

Abstract
Genetic resistance to disease incited by necrotrophic pathogens is not well understood in plants. Whereas resistance is often quantitative, there is limited information on the genes that underpin quantitative variation in disease resistance. We used a population genomic approach to identify genes in loblolly pine (Pinus taeda) that are associated with resistance to pitch canker, a disease incited by the necrotrophic pathogen Fusarium circinatum. A set of 498 largely unrelated, clonally propagated genotypes were inoculated with F. circinatum microconidia and lesion length, a measure of disease resistance, data were collected 4, 8, and 12 weeks after inoculation. Best linear unbiased prediction was used to adjust for imbalance in number of observations and to identify highly susceptible and highly resistant genotypes (“tails”). The tails were reinoculated to validate the results of the full population screen. Significant associations were detected in 10 single nucleotide polymorphisms (SNPs) (out of 3938 tested). As hypothesized for genes involved in quantitative resistance, the 10 SNPs had small effects and proposed roles in basal resistance, direct defense, and signal transduction. We also discovered associated genes with unknown function, which would have remained undetected in a candidate gene approach constrained by annotation for disease resistance or stress response.
GENETIC interactions between host and pathogen populations result in abundant natural variation in the genes involved in host disease resistance. Most of the studies leading to identification and cloning of disease resistance genes are focused on major gene disease resistance (Johal and Briggs 1992; Dangl and Jones 2001; Jones and Dangl 2006). In cases where resistance is associated with single genes, genetic effects are large in magnitude and detection is straightforward. In contrast, quantitative disease resistance is typically conditioned by many genes with relatively small effects. Quantitative resistance is generally considered to be more durable but also more difficult to investigate relative to major gene resistance, since the effects of individual genes are small and phenotyping experiments must be performed with high levels of precision. As a consequence, the genes and mechanisms of quantitative disease resistance are poorly understood, in part due to the smaller effect of individual genes on the resistance phenotype. Interactions between plants and necrotrophic pathogens often exhibit quantitative resistance (Balint-Kurti et al. 2008; Poland et al. 2009).
Pitch canker disease of loblolly pine and other pine species is incited by the necrotrophic pathogen Fusarium circinatum and is manifest as resinous lesions in stems and branches (Dwinell et al. 1985; Enebak and Stanosz 2003; Carey et al. 2005; Sakamoto and Gordon 2006). There is evidence for heritable resistance to pitch canker in loblolly pine (Kayihan et al. 2005) as well as other pine species (Hodge and Dvorak 2000, 2007). In this article we report the first population-wide phenotypic screen of a clonally propagated population of loblolly pine for association testing (Eckert et al. 2010). Clonal propagation of this population enabled precise phenotyping, which was required to obtain the resolution needed to identify candidates for quantitative disease resistance loci.
Pine species in general exhibit high levels of nucleotide variation and low linkage disequilibrium (LD) (Brown et al. 2004). An association genetic approach relies on the premise that historical, unrecorded recombination events over many generations have reduced LD between markers and quantitative trait loci such that only those marker-trait pairs that are tightly linked remain detectable; this may enable “fine mapping” to identify genes underlying quantitative variation (Flint-Garcia et al. 2003; Neale and Savolainen 2004). Association-based approaches have been used to identify candidate genes underlying traits in plants (Zhao et al. 2007; Stich et al. 2008; Wang et al. 2008; Yahiaoui et al. 2008; Inostroza et al. 2009; Stracke et al. 2009), based in part on applications in humans (D'alfonso et al. 2002; McGuffin et al. 2003; Easton et al. 2007; Lee et al. 2007), livestock (Martinez et al. 2006; Charlier et al. 2008; Goddard and Hayes 2009), and Drosophila (Kennington et al. 2007; Norry et al. 2007; Jiang et al. 2009). Recent association studies in tree species have evaluated single candidate genes or a modest number of candidate genes for association (Thumma et al. 2005; Gonzalez-Martinez et al. 2007, 2008; Ingvarsson et al. 2008; Eckert et al. 2009a). Association mapping has been used to identify disease resistance genes in several crop species including sugarcane, maize, barley, and potato (Flint-Garcia et al. 2005; Wei et al. 2006; Yu and Buckler 2006; Malosetti et al. 2007; Stich et al. 2008; Inostroza et al. 2009; Murray et al. 2009). The population analyzed in this study was genotyped at 3938 SNP loci that were selected without regard to the functional annotation of ESTs from which they were derived. Thus, we reasoned that the status of any particular marker as a candidate disease resistance gene would be determined by association testing, as opposed to previous studies in which markers were typically evaluated on the basis of their presumed roles in disease resistance in other species.
