

Abstract
ABC transporters are polytopic proteins. ATP hydrolysis and substrate transport take place in separate domains, and these activities must be coordinated through a signal interface. We previously characterized a mutation (S558Y) in the yeast multidrug transporter Pdr5 that uncouples ATP hydrolysis and drug transport. To characterize the transmission interface, we used a genetic screen to isolate second-site mutations of S558Y that restore drug transport. We recovered suppressors that restore drug resistance; their locations provide functional evidence for an interface in the cis rather than the trans configuration indicated by structural and crosslinking studies of bacterial and eukaryotic efflux transporters. One mutation, E244G, defines the Q-loop of the deviant portion of NBD1, which is the hallmark of this group of fungal transporters. When moved to an otherwise wild-type background, this mutation and its counterpart in the canonical ATP-binding site Q951G show a similar reduction in drug resistance and in the very high basal-level ATP hydrolysis characteristic of Pdr5. A double E244G, Q951G mutant is considerably more drug sensitive than either of the single mutations. Surprisingly, then, the deviant and canonical Q-loop residues are functionally overlapping and equivalent in a strikingly asymmetric ABC transporter.
The ATP-binding cassette (ABC)1 protein superfamily uses the energy from ATP binding and hydrolysis to import and export a diverse array of biologically important compounds. A functional transporter contains a pair of nucleotide-binding domains (NBDs) and transmembrane helical domains (TMDs). The NBDs contain conserved sequences that position ATP, catalyze its hydrolysis, and communicate this signal to drug-binding sites in the TMDs. These include, among others, the Walker A, Walker B, Q-loop, and signature motifs. Several crystal structures solved for bacterial NBDs and for the complete transporter Sav1866 support a model in which ATP forms a sandwich between the Walker A, B, and Q-loop sequences of one NBD and the signature and D-loop residues of the other (for review see 1). In this complex, the nucleotide is occluded in a nonexchangeable manner, and subsequent hydrolysis appears to be necessary for the disassembly of the sandwich (2).
Central to the mechanism of transport mediated by ABC proteins is the coupling of chemical energy liberated by binding/hydrolysis at the NBDs to transport of cargo that takes place at the TMDs. The crystal structure of Sav1866 (3) and crosslinking studies from several transporters (4,5) demonstrate a physical proximity between residues in or near the Q-loop of NBDs and residues in the intracellular loops (ICLs) that connect transmembrane helices (TMHs). This suggests a potential signaling interface presumably mediated by conformational changes. These studies, however, did not demonstrate the functional importance of the interface under dynamic conditions. Our earlier work with the yeast multidrug transporter Pdr5p presented evidence from genetic and biochemical studies that interactions between the TMH2 and the N-terminal NBD, NBD1, occur via the ICL1, which connects TMHs2 and 3 (6). This study exploited a mutation in the extracellular end of TMH2 (S558Y) that exhibited ATP hydrolysis and binding of drug substrate but drastically reduced Pdr5-mediated drug transport and allosteric inhibition of the ATPase. This mutation appears to abrogate coupling between ATP hydrolysis at the NBDs and various activities in the TMDs.
When we screened for second-site mutations that restore drug resistance, we identified another mutation, N242K. Based on the structure of Sav1866, Dawson and Locher (3) predicted that the Q- and x-loops upstream of the signature sequence of ABC transporters interact with the ICL1 to constitute a signaling interface. Similarly, Jones and George (7) argue that the Q-loop of ABC transporters may be implicated in NBD–TMD communication. As shown below, the Asn-242 residue lies two residues away from the Q-loop of the N-terminal NBD. Our data and molecular modeling led us to speculate that the S558Y mutation uncouples the contacts between ICL1 and NBD1. Because the lysine residue has greater bulk and conformational flexibility than asparagine, we hypothesized that these contacts would be reestablished in the S558Y, N242K double mutant. In this study, we analyzed additional second-site mutations that can restore drug resistance in the S558Y mutant Pdr5 strain.
Pdr5 defines a large subfamily of clinically important pathogenic fungi, such as Cryptococcus neoformans and Candida albicans. These multidrug transporters show a marked departure in critical domain sequences from their mammalian counterparts, notably P-glycoprotein (P-gp). The organization of the NBDs and TMDs in Pdr5 and significant variations in the conserved domains of the NBDs are depicted in Figure 1. Because a significant number of residues vary in the canonical sequence, it is important to determine whether the alternate residues perform the same function as the conserved residues in other ABC proteins.
FIGURE 1.

The architecture of Pdr5 and its ATP-binding sites: A 2-D topological model of Pdr5 is illustrated. The deviant (ATP-site1) and canonical (ATP-site 2) ATP binding sites of Pdr5 are shown. The deviant residues come from the Walker A and B of the N-terminal NBD and the signature region of the centrally-positioned NBD.
One second-site mutation in the S558Y background that restores drug resistance is E244G. This residue is of singular importance because in Pdr5, Glu-244 replaces the canonical Q that defines the Q-loop in NBDs of ABC proteins. We used site-directed mutagenesis to characterize this residue as well as the equivalent Gln-951 in NBD2. In all, nine suppressors of S558Y were detected, of which seven mapped either to the ICL1 or NBD1. The mutants located in the NBD1 were all close to the Asn-242 identified in our previous study(6). Our results are consistent with communication between the Walker A and the signature region (C region) of the NBD and the TMDs via the ICL and with the Q-loop acting as part of this interface. A conserved glutamine, however, does not necessarily define this loop in all ABC proteins. Perhaps most significantly, our work clearly demonstrates that the deviant Glu-244 and canonical Gln-951 Q-loop residues are functionally equivalent.
EXPERIMENTAL PROCEDURES
Genetic manipulations
Table 1 lists the strains used in this study are found in Table 1. We previously reported isolating suppressors of S558Y clotrimazole (clo) hypersensitivity (6). Our genetic system for isolating and analyzing Pdr5 suppressors is also outlined in detail elsewhere (6). The strain R-1 is closely related to AD1-7, pioneered by the Goffeau group (8). Thus, it lacks all ABC multidrug transporters and contains PDR1-3, which causes overexpression of PDR5 (9,10). In addition, in R-1 the PDR5 coding region was removed and replaced by a cassette (KANMX4) conferring geneticin resistance. When a site-directed mutation is introduced into R-1 via transformation with an integrating plasmid (pSS607), a duplication results (diagrammed in Figure 2A and B).
Table 1.
