salmonella typhimurium. J Immunol. 1999;163:6769–6776. [PubMed] [Google Scholar]
- 20.Mittrucker HW, Kursar M, Kohler A, Hurwitz R, Kaufmann SH. Role of CD28 for the generation and expansion of antigen-specific CD8+ T lymphocytes during infection with Listeria monocytogenes. J Immunol. 2001;167:5620–5627. doi: 10.4049/jimmunol.167.10.5620. [DOI] [PubMed] [Google Scholar]
- 21.Sigal LJ, Reiser H, Rock KL. The role of B7-1 and B7-2 costimulation for the generation of CTL responses in vivo. J Immunol. 1998;161:2740–2745. [PubMed] [Google Scholar]
- 22.Fuse S, Obar JJ, Bellfy S, Leung EK, Zhang W, Usherwood EJ. CD80 and CD86 control antiviral CD8+ T-cell function and immune surveillance of murine γherpesvirus 68. J Virol. 2006;80:9159–9170. doi: 10.1128/JVI.00422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 24.Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220–228. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- 25.Wang S, Chen L. Co-signaling molecules of the B7-CD28 family in positive and negative regulation of T lymphocyte responses. Microbes Infect. 2004;6:759–766. doi: 10.1016/j.micinf.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611–618. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 27.Orabona C, Grohmann U, Belladonna ML, Fallarino F, Vacca C, Bianchi R, Bozza S, Volpi C, Salomon BL, Fioretti MC, et al. CD28 induces immunostimulatory signals in dendritic cells via CD80 and CD86. Nat Immunol. 2004;5:1134–1142. doi: 10.1038/ni1124. [DOI] [PubMed] [Google Scholar]
- 28.Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441:890–893. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 30.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bevan MJ. Helping the CD8+ T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 32.Marzo AL, Vezys V, Klonowski KD, Lee SJ, Muralimohan G, Moore M, Tough DF, Lefrancois L. Fully functional memory CD8 T cells in the absence of CD4 T cells. J Immunol. 2004;173:969–975. doi: 10.4049/jimmunol.173.2.969. [DOI] [PubMed] [Google Scholar]
- 33.Prlic M, Hernandez-Hoyos G, Bevan MJ. Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J Exp Med. 2006;203:2135–2143. doi: 10.1084/jem.20060928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Croft M, Bradley LM, Swain SL. Naive versus memory CD4 T cell response to antigen: memory cells are less dependent on accessory cell costimulation and can respond to many antigen-presenting cell types including resting B cells. J Immunol. 1994;152:2675–2685. [PubMed] [Google Scholar]
- 35.London CA, Lodge MP, Abbas AK. Functional responses and costimulator dependence of memory CD4+ T cells. J Immunol. 2000;164:265–272. doi: 10.4049/jimmunol.164.1.265. [DOI] [PubMed] [Google Scholar]
- 36.Villegas EN, Elloso MM, Reichmann G, Peach R, Hunter CA. Role of CD28 in the generation of effector and memory responses required for resistance to Toxoplasma gondii. J Immunol. 1999;163:3344–3353. [PubMed] [Google Scholar]
- 37.Bertram EM, Dawicki W, Sedgmen B, Bramson JL, Lynch DH, Watts TH. A switch in costimulation from CD28 to 4-1BB during primary versus secondary CD8 T cell response to influenza in vivo. J Immunol. 2004;172:981–988. doi: 10.4049/jimmunol.172.2.981. [DOI] [PubMed] [Google Scholar]
- 38.Mirenda V, Jarmin SJ, David R, Dyson J, Scott D, Gu Y, Lechler RI, Okkenhaug K, Marelli-Berg FM. Physiological and aberrant regulation of memory T cell trafficking by the co-stimulatory molecule CD28. Blood. 2007;109:2968–2977. doi: 10.1182/blood-2006-10-050724. [DOI] [PubMed] [Google Scholar]