Several different, but not mutually exclusive hypotheses have been proposed regarding the genetic origins of quantitative resistance (Poland et al. 2009), providing a useful framework for understanding evolution of resistance to necrotrophic pathogens. These six hypotheses proposed by Poland et al. (2009) predict that quantitative disease resistance is conditioned by: (1) genes regulating morphological and developmental phenotypes; (2) mutations in genes involved in basal defense causing small, incremental levels of resistance; (3) components of chemical warfare, through the action of genes producing antibiotic or antifungal compounds; (4) genes involved in defense signal transduction pathways; (5) weak forms of defeated R genes; and/or (6) genes not yet known to be involved in disease resistance.
In this study, our main objective was to evaluate the genetic architecture of pitch canker disease resistance: to quantify the extent to which genes contribute to variation in the disease phenotype, to evaluate the hypothesis that disease resistance was quantitative, and to identify candidate genes for resistance as well as quantify their magnitude of effect. In the process of identifying candidate genes for resistance we were also able to evaluate support for hypotheses recently put forth by Poland et al. (2009) regarding the biological roles and origins of quantitative resistance genes.
MATERIALS AND METHODS
Plant material:
Loblolly pine (Pinus taeda L.) material was propagated by juvenile stem cuttings (Lebude et al. 2004), at the North Carolina State University (NCSU) Horticultural Field Laboratory, Raleigh, NC, obtained from repeatedly hedged stock plants representing a sample of 498 genotypes collected as wild selections of the NCSU Cooperative Tree Improvement Program (NCTIP), supplemented by a few unrelated genotypes from controlled crosses from the NCTIP and the Western Gulf Forest Tree Improvement Program (Figure 1). Depending on propagation efficiency and availability, one to four cuttings of each clone were transferred to the greenhouse facilities at the University of Florida, Gainesville, FL. The plants were placed on Ebb-n-Flow benches and subirrigated twice daily with a Peter's Professional Fertilizer (10-20-10; adjusted to 2 mm ammonium nitrate) supplemented with iron (Sequestrene; adjusted to 0.037 mm elemental iron). The cuttings were then hedged to stimulate flushing and were placed in the experimental design.
Figure 1.—

Geographical distribution of loblolly pine accessions sampled for this study. Size of the dots denotes the number of accessions collected in a particular county, as follows: •, 1–5; •, 6–10;  , 11–15; ○, 16+ accessions. Bar, 200 km.
, 11–15; ○, 16+ accessions. Bar, 200 km.
Experimental designs:
Two inoculation experiments were performed. An initial inoculation experiment consisted of up to four replicates of the entire population of 498 genotypes, placed in a randomized incomplete block design with 21 rows and 22 columns per replicate. A subset of genotypes from this first experiment was selected on the basis of response to pathogen challenge. This subset was composed of the 50 most susceptible and the 50 most resistant genotypes. The selected plants were hedged and transferred to 1-gallon pots and placed in a 9 × 9 partially balanced lattice design. Shoots selected for inoculation in the second experiment were individually identified for repeated measures.
Fungal inoculum:
F. circinatum strain S45 was cultured in PDA (potato dextrose agar) medium for 10–15 days, as described by Young et al. (2006). Microconidia, representing clonally derived spores of a single genetic isolate, were then harvested by flooding the culture plates with 5 ml of sterile distilled water and collecting the spore suspension with a pipette onto a glass beaker. The concentration of microconidia was estimated using a hemacytometer and dilutions were made until a final concentration of 500 spores/μl (Young et al. 2006) was obtained.
Phenotyping:
One to five shoots per plant were selected for inoculation and the tips were cut off to allow fungal penetration. Plants were sprayed with the spore solution described above using a manual pressure spray pump. For reinoculation experiments, selected genotypes from the resistant and susceptible tails were transferred to 1-gallon pots and hedged twice to induce multiple shoot growth. Plants were wounded at the shoot tip and 2 μl of inoculum (500 spores/μl) were manually placed on the wound with a pipettor. After inoculation, all plants were placed overnight in a humid chamber, constructed by sealing flood benches in clear plastic sheeting material. The following morning, the plastic was removed.
Lesion length measurements were taken at 4, 8, and 12 weeks after inoculation, using a digital caliper. Lesion lengths were recorded, in millimeters, from the shoot tip (wound site) to the lowest point where necrosis was observed. To keep data collection consistent, lesion lengths of a given block were recorded by the same person in all three measurements.
Estimates of clonal values were obtained using best linear unbiased predictions (BLUP) in ASReml (Gilmour et al. 2006) with the following model for the initial inoculation experiment:
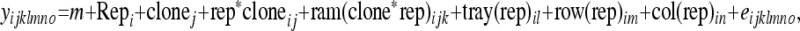 |
where:
yijklmno is the oth lesion log length observation for the kth ramet within the jth clone in the mth row and the nth column of the lth tray within the ith replicate for each time point.
μ is an overall mean.
repi is the fixed effect replication i = 1–4.
clonej is the random variable clone
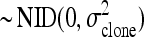 .
.rep*cloneij is the random variable rep by clone
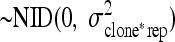 .