Yeast strains used in this study
All strains are isogenic to each other
| Strain designation | Pertinent genotype | Reference and comments |
|---|---|---|
| R-1 | MATα PDR1-3 pdr5::KANMX4 ura3 his1 yor1 pdr10 pdr11 ycf1 pdr3 | (6) |
| JG2011 | R-1 + pS558Y (5-FOA derivative) | This study |
| JG2016 | R-1 + pN242K (5-FOA derivative) | (6) |
| JG2030 | R-1 + pE244G (5-FOA derivative) | This study |
| JG2031 | R-1 + pE244G, S558Y (5-FOA derivative) | This study |
| JG2032 | R-1 + 2 copies of pE244G | This study |
| JG2033 | R-1 + 2 copies of pE244G, S558Y | This study |
| JG2034 | R-1 + pQ951G (5-FOA derivative) | This study |
| JG2037 | R-1 + 2 copies Q951G | This study |
| JG2038 | R-1 + pE244G, Q951G | This study |
| JG2039 | R-1+ D246del | This study |
| JG2040 | R-1+ 2 copies pE244G, Q951G | This study |
| JG2041 | R-1 + 2 copies D246del | This study |
FIGURE 2.

Experimental strategy designed to determine whether a suppressor is due to a second-site mutation in pdr5S558Y. A, B Suppressors were isolated as chromosomal mutations in JG2011 which has two PDR5 cassettes. The first has the coding sequence replaced with KANMX4 but retains the upstream and downstream flanking regions. This is separated by plasmid sequences including the selectable marker URA3 from a second copy of PDR5 containing S558Y. C, In this illustration, it is assumed that the new mutation (*) lies in S558Y rather than in a second gene (the case for all of the mutants recovered as S558Y suppressors). D, Loss of the plasmid sequences containing URA3 and one of two PDR5 cassettes (either pdr5::KANMX4 or pdr5S558Y ) occurs by homologous recombination. These events result in Ura- colonies that are selected on plates containing 1 mg/ml 5-FOA. These are tested on gen (200 mg/l) and clo-containing medium (7.5 μM). E, If the suppressor is attributable to a second mutation in the S558Y-bearing copy, clor gens, and clos genr recombinants will be recovered as shown.
Selection of ura3 segregants on 5-fluoroorotic acid (5-FOA) medium (1mg/ml) should yield geneticin-sensitive and -resistant segregants. The former should contain an intact PDR5 gene that is overexpressed. Several of the transformants we tested yielded only geneticin-resistant 5-FOA segregants or no 5-FOA segregants at all. These unexpected recombinants are presumed to represent aberrant recombination events of various kinds. Such transformants were discarded, but their existence demonstrates why the 5-FOA test is essential. PCR recovery of the mutant gene from the chromosome and DNA sequencing of the resulting product confirm the presence of the desired mutation. Once we have isolated a 5-FOA segregant bearing a desired mutation, we often introduce a second copy of the same mutation by a repeat transformation. These double-copy strains greatly increase the signal-to-noise ratio in our various biochemical assays (6).
PCR recovery of mutant sequences from chromosomal DNA
We recovered mutant suppressor alleles were recovered from chromosomal DNA by PCR, with Whatman FTA technology (GE Healthcare, Piscataway, NJ). We recently described the PCR cycling conditions and primer composition (6).
Site-directed mutagenesis of PDR5 in pSS607
The construction and characterization of the PDR5-bearing integrating plasmid pSS606 are described elsewhere (11). We designed primers for site-directed mutagenesis with the help of the PrimerX Web site (www.bioinformatics.org/primerx/). Mutagenesis was carried out with the Quick Change Kit (Agilent Technologies, Santa Clara, CA). Following bacterial transformation, we prepared plasmid DNA from putative mutant clones and sent it to Retrogen (San Diego, CA) for sequencing. Fourteen primers were used in sequencing ensuring substantial overlap in reads. The entire PDR5 gene was sequenced and in every case, we observed only the desired alteration.
Transformation of yeast
We introduced mutant integrative plasmids into R-1 with a transformation kit produced by Sigma-Aldrich (St. Louis, MO).
Chemicals
Chemicals were purchased from Sigma-Aldrich. We used nucleotides (ATP and GTP) of the highest purity in all NTPase assays. We dissolved all xenobiotic compounds in DMSO except for cycloheximide (cyh) which was dissolved in water. We added 5-FOA (1 mg/ml) as powder to SD complete medium after sterilization. We added geneticin (Gen) to YPD medium at a concentration of 200 mg/l. The medium was swirled to dissolve this reagent just prior to pouring the plates. We purchased Iodoarylazidoprazosin ( [125I]IAAP ) from PerkinElmer Life Sciences (Waltham, MA).
Determination of the IC50 in liquid culture
We quantitatively tested all of the mutants described here for cyh and clo as previously described (6).
Spot tests
We qualitatively tested mutant and control strains for their relative drug resistance by spotting 10-fold dilutions of cells in water as previously described (6).
Preparation of purified membrane vesicles and immunoblotting
We prepared purified plasma membrane vesicles and determined their protein levels with the BCA assay as previously described (11). Samples were solubilized in SDS-PAGE loading buffer for 25 min at 37°C before electrophoresis at 150 V on 7% NUPAGE gels (Invitrogen, Carlsbad, CA). Western blotting was carried out with the Pdr5-specific antibody yC-18 and the Pma1-specific antibody yN-20 (Santa Cruz Biotechnology, Santa Cruz, CA) as previously described (6).
ATPase and GTPase assay
We assayed ATP as previously described (9) with vesicles made from strains containing two copies of wild-type (WT) or mutant PDR5. We performed each assay with 12 μg of vesicle protein incubated for 8 min at 35°C in 100 mM MOPS buffer (pH 7.4), 50 mM KCl, 5 mM NaN3, 2 mM EGTA (pH 7.0), 2 mM DTT, and 10 mM MgCl2. We terminated reactions by addition of 2.5% (final volume) SDS. The assay for GTPase was identical except that we used 16 μg of vesicle protein.
Photo-affinity labeling of Pdr5 with[125I] IAAP
Purified PMs (15 μg protein) prepared from double-copy, overexpressing Pdr5 were incubated at room temperature in ATPase buffer with [125I]IAAP (7 nM) for 5 min under subdued light. The samples were photo-crosslinked at 365 nm for 5 min and solubilized in SDS-PAGE loading buffer for 25 min at 37°C. Gel electrophoresis was carried out on 7% NuPAGE gels (Invitrogen). We quantified the signal as previously described (6).