- 39.Ndejembi MP, Teijaro JR, Patke DS, Bingaman AW, Chandok MR, Azimzadeh A, Nadler SG, Farber DL. Control of memory CD4 T cell recall by the CD28/B7 costimulatory pathway. J Immunol. 2006;177:7698–7706. doi: 10.4049/jimmunol.177.11.7698. [DOI] [PubMed] [Google Scholar]
- 40.Dooms H, Abbas AK. Control of CD4+ T-cell memory by cytokines and costimulators. Immunol Rev. 2006;211:23–38. doi: 10.1111/j.0105-2896.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- 41.Obar JJ, Fuse S, Leung EK, Bellfy SC, Usherwood EJ. Gammaherpesvirus persistence alters key CD8 T-cell memory characteristics and enhances antiviral protection. J Virol. 2006;80:8303–8315. doi: 10.1128/JVI.00237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McFadden G. Poxvirus tropism. Nat Rev Microbiol. 2005;3:201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammarlund E, Lewis MW, Carter SV, Amanna I, Hansen SG, Strelow LI, Wong SW, Yoshihara P, Hanifin JM, Slifka MK. Multiple diagnostic techniques identify previously vaccinated individuals with protective immunity against monkeypox. Nat Med. 2005;11:1005–1011. doi: 10.1038/nm1273. [DOI] [PubMed] [Google Scholar]
- 44.Henderson DA, Moss B. Smallpox and vaccinia. In: Plotkin SA, Orenstein WA, editors. Vaccines. 3. Saunders; Philadelphia: 1999. pp. 74–97. [Google Scholar]
- 45.Nash AA, Dutia BM, Stewart JP, Davison AJ. Natural history of murine γherpesvirus infection. Philos Trans R Soc Lond B Biol Sci. 2001;356:569–579. doi: 10.1098/rstb.2000.0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sunil-Chandra NP, Efstathiou S, Arno J, Nash AA. Virological and pathological features of mice infected with murine gamma herpesvirus 68. J Gen Virol. 1992;73:2347–2356. doi: 10.1099/0022-1317-73-9-2347. [DOI] [PubMed] [Google Scholar]
- 47.Stevenson PG, Belz GT, Castrucci MR, Altman JD, Doherty PC. A γ-herpesvirus sneaks through a CD8+ T cell response primed to a lyticphase epitope. Proc Natl Acad Sci USA. 1999;96:9281–9286. doi: 10.1073/pnas.96.16.9281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Usherwood EJ, Ward KA, Blackman MA, Stewart JP, Woodland DL. Latent antigen vaccination in a model gammaherpesvirus infection. J Virol. 2001;75:8283–8288. doi: 10.1128/JVI.75.17.8283-8288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuse S, Bellfy S, Yagita H, Usherwood EJ. CD8+ T cell dysfunction and increase in murine gammaherpesvirus latent viral burden in the absence of 4-1BB ligand. J Immunol. 2007;178:5227–5236. doi: 10.4049/jimmunol.178.8.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Usherwood EJ. A new approach to epitope confirmation by sampling effector/memory T cells migrating to the lung. J Immunol Methods. 2002;266:135–142. doi: 10.1016/s0022-1759(02)00106-0. [DOI] [PubMed] [Google Scholar]
- 51.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 52.Williamson JD, Reith RW, Jeffrey LJ, Arrand JR, Mackett M. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J Gen Virol. 1990;71:2761–2767. doi: 10.1099/0022-1317-71-11-2761. [DOI] [PubMed] [Google Scholar]
- 53.Reading PC, Smith GL. A kinetic analysis of immune mediators in the lungs of mice infected with vaccinia virus and comparison with intradermal infection. J Gen Virol. 2003;84:1973–1983. doi: 10.1099/vir.0.19285-0. [DOI] [PubMed] [Google Scholar]
- 54.Tscharke DC, Karupiah G, Zhou J, Palmore T, Irvine KR, Haeryfar SM, Williams S, Sidney J, Sette A, Bennink JR, Yewdell JW. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J Exp Med. 2005;201:95–104. doi: 10.1084/jem.20041912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mazo IB, Honczarenko M, Leung H, Cavanagh LL, Bonasio R, Weninger W, Engelke K, Xia L, McEver RP, Koni PA, et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity. 2005;22:259–270. doi: 10.1016/j.immuni.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 56.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 58.Bachmann MF, Wolint P, Walton S, Schwarz K, Oxenius A. Differential role of IL-2R signaling for CD8+ T cell responses in acute and chronic viral infections. Eur J Immunol. 2007;37:1502–1512. doi: 10.1002/eji.200637023. [DOI] [PubMed] [Google Scholar]
