

Abstract
Inflammation plays a central role in the development and progression of coronary heart disease (CHD). The sex hormones estrogen and testosterone have been shown to modify the inflammatory response by influencing cytokine expression in human macrophage cells obtained from younger individuals. The effect of these hormones on the expression of proinflammatory markers in macrophages obtained from a CHD-age relevant population has not been studied. Human monocyte-derived macrophage cells (HMDM) were obtained from healthy normolipidemic men and postmenopausal women (age 50-70 years), and cultured in autologous serum along with both physiological and supraphysiological concentrations of estrogen or testosterone. HMDM were stimulated with oxidized low density lipoproteins (oxLDL) and the expression of the cytokines TNF-α, IL-6, and IL-1β, and of the acute-phase protein CRP was measured. Both physiological and supraphysiological concentrations of testosterone reduced the expression and secretion of TNF-α and reduced the expression of IL-1β, but did not affect IL-6 or CRP expression. Estrogen did not modify the expression of TNF-α, IL-6, and IL-1β. Estrogen caused a variable response in CRP expression that was positively associated with the donor’s plasma small dense LDL cholesterol concentration. There were no gender differences in any of the observed effects.
Our results indicate that testosterone may exert anti-inflammatory effects by reducing macrophage TNF-α expression while the effects of estrogen on macrophage CRP expression may depend upon the extracellular lipid environment.
Keywords: Sex Hormones, Inflammation, Coronary Heart Disease, Macrophage, Cytokine, CRP
Introduction
The pathology of coronary heart disease (CHD) is complex and multifaceted with inflammation playing a central part (Libby 2002). Peripheral blood monocytes recruited into the intima-media layer of an artery play a pivotal role in the local inflammation (Brown & Goldstein 1983). Recruited monocytes differentiate into macrophage cells and begin to take up oxidized lipoproteins, leading to the formation of foam cells. Foam cells are a primary component of early atheroma lesion formation (Lloyd-Jones et al. 2009) and are a significant source of proinflammatory cytokines (Tipping & Hancock 1993), which include tumor necrosis factor alpha (TNF-α), interleukin (IL)-6, and IL-1β. These cytokines actively participate in atherogenesis by promoting endothelial dysfunction, further monocyte recruitment, and smooth muscle cell apoptosis (Valente et al. 1992).
C-reactive protein (CRP) is an acute-phase protein that serves as a marker of systemic inflammation and has been shown to be an independent predictor of CHD risk (Torres & Ridker 2003). Most circulating CRP is secreted by the liver, yet a small amount of CRP is produced by macrophage cells present in atherosclerotic plaques (Yasojima et al. 2001). Levels of CRP mRNA and protein have been found to be up to 10-fold higher in arterial plaque tissue compared with the normal artery, suggesting that at least a portion of atheroma CRP content is locally produced (Kobayashi et al. 2003). In vitro and animal studies indicate that CRP may actively participate in plaque development by promoting endothelial dysfunction (Bisoendial et al. 2007), monocyte adhesion to the endothelium (Li et al. 2004), macrophage cholesterol accumulation (Singh et al. 2008), and fibrin breakdown (reduced plaque stability) (Williams et al. 2004).
The steroid hormones 17β-estradiol (E) and testosterone (T) are thought to play a role in modulating inflammation and thereby influencing atherogenesis. In postmenopausal women, E replacement therapy does not affect circulating TNF-α and IL-6 concentrations (Pradhan et al. 2002; Zegura et al. 2003). However, hormone treatment may cause changes in cytokine levels in the arterial lesions that may not be entirely predicted by plasma cytokine levels. Very little work has been conducted examining the effect of E on cytokine expression in human monocyte-derived macrophage cells (HMDM), with one study showing that E withdrawal in female pre-menopausal HMDM after 24 hours of treatment resulted in greater proinflammatory cytokine expression compared with HMDM continually exposed to the hormone for 48 hours (Kramer et al. 2004). With T, the expression of TNF-α, IL-6, and IL-1β was shown to be reduced both in rodent macrophage cell models and in human monocytes obtained from younger individuals (Chao et al. 1995; Kanda et al. 1996; Kanda et al. 1997; D’Agostino et al. 1999), yet no studies have been done in HMDM from older individuals. The effect of E and T on CRP expression in HMDM has not been studied. The purpose of this study was to assess the effect of E and T treatment on the expression of proinflammatory cytokines and CRP by macrophages obtained from a CHD age-relevant population.
Materials and Methods
Materials
E and T were purchased from Sigma-Aldrich (St. Louis, MO). Phenol-free RPMI1640 medium was purchased from Gibco (Carlsbad, CA). Ficoll-Paque was obtained from GE Healthcare (Piscataway, NJ). RNeasy mini kit was purchased from Qiagen (Germantown, MD). Penicillin, streptomycin, and Superscript III Reverse Transcriptase Kit were obtained from Invitrogen (Carlsbad, CA). Power SYBR-green master mix was purchased from Applied Biosystems (Carlsbad, CA). Bicinchoninic Acid Protein Assay was obtained from Pierce (Rockford, IL).