RESULTS
Isolation and sequence analysis of mutants that suppress the clo hypersensitivity of S558Y
The strategy for isolating independent suppressors of S558Y clo hypersensitivity was previously described (6). In that study, we demonstrated that a N242K mutation restores considerable drug resistance. We also isolated eight other suppressors of S558Y. The experimental strategy for determining whether a suppressor results from a second mutation in PDR5 or from a mutation elsewhere in the genome is illustrated in Figure 2. The recovery of Genr Clos and Gens Clor segregants provided qualitative proof that the responsible mutation arose in PDR5. In all cases, the suppressor phenotypes were attributable to a second mutation in the PDR5 gene.
We recovered the PDR5 coding region and sequenced the DNA to determine the nature of the mutants (Table 2). All of the mutants contained the original S558Y mutation plus a single additional alteration. A schematic representation of the mutants is shown in Figure 3 as a 2-D topological map of Pdr5. Of the nine mutants analyzed, including N242K, seven were in either the N-terminal NBD or ICL1. One mutation is an E244G substitution. When alignment of Pdr5 with other transporters was carried out (supporting information), the Glu-244 residue was found to be completely conserved in the Pdr5 fungal subfamily of multidrug transporters and to be a replacement for the Gln in the canonical Q-loop of other ABC transporters (13).
Table 2.
Location of suppressor mutations
| Residue alteration | Location | No. found | Comments |
|---|---|---|---|
| N242K | NBD1 near the Q loop (between Walker A and B) | 1 | See (6) |
| E244G | Defines Q-loop | 1 | Completely conserved residue equivalent of Q951G in NBD2 |
| D246Δ | NBD1 Q loop | 2 | Deletion of a triplet codon of a highly conserved residue |
| S597X | ICL1 | 3 | 2 alleles are S597I; 1 allele is S597T |
| M679L | TMH5 | 1 | |
| G1233D | ECL4 between TMH7 and TMH8 | 1 |
FIGURE 3.

Location of suppressor mutations in Pdr5. Mutations that suppress S558Y (TMH2) are shown on a 2-D topological diagram of Pdr5.
With this information, we identified a motif, shown in Figure 4, which illustrates comparable regions in the 2 NBDs. The motif is Y/F, Sp, X, E, X, D/E, X, H, where Spis a small polar residue (N, S, T, or C) and X is not conserved. Three mutations appeared in two of the conserved residues. We also found three independent alterations at residue Ser-597 in ICL1 (two were S597I, one was S597T). The remaining mutants were an M679L substitution in TMH5 and the G1233D mutation in ECL4 connecting TMH7 and 8. In this small sample, we recovered no mutations in NBD-2. It is striking that seven of the nine independently isolated suppressor mutants had an alteration near or at the Q-loop and in ICL1. Thus, the location of these mutants on both sides of the interface lends functional evidence to previous structural and crosslinking studies, which suggested that at least one signaling interface runs from the NBD through ICLs and into the TMDs. This arrangement, although reminiscent of the Sav1866 transporter, which functions as a homodimer, is strikingly different in one crucial feature. The crystallographic data from Sav1866 suggests an interface with a criss-cross or trans arrangement in which the Q-loop of 1 monomer interacts with the ICL of the other. Evidence for a similar arrangement is found in eukaryotic transporters Tap and P-gp (4, 5). Our results, however, strongly suggest that the cis arrangement is physiologically relevant in Pdr5. Additional, complementary evidence for the cis interface comes from an analysis of mutations that suppress the cyh hypersensitivity of N242K. These data are found in the supporting materials.
FIGURE 4.

The Q-loop region motif. The figure shows the deviant portion of the N-terminal NBD (left side) and the canonical counterpart from NBD2 (right side). A protein sequence logo depicting the conservation of residues in and around the Q-loops of NBDs 1 and 2 was constructed from the multiple sequence alignment of Pdr5p (Figure S1) as described previously (27). The height of each letter represents the probability of the residue occuring at that position in the alignment. The colors signify the chemistry of the amino acids: red, acidic; blue, basic; black, hydrophobic; green, polar.
The drug phenotypes of the suppressor mutants
We compared the relative resistance of the suppressor mutants to cyh and clo to positive (R-1 +pSS607 = WT) and negative (R-1 + pS558Y) controls (Figure 5A and B). Although they were selected on clo-containing medium, all of the suppressors restored significant cyh resistance as well. Furthermore, none of the mutants were as resistant as the WT control to either drug. Finally, the curves generated for each mutant were similar. In fact, several of the clo plots completely overlapped, making it appear—erroneously—as though fewer strains were tested on this drug. Thus, all of the mutants had IC50s for clo of ~ 4.0 – 8.0 μM and IC50s for cyh of ~ 3.5 – 4.0 μM. These values represent a significant increase in resistance when compared to the values for S558Y, which are 1.1 μM and 0.4 μM for clo and cyh, respectively.
FIGURE 5.

Quantitative analysis of drug resistance in suppressor mutants. Strains were inoculated into 2 ml YPD containing A, clo (1.25–25.0 μM) or B, cyh (0.25–21.6 μM) at an initial cell concentration of 25,000 cells/ml. Cultures were grown in a shaking bath at 30° C for 48 hr prior to determining the cell concentration using A600. Drug-free cultures of each strain served as a control. In each panel ▲ is WT, ■ is S558Y. Because many of the mutant curves superimpose, no attempt was made to distinguish the individual mutants. C, The E244G, S558Y double mutation was made in pSS607 (◆) for comparison with the original chromosomal suppressor mutation (▼).
Because the E244G mutation is the subject of a detailed study, we also recreated the E244G, S558Y suppressor mutation in pSS607 and compared the clo resistance of the plasmid-borne suppressor mutation to the chromosomal equivalent (Figure 5C). As with the reconstructed N242K, S558Y suppressor (6), the IC50s of the chromosomal and plasmid E244G, S558Y suppressor were, within experimental error, indistinguishable. In each case, they showed an ~8-fold increase in clo resistance when compared to S558Y. Although all of the suppressor mutations are interesting and most lie in a general interface suggested by structural and crosslinking studies, we focused on E244G.
The effect of NBD mutations in a S558 (otherwise WT) background
Previous work indicated that the N242K mutation, which together with S558Y restores significant drug resistance, actually creates drug hypersensitivity in an otherwise WT (S558) background (6). The sensitivity is more pronounced for cyh than for clo. This drug sensitivity establishes the importance of the deviant region of NBD-1 for complete transporter function. In the present study, we evaluated the effect of E244G and its neighboring residues on drug resistance, because Glu-244 defines the deviant Q-loop of NBD1. The Q-loop is thought to facilitate communication between the ATP-binding site and the ICLs (1). As a means of comparison, we constructed the corresponding mutation in NBD2; Q951G. Immunoblotting was carried out using Pma1 antibody as a loading control. As shown in Figure 6A, the levels of Pdr5p in purified membrane vesicles were similar.