.ram(clone*rep)ijk is the random variable ramet within clone by rep
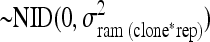 .
.tray(rep)il is the random variable tray within replicate
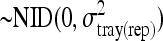 .
.row(rep)im is the random variable row within replicate
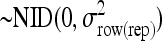 .
.col(rep)in is the random variable column within replicate
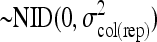 .
.eijklmo is the random variable error within the experiment
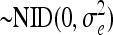 .
.The random variables rep*cloneij, tray(rep)il, and col(rep)in were later excluded from the model because of zero variance.
The genotypes were ranked according to their clonal BLUP estimates and the 50 most susceptible and resistant genotypes (tails) were selected. For the second inoculation experiment the BLUPs were obtained using the following model, with the same variables as described above:
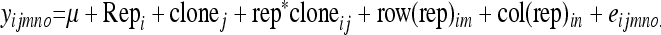 |
Clonal repeatability was estimated using the following formula:
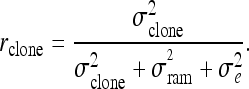 |
For the reinoculation experiment,  was omitted from the above formula.
was omitted from the above formula.
The variances of clone, ramet and residual effects were used to estimate the phenotypic standard deviation, as the square root of the sum of these three values.
Genotyping:
Genotyping of SNPs was performed using the Illumina Infinium assay (Illumina, San Diego, CA). Similar, yet lower throughput platforms have been shown to work well within the large and complex genome of conifers (Eckert et al. 2009b). The discovery, selection, and genotyping of these SNPs are described in Eckert et al. (2010). In brief, SNPs were detected in a discovery panel of 18 megagametophytes and genotyped for 7508 resequenced amplicons obtained from all available unique EST contigs representing all pine ESTs known to date using an Infinium genotyping chip. EST sequences were utilized without regard to gene annotation. In total, ∼22,000 SNPs were discovered, of which 7216 were chosen for genotyping. Results were analyzed using the BeadStudio ver. 3.1.3.0 software (Illumina), and 3938 SNPs were selected on the basis of the quality and reliability of reads as well as frequency of polymorphism across genotypes in the association population (i.e., common variants were selected). Genotypic data of the 3938 SNP markers were available for 404 of the 498 clones screened for pitch canker resistance.
Association analyses:
Patterns of population structure within this association population were assessed using 23 nuclear single sequence repeat markers in conjunction with STRUCTURE ver. 2.2 (Pritchard 2000). The association analyses performed in this study were done with a cluster number of five (K = 5). This value was the minimal value of K at which the log probability of the data leveled, and membership coefficients (i.e., q-values) illustrated geographical trends for most clusters (Eckert et al. 2010). Membership coefficients for these clusters were also in agreement with previous research, which identified significant structure (FST = 0.02–0.04) between samples spanning the Mississippi River Valley (Schmidtling 1999; Al-Rabab'ah and Williams 2002). These data were used to construct the X (structure) matrix described below.
Prior to testing for significant associations, SNPs were preselected on the basis of their significance for additive effects. The 400 SNPs with lowest P-values were used to test for significant associations in the entire population, since we anticipated these SNPs would exert the largest effects on the phenotype. A test for significance of SNP effect was performed by an analysis of variance on all 3938 SNPs, using R software version 2.8.1 (R Development Core Team 2005). A complete model, consisting of SNP, replicate, and interaction effects was compared to a reduced model with only replicate effects. The formulas for the analysis of variance are shown below, after which P-values for each individual SNP were obtained and ranked according to level of significance.
Complete model:
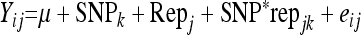
Reduced model:
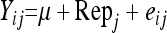 ,
,
where:
Yij is the ith log-transformed mean lesion length for the jth replicate,
μ is the overall mean,
SNPk is the fixed effect SNP k = 1–3938,
Repj is the fixed effect replicate j = 1–4, and
eij is the random variable error within the experiment
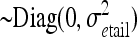 .
.
Significant associations were identified using the Bayesian association with missing data (BAMD) program developed in R software (R Development Core Team 2005), which incorporates a simultaneous solution for SNP effects, population structure, and imputation of missing SNP data (Li 2008; Gopal et al. 2009). This program is available free online at CRAN (http://cran.r-project.org/). The association model was the following:
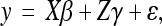 |
where y is the vector of clonal least-square means for the trait (mean log-transformed pitch canker lesion length), X is the structure matrix from the population, β is the coefficient for population structure effects, Z is the matrix for SNP effects, γ is the coefficient for SNP additive effects with a common variance, and ɛ is the residual  . A total of 50,000 iterations were performed on the program, of which the last 20,000 were kept. Mean SNP effects and 95% confidence intervals were obtained from the BAMD output, estimated from the gamma values of the last 20,000 iterations. This generated a 95% confidence interval of effect for each SNP that either did, or did not, include a value of zero. The 95% confidence interval reflects SNP effects calculated across all values (i.e., imputed multiple times) for missing SNP data points. SNPs were considered significant if they did not include a value of zero in the 95% confidence interval.