Subjects
Male (n=10) and postmenopausal female (n=10) volunteers between 50 and 70 years of age were recruited. Subjects were included if they reported no history of CHD, cancer, diabetes, or renal, liver, or thyroid disease. Subjects who smoked or had hypertension were excluded from the study. Volunteers were not taking any medications to control plasma lipid or glucose levels. Inclusion criteria were: low density lipoprotein cholesterol (LDL-C) <160 mg/dL, high density lipoprotein cholesterol (HDL-C) ≥40 mg/dL, triglycerides (TG) <150 mg/dL, and glucose ≤100 mg/dL. Women were considered postmenopausal if absence of menstrual periods exceeded 1 year. Most women (n=8) in this study had been postmenopausal for >5 years. Lastly, because it has been reported that the estrogen receptor α (ERα) gene polymorphism IVS1-401 T/C, located within the first intron, can affect the plasma lipid response to E (Herrington et al. 2002), volunteers were genotyped for this mutation and subjects with the IVSI C/C genotype were excluded. Characteristics of subjects are shown in Table 1.
Table 1.
Characteristics and fasting metabolic and lipid profile of study volunteers.
| Women (n=10) | Men (n=10) | p value * | |
|---|---|---|---|
| Age (years) | 59 (4) | 61 (6) | 0.423 |
| BMI (kg/m2) | 27.6 (6.2) | 26.0 (2.9) | 0.481 |
| Lipids | |||
| TC (mg/dL) | 206 (31) | 178 (34) | 0.069 |
| LDL-C (mg/dL) | 111 (33) | 87 (36) | 0.136 |
| sdLDL-C (mg/dL) | 26 (8) | 29 (13) | 0.555 |
| HDL-C (mg/dL) | 66 (16) | 46 (16) | 0.011* |
| TG (mg/dL) | 78 (28) | 99 (46) | 0.229 |
| Glucose (mg/dL) | 91 (7) | 91 (8) | 0.976 |
| Plasma CRP (μg/mL) | 3.9 (3.6) | 1.4 (0.91) | 0.053 |
| ERα IVS1 | T/C = 5, T/T = 5 | T/C = 8, T/T = 2 | 0.349 |
Values expressed as means (SD);
p value for gender difference.
Plasma Lipid Measurements, and LDL Isolation and Oxidation
Plasma lipid levels were determined by enzymatic assays (Roche Diagnostics, Indianapolis, IN). Plasma levels of small dense LDL-C (sdLDL-C) were assessed by an enzymatic assay (Denka Seiken Corporation, Tokyo, Japan) as previously described (Ai et al. 2008). Plasma CRP levels were measured using a high-sensitivity immunoturbidimetric assay (Roche Diagnostics, Indianapolis, IN).
LDL was isolated from pooled donor plasma by rapid single-step ultracentrifugation using a Beckman NVT90 rotor as described previously (Vieira et al. 1996). After desalting, LDL was oxidized by the addition of 100 μM CuSO4/100 μg protein. Oxidation extent was monitored by the formation of conjugated dienes at 234 nm. When absorbance began to increase exponentially (≈1.5 hours), LDL was placed on ice and immediately desalted using chromatography columns (BioRad Laboratories, Hercules, CA) to stop further oxidation. This typically produced a thiobarbituric acid-reactive substance (TBARS) value of 6-8 nM malondialdehyde/μg protein. TBARS measurements were performed as described previously (Cathcart et al. 1991). The moderately oxidized LDL (oxLDL) was stored at -80°C in the dark for up to 2 months, as TBARS values and 234 nm readings were found to remain stable for this duration. The same batch of oxLDL was used for all experiments.
Isolation and Culture of HMDMs
Blood was drawn in tubes containing 0.1% EDTA and centrifuged at 1000 rpm for 30 minutes (25°C) to remove plasma. Buffy coats were obtained by layering blood diluted 1:2 with RPMI culture media over Ficoll-Paque followed by centrifugation (37 min, 1600 rpm, 25°C). White blood cells were collected and washed twice in RPMI. Cells were plated in RPMI containing 100 U/mL penicillin and 100 μg/mL streptomycin. After 3-5 hours of incubation at 37°C in 5% CO2, nonadherent cells were washed off and the remaining monocytes were cultured in RPMI containing 10% autologous serum. Cells were allowed to differentiate for 10 days in the presence of vehicle (ethanol), E (2 nM or 20 nM), or T (10 nM or 100 nM), with medium changes every 3-4 days. E and T solutions were prepared fresh under sterile conditions every 2 weeks by dissolving in 100% ethanol, and stored in the dark at -80°C. HMDMs were treated with 50 μg/mL oxLDL for 48 hours (days 11-12) in the presence of 10% autologous serum and hormone treatment, and then exposed to hormones + medium without serum for 24 additional hours. The serum-free medium was collected, centrifuged to remove cell debris, and stored at -80°C. Cells were collected in lysis buffer (0.1 M KH2PO4, 0.05 M NaCl, 5 mM cholic acid, and 0.1% Triton®X-100) and cell protein was quantified using the bicinchoninic acid method with bovine serum albumin as a standard.