FIGURE 6.

Phenotypic features of the E244G and Q951G mutations. A, an immunoblot of purified plasma membrane (PM) vesicles prepared from various strains of yeast. N242K was previously shown to have WT levels of Pdr5 in the plasma membrane (6). Samples containing 20 μg PM vesicle protein were solubilized in SDS-PAGE for 30 min at 37 °. Conditions for gel electrophoresis and Immunoblotting were previously described (6). The blots were simultaneously treated with a 1:1000 dilution of antiPdr5 and anti Pma1 antibodies. The latter serves as a loading control. Pertinent molecular weights are given in kDa. Pdr5 is 160 kDa and Pma1 is 99.6 kDa. The lanes are as follows: lane 1, WT; lane 2, Δpdr5; lane 3, E244G; lane 4, Q951G; lane 5, E244G, Q951G; Quantitative analysis of B, cyh and C, clo resistance. Cultures containing drug were set up and grown as described (6). The plots and statistical analysis were carried out using GraphPad Prism software. The figures show the mean values of three independent experiments and the error bars represent the S.D. In these panels the strains are designated as follows: ▲ is WT, ■ is S558Y, ▼ is E244G, ◆ is Q951G, ● (---) is E244G, Q951G.
We determined the relative cyh and clo resistance of these strains and the double mutant E244G, Q951G, along with isogenic negative and positive controls (Figure 6C and D). The E244G phenotype was virtually identical to that previously observed with N242K (6). Compared to the WT, significant cyh sensitivity was observed but resistance to clo was only slightly lower (1.3-fold reduction in IC50 ). The phenotype of the corresponding NBD2 mutant, Q951G, was similar. This mutant was ~2-fold more sensitive to cyh than was the WT. The Q951G strain, however, exhibited significantly greater clo hypersensitivity (3.4-fold) than did the E244G mutant. Compared to either S558Y or Δpdr5, however, the phenotypes of these mutants were quite similar. Therefore, E244G is not a gain-of-function mutation in a nonessential residue. Rather, as was true for Asp-242, Glu-244 is required for WT levels of drug resistance.
Double-mutant analysis: redundancy of Q-loop function during ATP hydrolysis
Our single-mutant analysis led to several important observations. First, although E244G was a dramatic alteration in a highly conserved residue, it created only mild drug hypersensitivity. The same was true for Q951G in the canonical ATP-binding domain. Thus, all of these mutants were much like the previously characterized N242K mutant. The N242K and E244G mutants appeared more sensitive to cyh than clo, and Q951G appeared somewhat more sensitive to clo than cyh. All, however, were considerably more resistant than S558Y. The relatively mild loss in the drug resistance of the single mutants suggested that there might be functional overlap between the corresponding region in NBD-2. Essentially, then, only Glu-244 or Gln-951 is actually required for Pdr5 to mediate a significant portion of its drug resistance to cyh or clo. To further investigate the possible functional overlap between the 2 Q-loops, we constructed a E244G, Q951G double mutant and compared the drug sensitivity of the single and double mutants to the WT. We tested them quantitatively for their cyh and clo drug resistance. The results for E244G, Q951G are shown in Figure 6C (cyh) and 6D (clo). We calculated the IC50 values for cyh for the isogenic constructs and found that the difference in sensitivity relative to the WT strain was 17.2 for S558Y, 2.60 for E244G, 1.90 for Q951G, and 6.81 for E244G, Q951G. If the Q-regions were functioning independently and the 2 mutants were partially impaired in biochemical function, the double mutant would show simple additivity and thus be 4.50-fold more sensitive. The observed value of 6.81-fold was significantly greater than we expected. These initial results thus suggested that at least some functional redundancy exists between the 2 Q-loops during ATP hydrolysis. We tested the strains with clo and observed a similar pattern. In this set of experiments, the difference between WT (22.5μM) and E244G (17.5 μM) was only 1.3-fold; this result was virtually identical to the one obtained with N242K (6). The Q951G strain had an IC50 of 6.78 μM, a 3.40-fold difference. Thus, the expectation for the double mutant based on simple additivity is a difference from the WT strain of 4.7-fold. In fact, the E244G, Q951G mutant was 7.7-fold more sensitive than the WT.
The large difference in drug sensitivity between the single and double mutants was also observed qualitatively for 1.5 mM and 3.0 mM chloramphenicol, a moderately strong transport substrate used in whole-cell and vesicle transport assays (11, 14, 15). The single E244G and Q951G mutants were all qualitatively similar to WT. The E244G, Q951G double mutant was phenotypically very hypersensitive and similar to S558Y (see supporting information).
It was theoretically possible that the profound drug hypersensitivity observed with the double mutant was not entirely attributable to the combined E244G, Q951G alterations in Pdr5, but rather to an additional mutation in the genome that occurred spontaneously. We tested two additional, independent E244G, Q951G transformants and observed the drug hypersensitivity that was indistinguishable from that seen with the original double mutant. Thus, when analyzed together for their resistance to clo, all three E244G, Q951G independently constructed strains had identical minimum inhibitory concentrations of 5 μM and IC50s of ~2.9 μM.
The effect of Q-region mutations on NTPase activity
The effect of Q-loop mutations on ATP hydrolysis has not been investigated extensively, but two studies with ABC transporters suggest that these alterations do not affect catalysis. For instance, although the bacterial MsbA transporter Q-loop residue appears to be required for the proper conformational signaling change during ATP hydrolysis, a Q-to-C mutation does not affect ATPase activity per se (16). Analogous results were found with P-gp (17). In reporting on a study that used FRET (Forster resonance energy transfer) Rai et al. suggested a role for the equivalent Cdr1 residue (Glu-237) in coordinating the Mg ion during ATP hydrolysis, but this study was carried out with a single, purified NBD and did not look directly at ATPase activity or determine a drug phenotype (18).