. A total of 50,000 iterations were performed on the program, of which the last 20,000 were kept. Mean SNP effects and 95% confidence intervals were obtained from the BAMD output, estimated from the gamma values of the last 20,000 iterations. This generated a 95% confidence interval of effect for each SNP that either did, or did not, include a value of zero. The 95% confidence interval reflects SNP effects calculated across all values (i.e., imputed multiple times) for missing SNP data points. SNPs were considered significant if they did not include a value of zero in the 95% confidence interval.
Tests for linkage disequilibrium and departure from Hardy–Weinberg equilibrium:
All 3938 SNPs were tested for linkage disequilibrium and departure from Hardy–Weinberg equilibrium using the SAS PROC ALLELE procedure (SAS version 9.1, SAS Institute, Cary, NC). SNPs were considered significant for either test at a false discovery rate (FDR) of 5%.
Estimating SNP effects:
The effects of the significant SNPs on the clonal variances were determined by evaluating the model used to obtain the BLUPs, incorporating all significant SNPs as random effects and then rerunning a reduced model without SNP effects. A chi-square test was performed on the difference between the −2 log likelihood values from the two models to determine whether the effect of the SNPs was significant. The percentage of clonal variance explained by each individual SNP was obtained using the following formula:
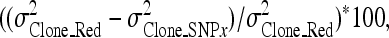 |
where 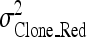 is the clonal variance of the reduced model (without SNP effects), and
is the clonal variance of the reduced model (without SNP effects), and 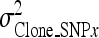 is the clonal variance obtained by including each individual SNP (x = 1–10) separately as a random variable in the model.
is the clonal variance obtained by including each individual SNP (x = 1–10) separately as a random variable in the model.
A similar approach was used to determine the percentage of the phenotypic variance accounted for by the effect of each individual SNP on the clonal variance. This was obtained by using
 |
where 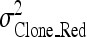 is the clonal variance of the reduced model (without SNP effect),
is the clonal variance of the reduced model (without SNP effect), 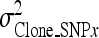 is the clonal variance obtained by including each SNP (x = 1–10) separately as a random variable in the model, and
is the clonal variance obtained by including each SNP (x = 1–10) separately as a random variable in the model, and  is the phenotypic variance obtained by summing all variance components other than environmental corrections from the reduced model.
is the phenotypic variance obtained by summing all variance components other than environmental corrections from the reduced model.
The phenotypic standard deviation was obtained by taking the square root of the sum of all variance components other than environmental corrections from the reduced model. The percentage of phenotypic standard deviation represented by each SNP was obtained by dividing the absolute value of the mean SNP effect from the association output by the phenotypic standard deviation and multiplied by 100, as shown below:
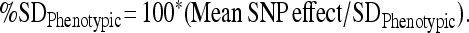 |
Blast analyses:
Sequences flanking SNPs as well as the corresponding EST contig sequences were obtained from the Dendrome database (http://dendrome.ucdavis.edu/interface) for each SNP that showed significant associations to pitch canker resistance. A BLASTx search was performed against the entire National Center for Biotechnology Information (NCBI) nonredundant protein database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to determine whether the sequences encoded proteins with known function. Hits with expect values lower than E-10 were selected, otherwise they were considered as no hits. The best hits were used as reference for interpretation of putative biological functions of the EST sequences from which the SNPs were obtained.
To determine whether a SNP was located in a coding region, the same cDNA sequences used for BLASTx were used as a BLASTn query against the NCBI database (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The pine genomic DNA sequences with highest similarity to the query sequence were used as guidelines to determine the location of the SNP. The EST contig sequence, genomic DNA sequence and the two versions of the SNP flanking sequences (each version with the corresponding nucleotide substitution), were aligned using ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html). The alignment of all four sequences was suggestive of the SNP being in a coding region.
To further verify whether the SNPs were located in a coding region, the EST contig sequence and the BLASTn best hit sequence were translated using ExPASy (http://ca.expasy.org/tools/dna.html). The translated sequences were compared to the protein sequence of the BLASTx best hit. The BLASTx best hit was also used as reference for the strand orientation of the translated EST contig sequence. BLASTx results of the EST contig sequence and BLASTn best hit sequences were also compared. In cases where the BLASTn best hit yielded no hits with the BLASTx results but its corresponding contig sequence did, was evidence that the SNP could be located in a noncoding region, possibly the 3′-UTR. Translation of the SNP flanking sequences and alignment with the translated EST contig and BLASTn best hit sequence allowed determining whether the SNP caused a synonymous or nonsynonymous substitution.