- 59.Schluns KS, Lefrancois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol. 2003;3:269–279. doi: 10.1038/nri1052. [DOI] [PubMed] [Google Scholar]
- 60.Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol. 2000;1:433–440. doi: 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- 61.Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nat Immunol. 2003;4:355–360. doi: 10.1038/ni908. [DOI] [PubMed] [Google Scholar]
- 62.Zammit DJ, Cauley LS, Pham QM, Lefrancois L. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity. 2005;22:561–570. doi: 10.1016/j.immuni.2005.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dubey C, Croft M, Swain SL. Naive and effector CD4 T cells differ in their requirements for T cell receptor versus costimulatory signals. J Immunol. 1996;157:3280–3289. [PubMed] [Google Scholar]
- 64.D’Souza WN, Lefrancois L. Frontline: an in-depth evaluation of the production of IL-2 by antigen-specific CD8 T cells in vivo. Eur J Immunol. 2004;34:2977–2985. doi: 10.1002/eji.200425485. [DOI] [PubMed] [Google Scholar]
- 65.Cheng LE, Ohlen C, Nelson BH, Greenberg PD. Enhanced signaling through the IL-2 receptor in CD8+ T cells regulated by antigen recognition results in preferential proliferation and expansion of responding CD8+ T cells rather than promotion of cell death. Proc Natl Acad Sci USA. 2002;99:3001–3006. doi: 10.1073/pnas.052676899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuekrek H, Schlingmann T, Valdez H, Boehm BO, Pollard RB, Mitsuyasu R, Goebel FD, Lederman MM, Lehmann PV, Tary-Lehmann M. Differential effect of interleukin-2 treatment on primary and secondary immunizations in HIV infected individuals. AIDS. 2005;19:1967–1974. doi: 10.1097/01.aids.0000189859.59559.9b. [DOI] [PubMed] [Google Scholar]
- 67.Lucas PJ, Negishi I, Negishi I, Nakayama K, Fields LE, Loh DY. Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J Immunol. 1995;154:5757–5768. [PubMed] [Google Scholar]
- 68.Sperling AI, Auger JA, Ehst BD, Rulifson IC, Thompson CB, Bluestone JA. CD28/B7 interactions deliver a unique signal to naive T cells that regulates cell survival but not early proliferation. J Immunol. 1996;157:3909–3917. [PubMed] [Google Scholar]
- 69.Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol. 2002;2:982–987. doi: 10.1038/nri959. [DOI] [PubMed] [Google Scholar]
- 70.Sunil-Chandra NP, Efstathiou S, Nash AA. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J Gen Virol. 1992;73:3275–3279. doi: 10.1099/0022-1317-73-12-3275. [DOI] [PubMed] [Google Scholar]
- 71.Weck KE, Kim SS, Virgin HI, Speck SH. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol. 1999;73:3273–3283. doi: 10.1128/jvi.73.4.3273-3283.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stewart JP, Usherwood EJ, Ross A, Dyson H, Nash T. Lung epithelial cells are a major site of murine gammaherpesvirus persistence. J Exp Med. 1998;187:1941–1951. doi: 10.1084/jem.187.12.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol. 2000;165:1074–1081. doi: 10.4049/jimmunol.165.2.1074. [DOI] [PubMed] [Google Scholar]
- 74.Fang M, Sigal LJ. Direct CD28 costimulation is required for CD8+ T cell-mediated resistance to an acute viral disease in a natural host. J Immunol. 2006;177:8027–8036. doi: 10.4049/jimmunol.177.11.8027. [DOI] [PubMed] [Google Scholar]