We therefore evaluated the effect of N242K, E244G, and Q951G on the Pdr5-ATPase activity along with S558Y, E244G. Results of our measurement of ATPase activity are in Figure 7A and 7B. For comparison, we also included Vmax and Km values for S558Y that were previously published (6). The average (n=3) WT value of ~220 nmol/min/mg was in the range we reported previously for such double-copy strains: ~179–275 nmol/min/mg (6). This was ~4 fold higher than the Vmax of E244G or Q951G, which were each ~50–60 nmol/min/mg (Figure 7B). It is particularly striking that mutation of the canonical Gln-951 residue and the deviant Glu-244 had an equivalent ATPase deficiency as well as similar drug hypersensitivity. The N242K mutant exhibited significantly higher ATPase activity. Although the reduction in ATPase activity of the Q-loop mutants was clear, significant activity remained. Therefore, it is highly unlikely that these residues are required for the reaction chemistry of NTP hydrolysis.
FIGURE 7.

The ATPase activity of single and double mutants. A, ATPase assays were conducted with purified plasma membrane vesicles recovered from strains bearing a double copy of the WT, single or double mutants. A representative experiment is shown for each strain. The data for S558Y were previously reported and serve as a reference (6). ATPase activity was determined as previously described (11) using 12 μg purified plasma membrane incubated for 8 min at 35° C before terminating the reaction with 2.5% SDS. The small amount (< 10%) of non-specific activity found in the Δpdr5 strain (R-1) is subtracted when calculating activity. Data were analyzed using GraphPad Prism software. The strains are represented as follows: ■ is WT, □ is N242K, ▲ is E244G, ▼ is Q951G, + is S558Y, E244G, * is S558Y and ◆ is E244G, Q951G B, and C, The kinetic parameters of ATPase activity in WT and mutant plasma membrane vesicles. The values represent at least 3 independent experiments carried out with two (Q951G) or three different plasma membrane vesicle preparations of each strain.
There was little or no difference in the observed Kms (Figure 7C). This suggests that none of the mutants has a significant effect on ATP-binding.
Data from our previous work (6) and the present study strongly suggest that Pdr5 has poor coupling of ATP hydrolysis to drug transport. For instance, both N242K and E244G restore considerable drug resistance to S558Y, yet they did not increase the basal level of ATPase activity. Furthermore, there was a relatively poor correlation between the ATPase activity of the E244G, N242K, and Q951G mutants and the IC50 data for cyh. This can be seen in Table 3, which summarizes the important phenotypes for the mutants. The N242K mutation had 3-fold greater ATPase activity than E244G but virtually identical resistance to cyh. These observations support the contention of Ernst et al. (19, 20) that much of the basal ATPase activity is uncoupled from transport—at least for the substrates that we employ.
Table 3.
Summary of important phenotypic characteristics
| Residue alteration | IC50 (μM cyh) | Δ1 | IC50 (μM clo) | Δ | Vmax (ATPase) | Δ |
|---|---|---|---|---|---|---|
| WT | 7.50 | 22.5 | 222 | |||
| S558Y | 0.43 | 17.4 | 1.11 | 20.3 | 74.82 | 2.9 |
| S558Y, N242K2 | 4.10 | 1.8 | Not done | 88.9 | 2.5 | |
| S558Y, E244G | 3.75 | 2.0 | 8.25 | 2.7 | 82.4 | 2.7 |
| N242K | 3.75 | 2.0 | 15.0 | 1.5 | 144 | 1.6 |
| E244G | 2.93 | 2.6 | 17.5 | 1.3 | 58.5 | 3.8 |
| Q951G | 3.90 | 1.9 | 6.78 | 3.4 | 57.3 | 3.7 |
| E244G, Q951G | 1.10 | 6.8 | 2.92 | 7.7 | 61.0 | 3.6 |
Δ value is the fold-difference between the particular mutant and the WT
Data are from (6)
In light of the equivalence of the deviant and canonical Q-loop residues and the greater than additive hypersensitivity of the E244G, Q951G strain, we expected that this double mutant would at least show an additive ATPase deficiency. If this were the case, the Vmax of the double mutant would be ~25 nmol/min/mg, a value that would be quite noticeable with our assay conditions. Significantly, the ATPase of the E244G, Q951G double-mutant strain was no lower than that of the single mutants, even though the difference in clo drug sensitivity between this strain and E244G was greater than 5-fold. This observation has several important implications, the most significant of which is that the severe transport deficiency in the double mutant is caused by something other than a further loss of ATP hydrolysis.
Pdr5 has a significant GTPase activity that is capable of fueling transport and is approximately 0.4 that of its ATPase counterpart (11). Data presented in Figure 8 demonstrate that both E244G and E244G, Q951G had a Vmax for the WT was only ~1.5- to 2-fold higher than that of the mutants. The reduction in GTPase activity of the mutants was therefore significantly less than that observed for ATPase activity. Furthermore, the plots of E244G and E244G, Q951G activity versus GTP concentration are indistinguishable. Thus, the sharp reduction in drug resistance observed for E244G, Q951G could not be attributed to a further loss of NTPase function.
FIGURE 8.

The GTPase activity of WT, E244G, and E244G, Q951G. The assay for GTPase activity is identical to the one used to measure ATP hydrolysis except that 16 μg of plasma membrane protein was used in each reaction GTPase activity of WT (■), E244G (▲), and E244G, Q951G (▼) were compared. Plots are the average of three independent experiments from three independent plasma membrane vesicle preparations of each strain (n=3).
The drug-binding capability of the E244G, Q951G mutant
Previous work in our laboratory demonstrated that Pdr5, like other transporters, has a multiplicity of drug-binding sites (15, 21). One of these mediates the transport of rhodamine 6G, clo, and 3,9-diacetylcarbazole (22). We previously demonstrated that rhodamine 6G and clo block the photoactivatable crosslinking of IAAP in a concentration-dependent manner (6, 22). This assay also showed that the clo-binding affinity of the S558Y mutant remained the same as in the WT(6). With the same assay, we evaluated drug-binding affinity in E244G, Q951G, because it is clo hypersensitive (Figure 9). The data clearly show that the WT and double mutant bind IAAP. We used clo in a competition experiment to block IAAP photoaffinity labeling and determined an IC50 value of ~20 μM for both strains. This value is similar to those obtained previously for WT and S558Y(6). Therefore, the large difference in drug sensitivity (6- to 8-fold) between the WT and E244G, Q951G strains is not attributable to a deficiency in drug binding at the transport site.
FIGURE 9.