RESULTS
The distribution of clonal predicted values validates the quantitative nature of pitch canker resistance:
The clonal predicted values for lesion length obtained using BLUP showed a continuous distribution, characteristic of quantitative traits (Figure 2). This supports previous observations on the nature of pitch canker resistance in loblolly pine (Kayihan et al. 2005). The 50 clones with the most extreme phenotypes at each end of the distribution (highly susceptible and highly resistant) that had three or more ramets were reinoculated and the lesion length values were compared to those from the original population.
Figure 2.—

Distribution of BLUP clonal estimates for pitch canker lesion length (log transformed), highlighting the 50 most resistant and susceptible clones. Inserts show phenotypes of resistant and susceptible genotypes.
Clonal lesion lengths progressively increased by measurement period and showed significant differences between extreme phenotypes:
Mean lesion lengths observed for the population in the first experiment increased from 5.75 mm at 4 weeks after inoculation to 9.5 mm at 12 weeks after inoculation. When measured in the resistant and susceptible tails, the respective mean lesion lengths ranged between 2.86 and 4.57 mm, and between 7.05 and 14.01 mm (Table 1). High levels of variation were apparent within the population and in both tails, as shown by large values for the standard deviation. Such variation is likely due to a highly unbalanced experimental design, because the number of ramets and available shoots for inoculation varied among genotypes. For this reason, we used best linear unbiased predicted values for selection of clones with extreme phenotypes, as this approach adjusts for the variable number of observations.
TABLE 1.
Clonal lesion length measurements increased with time period in the two inoculation experiments and were significantly different (P < 0.001) among tails in the second inoculation
| Second inoculation (tails) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| First inoculation (association population) |
Resistant |
Susceptible |
|||||||
| Time postinoculation (weeks) | |||||||||
| 4 | 8 | 12 | 4 | 8 | 12 | 4 | 8 | 12 | |
| Mean (mm) | 5.75 | 7.93 | 9.50 | 2.86 | 3.81 | 4.57 | 7.05 | 12.15 | 14.01 |
| Standard deviation | 2.63 | 5.01 | 7.56 | 1.10 | 1.31 | 3.00 | 3.84 | 7.72 | 9.57 |
| Median | 5.37 | 6.67 | 7.27 | 2.69 | 3.38 | 3.87 | 6.31 | 10.05 | 10.75 |
| N | 498 | 45 | 47 | ||||||
Measurements were performed at 4, 8, and 12 weeks after inoculation in the association population and in the reinoculated resistant and susceptible tails.
An analysis of variance was conducted to test for significant differences among the susceptible and resistant genotypes after the second inoculation. For this test, the results from the third measurement (12 weeks after inoculation) were used. The results showed that the differences among the susceptible and resistant genotypes were significant (P < 0.0001).
Clonal repeatability values were consistent with those from a population with known pedigree:
Clonal repeatability is a measure of heritability commonly estimated for populations where the family structure is unknown. Clonal repeatability values ranged from 0.21 to 0.28 in the first experiment, and from 0.35 to 0.38 in the second inoculation (Table 2). The results suggest that ∼30% of the variation on the disease resistance trait in the first inoculation can be attributed to genetic effects, whereas in the second inoculation experiment, genetic effects account for ∼40% of the phenotypic variation. The increase of 10% in clonal repeatability observed from one experiment to the next suggests that environmental effects were reduced. This could be due to better growth conditions and tissue uniformity of plant material and more effective inoculation procedures that caused clonal measurements to be less variable across replicates. Such repeatability values were consistent with the broad-sense heritabilities reported by Kayihan et al. (2005)
TABLE 2.
Clonal repeatability, a measure of heritability, was obtained from the variances for clone, ramet, and residual in the first inoculation experiment and from the variances for clone and residual in the second inoculation experiment
| Lesion log length | Variances |
|||
|---|---|---|---|---|
| Clone | Ramet | Residual | Repeatability | |
| First inoculation | ||||
| 4 weeks | 0.11 | 0.098 | 0.32 | 0.21 |
| 8 weeks | 0.16 | 0.077 | 0.41 | 0.25 |
| 12 weeks | 0.21 | 0.069 | 0.47 | 0.28 |
| Second inoculation | ||||
| 4 weeks | 0.29 | — | 0.54 | 0.35 |
| 8 weeks | 0.34 | — | 0.53 | 0.39 |
| 12 weeks | 0.35 | — | 0.56 | 0.38 |
Respective repeatability values totaled 0.28 in the first inoculation and 0.38 in the second inoculation at 12 weeks after inoculation.