Real Time-PCR
Total cellular RNA was isolated using the RNeasy mini kit according to the manufacturer’s instructions. 100 nanograms of RNA was reverse transcribed using the Superscript III reverse transcription kit and amplified on a real-time PCR system 7300 using specific primers. The primer sequences were as follows: β-actin (F’ TGAAGTGTGACGTGGACATCC, R’ CTCAGGAGGAGCAATGATCTTG), TNF-α (F’ TGGAGAAGGGTGACCGACTC, R’ TCCTCACAGGGCAATGATCC), IL-6 (F’ GTGGCTGCAGGACATGACAA, R’ TGAGGTGCCCATGCTACATTT), IL-1β (F’ TTATTACAGTGGCAATGAGGATGAC, R’ CGCCATCCAGAGGGCAG), CRP (F’ ATTCAGGCCCTTGTATCACTGG, R’ AACAGCTTCTCCATGGTCACG), androgen receptor (AR) (F’ GACTCCGTGCAGCCTATTGC, R’ TCTGCCATCATTTCCGGAA), estrogen receptor α (ERα) (F’ CGGCATTCTACAGGCCAAA, R’ GCGAGTCTCCTTGGCAGATTC), and ERβ (F’ TACAATCGATAAAAACCGGCG, R’ GGGAGCCACACTTCACCATT). Primers were designed using Primer Express software, included intron/exon boundaries, and were validated for efficiency and specificity by standard curve dilution and melting point analysis. Real Time-PCR using power SYBR-green master mix was carried out for 40 cycles of 95°C (15 sec) + 60°C (1 min). Changes in gene expression were assessed by ΔΔCt analysis with β-actin as the control/housekeeping gene. Changes were expressed as a percent of control (vehicle only).
TNF-α ELISA Assay
TNF-α concentration in cell culture medium was measured using an ultrasensitive human TNF-α ELISA kit, according to the manufacturer’s instructions (Alpco Diagnostics, Salem NH). Final TNF-α concentrations were adjusted for cell protein.
Statistical Analysis
Statistical analyses were performed using SAS software (version 9.1), while correlations were determined using graph pad prism software (version 4). Gene expression results are expressed as a percentage of control (vehicle treatment only). Means and SD are representative of the treatment response in macrophage cultures donated from 10 females or 10 males. Statistical differences were determined by 2-way ANOVA for both treatment effect and sex effect using Tukey’s Student Range test.
Results
Both hormone receptors AR and ERα were expressed in HMDM (average Ct of 33 and 32, respectively, on real time PCR, compared to average Ct of 24 for β-actin). There was no difference in AR expression between female and male HMDM, while the expression of ERα was 3-fold higher in female than in male HMDM. The expression of ERβ was too low to accurately quantify (Ct > 36).
E treatment did not significantly affect the expression of TNF-α, IL-6, and IL-1β in HMDM (Figure 1). Relative to control, both physiological and pharmacological concentrations of T significantly reduced TNF-α expression in HMDM from both males and females (Figure 1). These reductions averaged 20-25% at 10 nM and 25-30% at 100 nM (Figure 1). Medium TNF-α concentration was measured in vehicle, E2 nM, and T10 nM treated cells (Figure 2). Similar to the effect on gene expression, T treatment, but not E treatment, significantly reduced HMDM TNF-α secretion, compared to control. There was no statistical difference between genders. T treatment significantly reduced IL-1β expression at 10 nM in males and at 100 nM in females, but did not affect IL-6 expression (Figure 1).
Figure 1. Effect of E or T treatment on proinflammatory cytokine expression in HMDM.
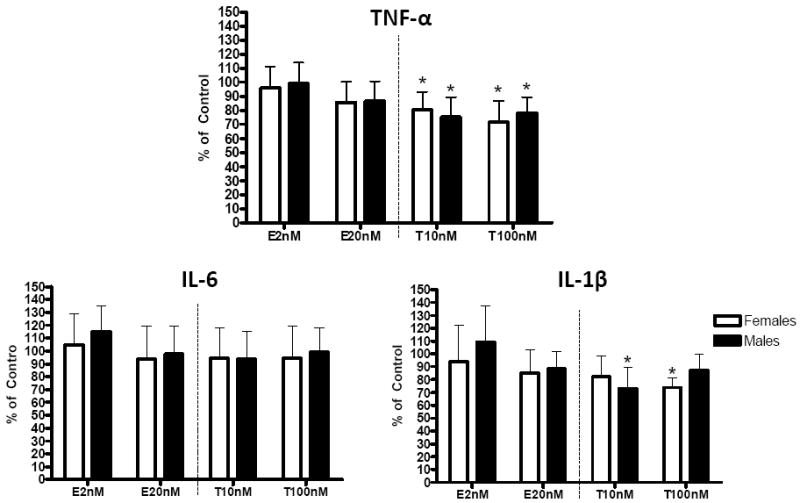
HMDMs were cultured in 10% autologous serum and differentiated over 10 days in the presence of vehicle (control), E 2 nM, E 20 nM, T 10 nM, or T 100 nM. HMDMs were then exposed for 48 hours to oxLDL with hormone treatment. RNA was isolated, and the expression of proinflammatory cytokines was measured. Data are expressed as % of control; * p<0.05.