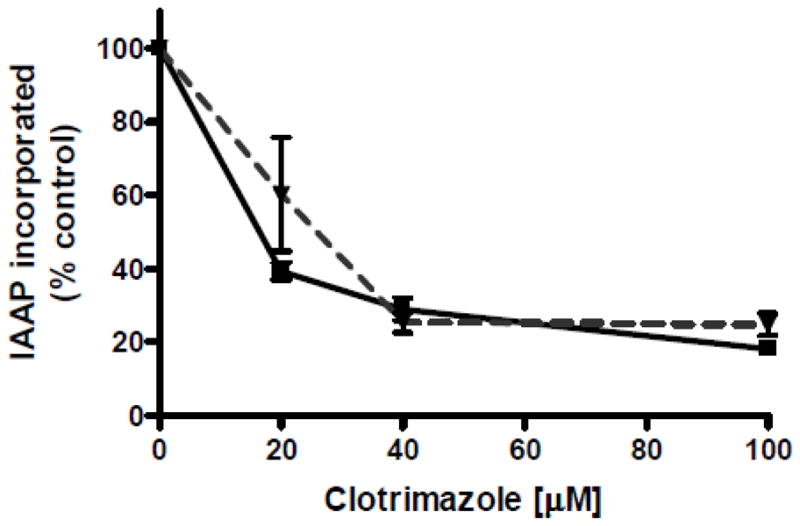
Drug binding in vesicles from the E244G, Q951G mutant. The IAAP-binding capability of the WT (■) and E244G, Q951G double mutant (▼) were evaluated using the photo-affinity labeling protocol as previously described (6). The plots are the averages of readings from three gels.
Taken together, our results strongly implicate the Q-loop residues in signaling between the ATP-binding sites and the TMDs. Furthermore, they strongly imply that the atypical Glu-244 and canonical Gln-951 residues are functionally equivalent and overlapping.
DISCUSSION
The recently resolved structures of Sav1866 (3) and crosslinking studies (4, 5) provide insights into the coupling interface of ABC proteins. The coupling of ATP hydrolysis (at the NBDs) to transport (at the TMDs) is one of the most critical steps of the transport cycle of ABC proteins. Characterizing the coupling interface in a functional protein, however, is challenging. We previously developed a genetic screen coupled with biochemical assays to identify residues associated with the coupling interface in the yeast ABC transporter Pdr5 (6). In that study, we exploited a mutation at the extracellular end of TMH2 (S558Y). ATP hydrolysis and molecular movements in the TMHs (which bring about transport) are uncoupled in the mutant Pdr5, which shows both ATP hydrolysis and binding of the transport substrate, IAAP, but no drug resistance. Our strategy was to screen for second-site mutations that restore drug resistance, because these suppressors should identify interacting residues. We reported that the double-mutant S558Y, N242K exhibited almost a complete reversal of the null phenotype of S558Y. The location of N242K was consistent with the NBD face of the coupling interface surmised from the X-ray crystallographic structure of the bacterial ABC multidrug transporter Sav1866 (3) as well as with crosslinking studies on P-glycoprotein (4). In the current study we extended our work to further elucidate the coupling interface of Pdr5, identifying key residues in both the NBD face of the protein and ICL1, which connects TMHs 2 and 3.
We used the mutant S558Y to screen for clones that reverse the drug-sensitive phenotype of this mutant as described in detail previously (6). Upon sequencing the Pdr5 gene in the revertants, we identified nine second-site mutations (including N242K from the previous study), representing 5 unique point mutations and one triplet-codon deletion. Four of these occurred in the NBD and were located almost contiguous to each other at positions 242, 244, and 246 (one independent mutant each of N242K and E244G and two independent mutants with D246del). The residue Glu-244 is equivalent to the conserved glutamine that is used to identify the Q-loop within the NBDs of ABC transporters. Thus, four of the nine second-site mutations appear to define the NBD face of the coupling interface and are proximal to or at the Q-loop. An additional 3 are in Ser-597, which is in the ICL1. The structure of Sav1866 suggested that conserved Q might interact with the ICL1 to couple ATP hydrolysis to drug transport. The residues we identified lie in the same region of Pdr5. Moreover, the residue Glu-244 in Pdr5 lies at the same position as the conserved Q in other ABC transporters, and the glutamic acid at this position is completely conserved in the Pdr5 family (see sequence alignment, Figure S1 in the supporting information). Our results are consistent with the suggestion put forward by several authors that the Q-loop is implicated in intradomain coupling (1, 7). Beside the second-site mutations in the NBD, we identified three additional point mutations in ICL1, and all were in the same residue, Ser-597. Thus, both faces of the interface—the NBD as well as the ICL—were recovered in our collection. Although ICL1 is predicted to be approximately 21 amino acids long, the identification of Ser-597 as a second-site mutant in three independently derived colonies suggests that relatively few residues in this loop actually participate in critical contact with the ATP-binding sites. Alternatively, the codons specifying Ser-597 may have a higher likelihood of undergoing mutation than those specifying the other ICL 1 residues.
The genetic screen therefore provides a functional confirmation of the suggestion prompted by the structure of Sav1866 that the NBDs interact with the TMDs via ICLs. This is found in all ABC transporters analyzed to date. However, the functional, genetic evidence from the current study suggests that the Pdr5 signaling interface is in the cis configuration—at least when transporting clo. The inferred pathway is N-terminal NBD Q-loop region (Asn-242, Glu-244, Asp-246) to ICL-1 (Ser-597) to TMD1 (Ser-558, Met-679). An atomic model of Pdr5 shows that E244 lies very close to ICL1, but it is also directly under ICL4, which connects TMH 10 and 11 in TMD2 (R. Rutledge, unpublished observations). Thus, a trans configuration is also theoretically feasible. However, we did not recover any NBD2 (trans) suppressors of S558Y, but we found 4 independent mutations in NBD1. Similar results were obtained by selecting cyhr suppressors of N242K in NBD1 (see Figure S2 in the supporting materials). It remains to be seen whether the trans configuration is ever used physiologically. An interesting future experiment would be to determine whether selection of S558Y suppressors on other Pdr5 transport substrates leads to second-site mutations in NBD2.
Two possible explanations are strongly implied by these findings. Pdr5 may differ from all prokaryotic and eukaryotic ABC efflux pumps analyzed to date. Structural studies of several importers, however, such as E. coli ButCD, which influxes vitamin B, and ModBC, which is an archea molybdate pump, appear to have a signal interface in the cis configuration (23, 24). Because Pdr5 is evolutionarily distant from these, it would be important to determine whether other eukaryotic members of the ABC family have this arrangement. Other studies were conducted in the absence of transport substrate, whereas our mutants were selected on drug plates, so it is also quite possible that both cis and trans exist physiologically. For instance, it is plausible that drug binding results in conformational switching.