Associations suggest pitch canker resistance involves many genes with small effects:
Out of 3938 SNPs, 10 were significant at a 95% Bayesian confidence interval (α = 0.05). Mean SNP effects ranged between 0.040 and 0.061 log mm (Table 3). The phenotypic standard deviation was 0.82 log mm and was estimated as the square root of the sum of the variances of clone, ramet, and residual effects. The percentage of the phenotypic standard deviation affected by a given SNP ranged between 4.78 and 7.21 (Table 3), suggesting that there are no major genes that are involved in pitch canker resistance, but rather resistance could be due to the action of several genes with small effects. The effect of all significant SNPs on the clonal variance was estimated by running a full model (with all SNPs included as random variables in the model), as well as a reduced model (without SNPs in the model). The percentage of clonal variance accounted for by all the SNPs together was 13.19%. A likelihood ratio test on the full model against the reduced model was performed to determine the significance of the effect of SNPs on the clonal variance. The observed chi-square value was 18.26, resulting in a P-value significant at α < 0.05 with 10 degrees of freedom. Thus, the SNPs appear to have a significant, although not large, effect on the clonal variance. The individual SNP effects on the clonal variance ranged from 0.29 to 3.83% (Table 3). The sum of all values accounts to 14.6% (data not shown), which is close to the 13.19% accounted for by the effect of all SNPs together in the clonal variance. In terms of the effect of the SNPs on the phenotypic variance, these are very low, with the highest being 0.98%. Overall, the sum of the SNP effects on the phenotypic variance account for ∼3.74%. To evaluate potential consequences of preprocessing, we used the 400 SNPs with the greatest effect on the clonal variance as a preprocessing criterion. This identified 18 significant SNPs, of which 5 were identical to those described above (supporting information, Table S1).
TABLE 3.
List of SNPs significantly associated with pitch canker resistance (α < 0.05) and their effects on genotypic (clonal) and phenotypic (lesion length) variation
| SNP_ID | Allele A | Allele B | Mean (log mm) | 95% confidence interval (log mm) | % phenotypic standard deviation | % diff in clonal variance | % σ2Phenotypic due to SNP effect in clonal variance |
|---|---|---|---|---|---|---|---|
| 0_15227_01_160 | T | C | 0.040 | [0.002, 0.078] | 4.775 | 0.924 | 0.236 |
| 0_15382_01_104 | G | A | 0.061 | [0.007, 0.115] | 7.210 | 3.832 | 0.980 |
| 0_2234_01_128 | G | T | 0.048 | [0.009, 0.087] | 5.697 | 1.745 | 0.446 |
| 0_6323_01_248 | G | C | 0.045 | [0.002, 0.088] | 5.307 | 0.889 | 0.227 |
| 0_9288_01_372 | A | G | 0.048 | [0.011, 0.085] | 5.662 | 0.288 | 0.074 |
| 1_3327_01_116 | A | G | 0.054 | [0.005, 0.103] | 6.383 | 2.187 | 0.559 |
| 2_4484_02_622 | T | C | 0.057 | [0.004, 0.110] | 6.738 | 1.101 | 0.282 |
| 2_6181_02_400 | T | G | 0.057 | [0.005, 0.110] | 6.761 | 1.129 | 0.289 |
| 2_8946_02_437 | G | C | 0.056 | [0.003, 0.107] | 6.560 | 1.031 | 0.264 |
| CL4336Contig1_01_180 | T | C | 0.053 | [0.001, 0.106] | 6.277 | 1.519 | 0.389 |
Mean SNP effects on the phenotype were estimated from the last 20,000 iterations in the BAMD program, along with the corresponding 95% confidence intervals. The contribution of the SNP to the phenotype is shown as the percentage of the phenotypic standard deviation, whereas genotypic effects are indicated as the percentage of difference in the clonal variance. The percentage of the phenotypic variance due to the SNP effect in clonal variance is an indicator of the genetic contribution of the SNP to the phenotype.
Annotation of genes containing significant SNPs:
BLASTx analysis of the contig EST sequences was performed against the complete NCBI database for those SNPs that showed significant associations with the disease resistance phenotype. Out of the 10 EST sequences from the corresponding significant SNPs, two gave no hits and one resulted in an “unknown protein” when a maximum expected value of E-10 was used (Table 4). Without any cutoff value, the sequences that were detected as “no hits” had similarities with unknown predicted or unnamed proteins.
TABLE 4.