Figure 2. Effect of E or T treatment on TNF-α secretion by HMDM.

As described in Figure 1, HMDMs were differentiated for 10 days in 10% autologous serum and corresponding treatment, and then exposed to oxLDL for 48 hours, followed by exposure to medium without serum for 24 hours. Indicated treatment was present for the entire time. Medium was collected and TNF-α concentration was measured and adjusted for cell protein. Data are expressed as % of control; * p<0.05.
The effect of E on CRP expression was variable in both women and men (Figure 3). The change in CRP expression by HMDM in response to E, but not T treatment was positively associated with the concentration of sdLDL-C in the subject’s plasma (Figure 4a and 4b). Donors with plasma sdLDL-C >30 mg/dL showed a significant increase in HMDM CRP expression with estrogen treatment whereas there was no effect in HMDM from donors with sdLDL-C < 30 mg/dL (Figure 4c). This effect was gender independent. The change in CRP expression with E was not associated with other parameters (age, BMI, other plasma lipid or CRP levels).
Figure 3. Effect of E or T treatment on HMDM CRP expression.

HMDMs were cultured as described in Figure 1 and the expression of CRP was measured. Data are expressed as % of control.
Figure 4. Association between volunteer fasting plasma sdLDL-C concentration and HMDM CRP gene expression response to E.
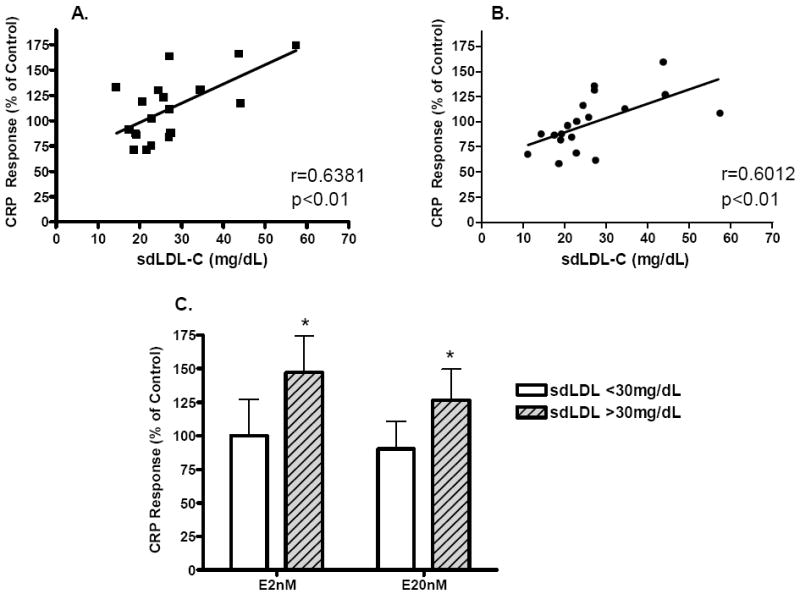
A: Association between the change in CRP expression during E 2 nM treatment, relative to control (vehicle), and the donor’s sdLDL-C levels. B: Association between the change in CRP expression during E 20 nM treatment, relative to control (vehicle), and the donor’s sdLDL-C levels Both correlations were significant (p<0.01) and were gender independent. C: Effect of E on CRP expression as a function of volunteer plasma sdLDL-C level. The number of subjects with sdLDL-C < 30 mg/dL is 12, and the number of subjects with sdLDL-C >30 mg/dL is 8. Data are expressed as % of control; * p<0.05.
Discussion
The effect of sex hormones on macrophage inflammation is an important area of investigation because of the contribution of inflammation to CHD. The hormone receptors AR and ER, which are expressed in both male and female HMDM cells, are the likely mediators of the effects of sex hormones on macrophage function (Cutolo, Accardo et al, 1996). Low serum T levels in men have been associated with increased atheroma formation (Hak et al. 2002). In addition, hypogonadal men have been found to have higher serum cytokine levels compared with healthy men, and androgen supplementation was found to reduce these levels (Yesilova et al. 2000). T treatment has been shown to suppress cytokine expression in rodent macrophages and in human monocytes from younger individuals (Chao, Van Alten et al. 1995; Kanda, Tsuchida et al. 1996; Kanda, Tsuchida et al. 1997; D’Agostino, Milano et al. 1999). Therefore we hypothesized similar changes would occur in HMDM from older individuals. In cells exposed to moderately oxidized LDL, T significantly reduced proinflammatory cytokine expression, specifically TNF-α and IL-1β, in agreement with these previous studies, (Chao, Van Alten et al. 1995; Kanda, Tsuchida et al. 1996; Kanda, Tsuchida et al. 1997; D’Agostino, Milano et al. 1999). The effects of T on TNF-α and IL-1β were observed at physiological and supraphysiological concentrations. Macrophage cells express the enzyme aromatase and are therefore able to convert T to E, (Mor et al. 2001). Since reductions in TNF-α and IL-1β were only observed with T, it is reasonable to assume that the effect seen with T treatment was not due to aromatization to E but to an androgen specific action of T.