As illustrated in Figures 1 and S1, the Walker A and Walker B domains of the N-terminal NBD and the signature region of the second NBD vary significantly in the identity of key conserved residues vis-à-vis other ABC transporters. Thus, together they make up a deviant ATP-binding site. Because Glu-244 replaces the canonical Gln in the Q-loop, we characterized it in an otherwise WT background as well as the equivalent Gln-951 in NBD2.
The effect of site-directed mutagenesis on Q-loop residues was studied in mouse P-gp by Urbatsch et al. (17). In mouse P-gp, these residues are Gln-471 and Gln-1114 in the NBD1 and 2, respectively. The mutants Gln-471A and Gln-1114A showed a reduction in ATPase activity. However, in no case was the impairment greater than two orders of magnitude, and the Km (ATP) was not altered, leading the authors to conclude that the mutations had no major effect on the substrate binding or on reaction chemistry (17). Furthermore, mutations in either of the two P-gp catalytic sites produced the same effects, implying functional symmetry.
Because the most obvious effect of the Gln-471A or Gln-1114A mutation was to reduce stimulation of ATPase activity by transport substrates, it was hypothesized that such residues play a critical role in interdomain communication. Mutations in either the Glu-244 or Gln-951 residue of Pdr5 had similar effects on ATP hydrolysis (Figure 6 and Table 3); although ATP hydrolysis was diminished, a significant residual activity remained, and the Km was unaffected. We also studied E244G, Q951G, which showed no less ATPase activity than that observed in either of the two single mutants (Figure 6B). GTPase activity, which our previous work suggests has a physiological role to play in Pdr5-mediated transport, decreased even less than ATPase activity. Thus, our data are consistent with these residues being nonessential to the reaction chemistry (such as activation of the attacking water for ATP hydrolysis). However, unlike the Km of ATP hydrolysis, the IC50 for the Pdr5 transport substrates clo and cyh is significantly decreased in both the E244G or Q951G mutant. Moreover, Pdr5-mediated drug resistance is largely abrogated in the double mutant, E244G, Q951G. Its phenotype is almost equivalent to the S558Y strain originally used to isolate the suppressor collection (Figure 6).
It was evident that the reduced resistance to drug substrates in the E244G, Q951G double mutant was not a consequence of impaired binding of the transport substrate. We demonstrated that the photoaffinity transport substrate analog IAAP crosslinked to WT and mutant Pdr5s with comparable efficiency (Figure 9). E244G was initially isolated as a suppressor of the faulty-signaling S558Y mutant, and the results from the current study further indicate that the primary role of the conserved residues Glu-244 and Gln-951 is to communicate signals from the ATP sites to the TMDs via the ICLs. Furthermore, the much greater drug sensitivity of the double mutant and the similarity in phenotype of the single mutants strongly implies that these residues are functionally overlapping even though one is from a deviant portion of NBD-1.
Equivalence of function is, of course, a striking feature of symmetric transporters such as P-gp; hence the phenotypic similarity of the Q-loop residue mutants (Q471A and Q1114A). Asymmetric transporters may differ, however. For instance, mutation of the conserved Walker A lysine in Tap1 and the analogous residue in Tap2 (Tap1 and Tap2 make up a peptide-translocating heterodimer) results in different effects on ATP binding and peptide transport (25). A similar nonequivalence is observed in MRP1. Mutation of the same residue in the Walker A motif of each NBD has disparate effects on nucleotide binding (24). The observation, therefore, that the Q-loops of Pdr5 are equivalent and overlapping strongly implies that both ATP-binding sites are sending signals to the TMDs.
Pdr5 has a high basal ATPase that is not stimulated further by the addition of exogenous transport substrates (11, 19, 26). In this regard, it is strikingly different from P-gp. Our data also strongly suggest that Pdr5 is not especially efficient at coupling ATP hydrolysis to transport and they support the contention that much of this activity is uncoupled (19, 20). For instance, although the S558Y, E244G and S558Y, N242K suppressors restore cyh resistance 8-fold, they show no restoration of ATPase activity. The Pdr5 family of fungal transporters appears distinct from all other eukaryotes and may be a unique, but clinically important evolutionary variant.
Supplementary Material
Acknowledgments
We thank Jenine Musa for constructing E244G, Lee May for many useful discussions and Trish Weisman for excellent copy editing of the main text.
Footnotes
Abbreviations: ABC, ATP-binding cassette; NBD, nucleotide-binding domain; TMD, transmembrane domain; ICL, intracellular loop; TMH, transmembrane helix, clo, clotrimazole; cyh, cycloheximide; 5-FOA, 5 fluoroorotic acid; Gen, geneticin; IAAP, iodoarylazidoprazosin; WT, wild type; P-gp, P-glycoprotein
This work was supported by NIH Grant GM07721 to J.G., the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (R.R., D.X., S.V.A.), and the FDA (Z.E.S.)
SUPPORTING INFORMATION
These data include the alignment of the NBDs of the Pdr5 family (Figure S1), additional evidence for a cis arrangement of Q-loops and TMDs (Figure S2 and Table S1) and additional testing of drug phenotypes for the Q-loop mutations (Figure S3). This is available free of charge at http://pubs.acs.org.