SNPs significant for association with pitch canker resistance and best hits based on BLASTx search using the contig sequence as query
| SNP_ID | Best hit (expect < 1e-10) | Best hit (no cutoff) | Predicted SNP location | Effect on aa sequence | No. SNPs in LDa |
|---|---|---|---|---|---|
| 0_15227_01_159 | ATP binding protein, lectin-like protein kinase | ATP binding protein, lectin-like protein kinase (expect = 7e-27) | Coding region | Synonymous | 0 |
| 0_15382_01_99 | Geranylgeranyl transferase type I beta subunit | Geranylgeranyl transferase type I beta subunit (expect = 2e-30) | Coding region | V to A | 0 |
| 0_2234_01_128 | Putative long-chain acyl-CoA synthetase | Putative long-chain acyl-CoA synthetase (expect = 5e-63) | Coding region | D to Y | 0 |
| 0_6323_01_240 | DELLA protein | DELLA protein (expect = 3e-59) | Coding region | Synonymous | 0 |
| 0_9288_01_370 | No hits found | Predicted protein Populus trichocarpa (expect = 4e-06) | Coding region | Synonymous | 0 |
| 1_3327_01_113b | No hits found | Unnamed protein product (Vitis vinifera) (expect = 0.23) | Coding region | C to Y | 8 |
| 2_4484_02_622 | Plastid hexose transporter | Plastid hexose transporter (expect = 8e-64) | Coding region | C to Y | 0 |
| 2_6181_02_400 | Hexokinase | Hexokinase (expect = 2e-31) | Noncoding, putative 3′-UTR | NA | 0 |
| 2_8946_02_435 | Cucumber peeling cupredoxin | Cucumber peeling cupredoxin (expect = 3e-10) | Non-coding, Putative 3′UTR | NA | 1 |
| CL4336Contig1_01_180 | Unknown (Picea sitchensis) | Unknown (Picea sitchensis) (expect = 2e-72) | Coding region | Synonymous | 1 |
Predicted SNP location and effect on amino acid sequence are also shown on the basis of sequence alignments with genomic DNA sequences. NA, not applicable.
Tests for departure from Hardy–Weinberg equilibrium and linkage disequilibrium were performed for all pairwise combinations of the 3938 SNPs available for this study. None of the significant SNPs detected were in LD with each other.
Departure from Hardy–Weinberg equilibrium was significant at 5% FDR. Linkage disequilibrium was also considered significant at 5% FDR.
The remaining EST sequences showed similarity to known proteins, such as lectin-like protein kinase, geranylgeranyl transferase beta I subunit, DELLA protein, hexokinase, plastid hexose transporter, and blue copper protein (Table 4). The flanking sequences of 8 of the 10 significant SNPs were located to a coding region. The remaining 2 appear to be in the 3′-UTR end, based on sequence alignments with the EST contig and their corresponding pine genomic DNA (Table 4).
Six of the significant SNPs result in nonsynonymous substitutions and, although some amino acid changes observed appear minor (V to A substitution), others may cause nonsynonymous substitutions or truncation of the coding sequence, which may result in more dramatic changes to protein structure (Table 4). In addition, only 1 of the 10 significant SNPs showed departure from Hardy–Weinberg equilibrium (Table 4).
DISCUSSION
In this study we exploited vegetative propagation to quantify the extent to which genetic factors condition disease resistance, and to enable the precision required to detect quantitative disease resistance genes that exert small effects on the phenotype. Lesion lengths increased among time points with significant differences between two groups of clones that showed extreme phenotypes (tails). High standard deviation values were suggestive of high levels of variation within the population, partly because of the broad geographical range from which the clones used in this study were collected, but also because the number of observations for each genotype was not uniform. When this occurs, individuals with fewer observations have a tendency to be overestimated (Beavis 1998; Goring et al. 2001). Therefore, clonal values were adjusted for different numbers of observations using BLUP. This provided more reliable clonal values for the experiment and allowed a more unbiased detection of the extreme phenotypes.
Genetic resistance to necrotrophic pathogens is frequently found to have a quantitative basis, although exceptions to this general rule have been noted in crops (e.g., Johal and Briggs 1992). Pitch canker resistance in loblolly pine appears to be quantitative on the basis of the observed continuous distribution of resistance phenotypes within a large family-based population (Kayihan et al. 2005) and the results reported in the present study. Quantitative traits typically are defined by relatively small contributions from several genes, or by one or two genes with large effect and several additional genes with small effects (Flint and Mackay 2009). Our detection of 10 loci associated with disease resistance that collectively account for ∼15% of the clonal variance is consistent with an “infinitessimal model” in which all of the clonal variance could be explained by many genes with small effects, similar to flowering traits in maize (Buckler et al. 2009). However it should be noted that the discovery panel was intended to discover common variants, and furthermore the proportion of all genes in the pine genome marked by SNPs in the present study is not known, since the total number of genes encoded in the pine genome is not known. Perhaps major genes for pitch canker resistance exist but remain undetected in this study, as well as potentially severe alleles in loci that could account for large fractions of the remaining clonal variance that is not currently explained by SNPs. Thus the evidence for lack of major genes reported in this study, which is consistent with results of other studies (Gonzalez-Martinez et al. 2007, 2008; Eckert et al. 2009a), should not be taken as evidence that quantitative disease resistance is conditioned entirely by genes that exert minor effects.
The results from BLASTx analyses showing that 3 of the 10 SNPs significant for associations corresponded to unknown or predicted proteins suggests that such sequences correspond to taxonomically restricted genes that have not been detected in other plants. Given the observation that these genes lack detectable orthologs in angiosperms, this illustrates the value of testing all possible loci for association with phenotypes of interest—these loci would not have been detected had we restricted the pool of tested SNPs to those annotated with roles in disease resistance (Morse et al. 2004).