NF-κB, a transcription factor that plays a direct role in the expression of numerous proinflammatory cytokines including TNF-α and IL-1β (Li & Verma 2002), may be the mediator of the repression of cytokine expression by T. Several studies have shown that androgen receptor activation suppresses NF-κB activity (Hatakeyama et al. 2002; Libby 2002; Itoh et al. 2007), presumably by increasing the expression of IkB-α (Death et al. 2004). NF-κB inhibition by T is also associated with enhanced macrophage apoptosis (Cutolo et al. 2005) suggesting that the reduction in TNF-α expression is accompanied by an immunosuppressive effect.
The effect of E on cytokine expression is less clear. Short-term exposure to E (<1 hour) in vitro has been reported to decrease the production of proinflammatory cytokines in LPS-activated rodent macrophages (Ghisletti et al. 2005). In contrast, several studies have reported that long-term exposure to E in vivo enhanced the proinflammatory cytokine production from LPS-activated rodent macrophages (Soucy et al. 2005; Calippe et al. 2008). In our study, in which HMDM obtained from 50-70 year old male and postmenopausal female donors were cultured in the continual presence of E, we observed no affect on proinflammatory cytokine expression compared with vehicle-treated cells.
Randomized, placebo-controlled trials in postmenopausal women have shown that oral, but not transdermally-delivered, estrogen therapy increases plasma CRP concentrations (Cushman et al. 1999; Hodis et al. 2008). It has been speculated that hepatic metabolism of the orally delivered therapy is responsible for this rise in plasma CRP levels (Zegura, Keber et al. 2003). However, since CRP is also produced by macrophages in the aortic lesions, plasma CRP level changes in response to E treatment may not predict CRP changes in the arterial wall. Because CRP has been shown to elicit proatherogenic effects such as promoting macrophage cholesterol accumulation (Singh, Dasu et al. 2008), CRP secreted by macrophages may play a role in lesion development. The CRP expression in response to E treatment was quite variable in both female and male HMDM donors. It has been previously shown that the lipoprotein composition of culture serum may influence the cellular response (de la Llera Moya et al. 1994). Since HMDM were cultured in 10% autologous serum, we tested the hypothesis that changes in CRP expression by E may be dependent on the donor plasma levels of lipoproteins. The CRP gene response to E was significantly correlated with plasma sdLDL-C levels, with a greater increase in CRP expression in subjects with higher sdLDL-C levels. Individuals with a high concentration of sdLDL-C often display some degree of dyslipidemia and a greater degree of chronic inflammation and CHD (Vakkilainen et al. 2003; Krauss & Siri 2004). This would indicate that, in a proatherogenic lipid environment, E may promote arterial disease, an effect not present under a healthy lipid environment. This concept is supported by clinical evidence suggesting E therapy is beneficial in younger premenopausal women but harmful in older postmenopausal women (Rossouw et al. 2007). In support of our findings, a recent study by Norata et al. (Norata et al. 2009) has shown a greater expression of several proinflammatory molecules in HMDMs obtained from individuals with high sdLDL-C levels compared with individuals with lower sdLDL-C levels. Furthermore, in postmenopausal women, plasma sdLDL-C levels have been found to be significantly associated with plasma CRP concentration further indicating the link between sdLDL-C and inflammation (Muzzio et al. 2007). We did not observe a significant correlation between these two factors in our study (data not shown). However, this may due to the small sample size.
To our knowledge, our study is the first to report a modulation of CRP expression by E in HMDM. The mechanism by which high sdLDL-C levels may alter the effect of E on CRP expression is not currently known. IL-6 and IL-1β are known stimulators of CRP expression (Calabro et al. 2003). In the current study, and in the study by Norata et al. (Norata, Raselli et al. 2009), sdLDL-C concentrations were not associated with HMDM IL-6 expression. Similarly, the expression of IL-6 and IL-1β did not change with E treatment. Therefore, the observed up-regulation of macrophage CRP expression by E is not being driven by greater macrophage IL-6 or IL-1β expression. An IL-6-independent stimulation of CRP by HRT has also been suggested by observational and intervention studies (Lakoski & Herrington 2005). The expression of STAT3, a transcription factor regulating the expression of CRP, has been shown to be increased by estrogen in the ob/ob mouse model of obesity, possibly through ERα binding to the promoter region of STAT3 (Gao et al. 2006). How sdLDL may modulate the effect of E on CRP expression is currently not known. sdLDL are known to be in circulation longer than LDL and therefore are more likely to undergo oxidation (Millar & Packard, 1998).