References
- 1.Seeger MA, van Veen HW. Molecular basis of multidrug transport by ABC transporters. BBAPAP. 2009;1794:725–737. doi: 10.1016/j.bbapap.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Sauna ZE, Nandigama K, Ambudkar SV. Exploiting Reaction Intermediates of the ATPase Reaction to Elucidate the Mechanism of Transport by P-glycoprotein. (ABCB1) J Biol Chem. 2006;281:26501–26511. doi: 10.1074/jbc.M601917200. [DOI] [PubMed] [Google Scholar]
- 3.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 4.Zolnerciks JK, Wooding C, Linton KJ. Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. FASEB J. 2007;21:3937–3948. doi: 10.1096/fj.07-8610com. [DOI] [PubMed] [Google Scholar]
- 5.Oancea G, O’Mara ML, Drew-Bennett WF, Tielman P, Abeles R, Tampe R. Structural arrangement of the the transmission interface in the antigen ABC transport complex TAP. Proc Natl Acad Sci (USA) 2009;106:5551–5556. doi: 10.1073/pnas.0811260106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sauna ZE, Bohn SS, Rutledge R, Dougherty MP, Cronin S, May L, Xia D, Ambudkar SV, Golin J. Mutations Define Cross-talk between the N-terminal Nucleotide-binding Domain and Transmembrane Helix-2 of the Yeast Multidrug Transporter Pdr5. J Biol Chem. 2008;283:35010–35022. doi: 10.1074/jbc.M806446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones PM, George AM. The ABC transporter structure and mechanism: perspectives on recent researchCMLS. Cell Mol Life Sci. 2004;61:682–699. doi: 10.1007/s00018-003-3336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogers B, Decottignies A, Kolaczkowski M, Carvajal E, Balzi E, Goffeau A. The pleiotropic drug ABC transporters from Saccharomyces cerevisiae. J Mol Microbiol Biotechnol. 2001;3:207–214. [PubMed] [Google Scholar]
- 9.Meyers S, Schauer W, Balzi E, Wagner M, Goffeau A, Golin J. Interaction of the yeast pleiotropic drug resistance genes PDR1 and PDR5. Curr Genet. 1992;21:431–436. doi: 10.1007/BF00351651. [DOI] [PubMed] [Google Scholar]
- 10.Balzi E, Wang M, Leterme S, Dyck LV, Goffeau A. PDR5, a Novel Yeast Multidrug Resistance Conferring Transporter Controlled by the Transcription Regulator PDR1. J Biol Chem. 1994;269:2206–2214. [PubMed] [Google Scholar]
- 11.Golin J, Kon ZN, Wu CP, Martello J, Hanson L, Supernavage S, Ambudkar SV, Sauna ZE. Complete inhibition of the Pdr5p multidrug efflux pump ATPase by its transport substrate clotrimazole suggests that GTP as well as ATP may be used as an energy source. Biochemistry. 2007;46:13109–13119. doi: 10.1021/bi701414f. [DOI] [PubMed] [Google Scholar]
- 12.Rutledge RM, Ghislain M, Mullins JM, de Thozée CP, Golin J. Pdr5-mediated multidrug resistance requires the CPY-vacuolar sorting protein Vps3: are xenobiotic compounds routed from the vacuole to plasma membrane transporters for efflux. Mol Genet Genomics. 2008;279:573–583. doi: 10.1007/s00438-008-0334-5. [DOI] [PubMed] [Google Scholar]
- 13.Rutledge R, Esser L, Ma, Jichun, Xia D. Toward understanding the mechanism of the yeast multidrug resistance transporter Pdr5p: a molecular modeling study. 2010 doi: 10.1016/j.jsb.2010.10.012. manuscript in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonard PJ, Rathod PK, Golin J. Loss of function mutation in the yeast multiple drug resistance gene PDR5 causes a reduction in chloramphenicol efflux. Antimicrob Agents Chemother. 1994;38:2492–2494. doi: 10.1128/aac.38.10.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golin J, Ambudkar S, Gottesman M, Habib A, Sczepanski J, Ziccardi W, May L. Studies with novel Pdr5p substrates demonstrate a strong size dependence for xenobiotic efflux. J Biol Chem. 2003;278:5963–5969. doi: 10.1074/jbc.M210908200. [DOI] [PubMed] [Google Scholar]
- 16.Dalmas O, Orelle C, Foucher AE, Geourjon C, Crouzy S, Pietro AD, Jault JM. The Q-loop Disengages from the First Intracellular Loop during the Catalytic Cycle of the Multidrug ABC Transporter BmrA. J Biol Chem. 2005;280:36857–36864. doi: 10.1074/jbc.M503266200. [DOI] [PubMed] [Google Scholar]
- 17.Urbatsch IL, Gimi K, Wilke-Mounts S, Senior AE. Investigation of the Role of Glutamine-471 and Glutamine-1114 in the Two Catalytic Sites of P-Glycoprotein. Biochemistry. 2000;39:11921–11927. doi: 10.1021/bi001220s. [DOI] [PubMed] [Google Scholar]
- 18.Rai V, Gaur M, Kumar A, Shukla S, Komath SS, Prasad R. A novel catalytic mechanism for ATP hydrolysis employed by the N-terminal nucleotide-binding domain of Cdr1p, a multidrug ABC transporter of Candida albicans. Biochim Biophys Acta. 2008;1778:2143–2153. doi: 10.1016/j.bbamem.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 19.Ernst R, Kueppers P, Klein CM, Schwarzmueller T, Kuchler K, Schmitt L. A mutation of the H-loop selectively affects rhodamine transport by the yeast multidrug ABC transporter Pdr5. Proc Natl Acad Sci. 2008;105:5069–5074. doi: 10.1073/pnas.0800191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ernst R, Kueppers P, Stindt J, Kuchler K, Schmitt L. Multidrug efflux pumps: Substrate selection in ATP-binding cassette multidrug efflux pumps – first come, first served. FEBS J. 2010;277:540–549. doi: 10.1111/j.1742-4658.2009.07485.x. [DOI] [PubMed] [Google Scholar]
- 21.Golin J, Ambudkar SV, May L. The yeast Pdr5p multidrug transporter: How does it recognize so many substrates. Biochem Biophys Res Commun. 2007;356:1–5. doi: 10.1016/j.bbrc.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 22.Hanson L, May L, Tuma P, Keeven J, Mehl P, Ferenz M, Ambudkar SV, Golin J. The role of hydrogen bond acceptor groups in the interaction of substrates with Pdr5p, a major yeast drug transporter. Biochemistry. 2005;44:9703–9713. doi: 10.1021/bi0502994. [DOI] [PubMed] [Google Scholar]
- 23.Locher KP, Lee AT, Rees DC. The E. coli ButCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 24.Hollenstein K, Frei DC, Locher KP. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 25.Lapinski PE, Neubig R, Raghavan M. Walker A lysine mutations of Tap1 and Tap2 interfere with peptide translocation, but not peptide binding. J Biol Chem. 2001;276:7526–7533. doi: 10.1074/jbc.M009448200. [DOI] [PubMed] [Google Scholar]
- 26.Decottignies A, Kolaczkowski M, Balzi E, Goffeau A. Solubilization and characterization of the overexpressed PDR5 multidrug resistance nucleotide triphosphatase of yeast. J Biol Chem. 1994;269:12797–12803. [PubMed] [Google Scholar]
- 27.Kim I-W, Peng X-H, Sauna ZE, FitzGerald PC, Xia D, Muller M, Nandigama K, Ambudkar SV. The conserved tyrosine residues 401 and 1044 in the ATP sites of human P-glycoprotein are critical for ATP binding and hydrolysis: evidence for a conserved subdomain, the A-loop in the ATP-binding cassette. Biochemistry. 2006;45:7605–7616. doi: 10.1021/bi060308o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.