Overall, these data suggest that T may protect against the progression of atherosclerosis by inhibiting the expression of select proinflammatory cytokines in human macrophage cells, while E may not be as potent in this regard. Furthermore, E may actually encourage macrophage CRP production under conditions of high sdLDL-C, thereby potentially exacerbating atherosclerosis in individuals at risk for the disease. Understanding the mechanism of sdLDL-C induced macrophage inflammation and how E may modulate this is an important step in defining the role of both sdLDL-C and E in the progression of CHD.
Acknowledgments
The authors would like to thank Katalin V. Horvath for measuring the volunteer plasma lipid and CRP concentrations.
Funding
This work was supported by the United States Department of Agriculture, under agreement No. 58-1950-40401 and T32 HL69772-01A1 (MPC). Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the authors, and do not necessarily reflect the view of the USDA. Michael P Corcoran was supported by the Unilever Health Science Scholarship.
Footnotes
Declaration of Interest
The authors declare no conflict of interest.
References
- Ai M, Otokozawa S, Asztalos BF, Nakajima K, Stein E, Jones PH, Schaefer EJ. Effects of maximal doses of atorvastatin versus rosuvastatin on small dense low-density lipoprotein cholesterol levels. Am J Cardiol. 2008;101:315–8. doi: 10.1016/j.amjcard.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Bisoendial RJ, Kastelein JJ, Peters SL, Levels JH, Birjmohun R, Rotmans JI, Hartman D, Meijers JC, Levi M, Stroes ES. Effects of CRP infusion on endothelial function and coagulation in normocholesterolemic and hypercholesterolemic subjects. J Lipid Res. 2007;48:952–60. doi: 10.1194/jlr.P600014-JLR200. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. 1983;52:223–61. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- Calabro P, Willerson JT, Yeh ET. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation. 2003;108:1930–2. doi: 10.1161/01.CIR.0000096055.62724.C5. [DOI] [PubMed] [Google Scholar]
- Calippe B, Douin-Echinard V, Laffargue M, Laurell H, Rana-Poussine V, Pipy B, Guery JC, Bayard F, Arnal JF, Gourdy P. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: involvement of the phosphatidylinositol 3-kinase pathway. J Immunol. 2008;180:7980–8. doi: 10.4049/jimmunol.180.12.7980. [DOI] [PubMed] [Google Scholar]
- Cathcart MK, McNally AK, Chisolm GM. Lipoxygenase-mediated transformation of human low density lipoprotein to an oxidized and cytotoxic complex. J Lipid Res. 1991;32:63–70. [PubMed] [Google Scholar]
- Chao TC, Van Alten PJ, Greager JA, Walter RJ. Steroid sex hormones regulate the release of tumor necrosis factor by macrophages. Cell Immunol. 1995;160:43–9. doi: 10.1016/0008-8749(95)80007-6. [DOI] [PubMed] [Google Scholar]
- Cushman M, Legault C, Barrett-Connor E, Stefanick ML, Kessler C, Judd HL, Sakkinen PA, Tracy RP. Effect of postmenopausal hormones on inflammation-sensitive proteins: the Postmenopausal Estrogen/Progestin Interventions (PEPI) Study. Circulation. 1999;100:717–22. doi: 10.1161/01.cir.100.7.717. [DOI] [PubMed] [Google Scholar]
- Cutolo M, Capellino S, Montagna P, Ghiorzo P, Sulli A, Villaggio B. Sex hormone modulation of cell growth and apoptosis of the human monocytic/macrophage cell line. Arthritis Res Ther. 2005;7:R1124–32. doi: 10.1186/ar1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutolo M, Accardo S, Villaggio B, Barone A, Sulli A, Coviello DA, Carabbio C, Felli L, Miceli D, Farruggio R, Carruba G, Castagnetta L. Androgen and estrogen receptors are present in primarycultures of human synovial macrophages. J Clin Endocrinol Metab. 1996;81:820–827. doi: 10.1210/jcem.81.2.8636310. [DOI] [PubMed] [Google Scholar]
- D’Agostino P, Milano S, Barbera C, Di Bella G, La Rosa M, Ferlazzo V, Farruggio R, Miceli DM, Miele M, Castagnetta L, et al. Sex hormones modulate inflammatory mediators produced by macrophages. Ann N Y Acad Sci. 1999;876:426–9. doi: 10.1111/j.1749-6632.1999.tb07667.x. [DOI] [PubMed] [Google Scholar]
- de la Llera Moya M, Atger V, Paul JL, Fournier N, Moatti N, Giral P, Friday KE, Rothblat G. A cell culture system for screening human serum for ability to promote cellular cholesterol efflux. Relations between serum components and efflux, esterification, and transfer. Arterioscler Thromb. 1994;14:1056–65. doi: 10.1161/01.atv.14.7.1056. [DOI] [PubMed] [Google Scholar]
- Death AK, McGrath KC, Sader MA, Nakhla S, Jessup W, Handelsman DJ, Celermajer DS. Dihydrotestosterone promotes vascular cell adhesion molecule-1 expression in male human endothelial cells via a nuclear factor-kappaB-dependent pathway. Endocrinology. 2004;145:1889–97. doi: 10.1210/en.2003-0789. [DOI] [PubMed] [Google Scholar]
- Gao H, Bryzgalova G, Hedman E, Khan A, Efendic S, Gustafsson JA, Dahlman-Wright K. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol. 2006;20:1287–99. doi: 10.1210/me.2006-0012. [DOI] [PubMed] [Google Scholar]
- Ghisletti S, Meda C, Maggi A, Vegeto E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol Cell Biol. 2005;25:2957–68. doi: 10.1128/MCB.25.8.2957-2968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hak AE, Witteman JC, de Jong FH, Geerlings MI, Hofman A, Pols HA. Low levels of endogenous androgens increase the risk of atherosclerosis in elderly men: the Rotterdam study. J Clin Endocrinol Metab. 2002;87:3632–9. doi: 10.1210/jcem.87.8.8762. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H, Nishizawa M, Nakagawa A, Nakano S, Kigoshi T, Uchida K. Testosterone inhibits tumor necrosis factor-alpha-induced vascular cell adhesion molecule-1 expression in human aortic endothelial cells. FEBS Lett. 2002;530:129–32. doi: 10.1016/s0014-5793(02)03440-3. [DOI] [PubMed] [Google Scholar]
- Herrington DM, Howard TD, Hawkins GA, Reboussin DM, Xu J, Zheng SL, Brosnihan KB, Meyers DA, Bleecker ER. Estrogen-receptor polymorphisms and effects of estrogen replacement on high-density lipoprotein cholesterol in women with coronary disease. N Engl J Med. 2002;346:967–74. doi: 10.1056/NEJMoa012952. [DOI] [PubMed] [Google Scholar]
- Hodis HN, St John JA, Xiang M, Cushman M, Lobo RA, Mack WJ. Inflammatory markers and progression of subclinical atherosclerosis in healthy postmenopausal women (from the Estrogen in the Prevention of Atherosclerosis Trial) Am J Cardiol. 2008;101:1131–3. doi: 10.1016/j.amjcard.2007.09.120. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Hayashi H, Xu J, Takii T, Miyazawa K, Ariga H, Akahoshi T, Waguri-Nagaya Y, Otsuka T, Okamoto T, et al. Dihydrotestosterone inhibits tumor necrosis factor alpha induced interleukin-1alpha mRNA expression in rheumatoid fibroblast-like synovial cells. Biol Pharm Bull. 2007;30:1140–3. doi: 10.1248/bpb.30.1140. [DOI] [PubMed] [Google Scholar]
- Kanda N, Tsuchida T, Tamaki K. Testosterone inhibits immunoglobulin production by human peripheral blood mononuclear cells. Clin Exp Immunol. 1996;106:410–5. doi: 10.1046/j.1365-2249.1996.d01-842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda N, Tsuchida T, Tamaki K. Testosterone suppresses anti-DNA antibody production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1997;40:1703–11. doi: 10.1002/art.1780400921. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Inoue N, Ohashi Y, Terashima M, Matsui K, Mori T, Fujita H, Awano K, Kobayashi K, Azumi H, et al. Interaction of oxidative stress and inflammatory response in coronary plaque instability: important role of C-reactive protein. Arterioscler Thromb Vasc Biol. 2003;23:1398–404. doi: 10.1161/01.ATV.0000081637.36475.BC. [DOI] [PubMed] [Google Scholar]
- Kramer PR, Kramer SF, Guan G. 17 beta-estradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum. 2004;50:1967–75. doi: 10.1002/art.20309. [DOI] [PubMed] [Google Scholar]
- Krauss RM, Siri PW. Metabolic abnormalities: triglyceride and low-density lipoprotein. Endocrinol Metab Clin North Am. 2004;33:405–15. doi: 10.1016/j.ecl.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Lakoski SG, Herrington DM. Effects of hormone therapy on C-reactive protein and IL-6 in postmenopausal women: a review article. Climacteric. 2005;8:317–26. doi: 10.1080/13697130500345109. [DOI] [PubMed] [Google Scholar]
- Li L, Roumeliotis N, Sawamura T, Renier G. C-reactive protein enhances LOX-1 expression in human aortic endothelial cells: relevance of LOX-1 to C-reactive protein-induced endothelial dysfunction. Circ Res. 2004;95:877–83. doi: 10.1161/01.RES.0000147309.54227.42. [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, et al. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–6. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- Millar JS, Packard CJ. Heterogeneity of apolipoprotein B-100-containing lipoprotein: what we have learnt from kinetic studies. Curr Opin Lipidol. 1998;9:197–202. doi: 10.1097/00041433-199806000-00003. [DOI] [PubMed] [Google Scholar]
- Mor G, Eliza M, Song J, Wiita B, Chen S, Naftolin F. 17alpha-methyl testosterone is a competitive inhibitor of aromatase activity in Jar choriocarcinoma cells and macrophage-like THP-1 cells in culture. J Steroid Biochem Mol Biol. 2001;79:239–46. doi: 10.1016/s0960-0760(01)00162-5. [DOI] [PubMed] [Google Scholar]
- Muzzio ML, Berg G, Zago V, Basilio F, Sanguinetti S, Lopez G, Brites F, Wikinski R, Schreier L. Circulating small dense LDL, endothelial injuring factors and fibronectin in healthy postmenopausal women. Clin Chim Acta. 2007;381:157–63. doi: 10.1016/j.cca.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Norata GD, Raselli S, Grigore L, Garlaschelli K, Vianello D, Bertocco S, Zambon A, Catapano AL. Small dense LDL and VLDL predict common carotid artery IMT and elicit an inflammatory response in peripheral blood mononuclear and endothelial cells. Atherosclerosis. 2009 doi: 10.1016/j.atherosclerosis.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Pradhan AD, Manson JE, Rossouw JE, Siscovick DS, Mouton CP, Rifai N, Wallace RB, Jackson RD, Pettinger MB, Ridker PM. Inflammatory biomarkers, hormone replacement therapy, and incident coronary heart disease: prospective analysis from the Women’s Health Initiative observational study. Jama. 2002;288:980–7. doi: 10.1001/jama.288.8.980. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, Ko M, LaCroix AZ, Margolis KL, Stefanick ML. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. 2007;297:1465–77. doi: 10.1001/jama.297.13.1465. [DOI] [PubMed] [Google Scholar]
- Singh U, Dasu MR, Yancey PG, Afify A, Devaraj S, Jialal I. Human C-reactive protein promotes oxidized low density lipoprotein uptake and matrix metalloproteinase-9 release in Wistar rats. J Lipid Res. 2008;49:1015–23. doi: 10.1194/jlr.M700535-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy G, Boivin G, Labrie F, Rivest S. Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J Immunol. 2005;174:6391–8. doi: 10.4049/jimmunol.174.10.6391. [DOI] [PubMed] [Google Scholar]
- Tipping PG, Hancock WW. Production of tumor necrosis factor and interleukin-1 by macrophages from human atheromatous plaques. Am J Pathol. 1993;142:1721–8. [PMC free article] [PubMed] [Google Scholar]
- Torres JL, Ridker PM. Clinical use of high sensitivity C-reactive protein for the prediction of adverse cardiovascular events. Curr Opin Cardiol. 2003;18:471–8. doi: 10.1097/00001573-200311000-00008. [DOI] [PubMed] [Google Scholar]
- Vakkilainen J, Steiner G, Ansquer JC, Aubin F, Rattier S, Foucher C, Hamsten A, Taskinen MR. Relationships between low-density lipoprotein particle size, plasma lipoproteins, and progression of coronary artery disease: the Diabetes Atherosclerosis Intervention Study (DAIS) Circulation. 2003;107:1733–7. doi: 10.1161/01.CIR.0000057982.50167.6E. [DOI] [PubMed] [Google Scholar]
- Valente AJ, Rozek MM, Sprague EA, Schwartz CJ. Mechanisms in intimal monocyte-macrophage recruitment. A special role for monocyte chemotactic protein-1. Circulation. 1992;86:III20–5. [PubMed] [Google Scholar]
- Vieira OV, Laranjinha JA, Madeira VM, Almeida LM. Rapid isolation of low density lipoproteins in a concentrated fraction free from water-soluble plasma antioxidants. J Lipid Res. 1996;37:2715–21. [PubMed] [Google Scholar]
- Williams TN, Zhang CX, Game BA, He LHuang Y. C-reactive protein stimulates MMP-1 expression in U937 histiocytes through Fc[gamma]RII and extracellular signal-regulated kinase pathway:: an implication of CRP involvement in plaque destabilization. Arterioscler Thromb Vasc Biol. 2004;24:61–6. doi: 10.1161/01.ATV.0000104014.24367.16. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL. Generation of C-reactive protein and complement components in atherosclerotic plaques. Am J Pathol. 2001;158:1039–51. doi: 10.1016/S0002-9440(10)64051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesilova Z, Ozata M, Kocar IH, Turan M, Pekel A, Sengul A, Ozdemir IC. The effects of gonadotropin treatment on the immunological features of male patients with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2000;85:66–70. doi: 10.1210/jcem.85.1.6226. [DOI] [PubMed] [Google Scholar]
- Zegura B, Keber I, Sebestjen M, Koenig W. Double blind, randomized study of estradiol replacement therapy on markers of inflammation, coagulation and fibrinolysis. Atherosclerosis. 2003;168:123–9. doi: 10.1016/s0021-9150(03)00088-1. [DOI] [PubMed] [Google Scholar]