Most loci associated with pitch canker disease resistance were related to known genes, many of which had supporting evidence of possible involvement, directly or indirectly, in disease resistance or stress response. We interpret these associations in the context of hypotheses recently proposed by Poland et al. (2009) to explain the genetic basis of quantitative disease resistance. These are not expected to be mutually exclusive, and our observations regarding the nature of the genes containing significant SNPs associated with pitch canker resistance suggest that different genes support several of those hypotheses. DELLA proteins and geranylgeranyl transferases are both involved in modulating the salicylic acid, and the jasmonic acid/ethylene pathways (Goritschnig et al. 2008; Llorente et al. 2008; Navarro et al. 2008a,b; Courdavault et al. 2009), supporting the hypotheses that quantitative resistance loci are involved in defense signal transduction. Similarly, hexokinases and hexose transporters could also support this hypothesis, if their role in plant pathogen response were mediated by their roles in sugar signaling and sensing (Herbers et al. 1995, 1996; Yoshida et al. 2002).The role of blue copper proteins in redox reactions (Nersissian et al. 1998), known to be involved in detoxifying pathogen-produced phytotoxins, could be interpreted as supporting the chemical warfare hypothesis, or as supporting the hypothesis of developmental phenotypes given a potential role for blue copper proteins in lignin formation (Loopstra and Sederoff 1995). The lectin-like protein kinase supports the hypothesis that mutations in genes involved in basal defense occur through putative recognition of pathogen elicitors (Kanzaki et al. 2008). Finally, those SNPs within unknown or unclassified genes favor the hypothesis that quantitative resistance genes represent a set of genes that have not previously been associated with disease resistance and are therefore not annotated with any known function (Poland et al. 2009).
The results of this study raise important and unresolved questions regarding durability of quantitative resistance. Evidence was obtained for significant provenance X isolate interactions between F. circinatum and P. patula but not P. tecunumanii (Hodge and Dvorak 2007), which may suggest specificity in pitch canker disease resistance. Specific interactions would imply that subsets of quantitative resistance loci may have been overcome by the pathogen during coevolution with P. patula. In this context, it would be informative to inoculate the loblolly pine genotypes in the extreme tails with diverse isolates of the pitch canker fungus to test the hypothesis that subsets of quantitative resistance loci may be involved in isolate-specific resistance in this host species, as well. Genetic variation in Fusarium is conditioned by sexual reproduction (Covert et al. 1999; Britz et al. 2005) as well as horizontal transfer of chromosomes that confer pathogenicity (Ma et al. 2010). This increases the importance of distinguishing whether resistance loci are associated with general (i.e., more durable) or specific (i.e., less durable) interactions to inform breeding and selection in genetic improvement programs aimed at increasing disease resistance (Poland et al. 2009).
Acknowledgments
The authors thank Lauren McIntyre and Alison Morse for their helpful discussions on the design of the project, as well as all the members of the Forest Genomics Group who kindly donated their time to help in the inoculation and data collection processes. In addition, two anonymous reviewers gave insightful suggestions. This work was funded by the National Science Foundation (project no. 0501763).
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.117549/DC1.
References
- Al-Rabab'ah, M. A., and C. G. Williams, 2002. Population dynamics of Pinus taeda L. based on nuclear microsatellites. For. Ecol. Manage. 163 263–271. [Google Scholar]
- Balint-Kurti, P. J., J. C. Zwonitzer, M. E. Pe, G. Pea, M. Lee et al., 2008. Identification of quantitative trait loci for resistance to southern leaf blight and days to anthesis in two maize recombinant inbred line populations. Phytopathology 98 315–320. [DOI] [PubMed] [Google Scholar]
- Beavis, W. D., 1998. QTL analysis: power, precision, and accuracy, pp. 145–162 in Molecular Dissection of Complex Traits, edited by A. H. Paterson. CRC Press, Boca Raton, FL.
- Britz, H., T. A. Coutinho, B. D. Wingfield, W. F. O. Marasas and M. J. Wingfield, 2005. Diversity and differentiation in two populations of Gibberella circinata in South Africa. Plant Pathol. 54 46–52. [Google Scholar]
- Brown, G. R., G. P. Gill, R. J. Kuntz, C. H. Langley and D. B. Neale, 2004. Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. USA 101 15255–15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler, E. S., J. B. Holland, P. J. Bradbury, C. B. Acharya, P. J. Brown et al., 2009. The genetic architecture of maize flowering time. Science 325 714–718. [DOI] [PubMed] [Google Scholar]
- Carey, W. A., S. W. Oak and S. A. Enebak, 2005. Pitch canker ratings of longleaf pine clones correlate with Fusarium circinatum infestation of seeds and seedling mortality in containers. For. Pathol. 35 205–212. [Google Scholar]
- Charlier, C., W. Coppieters, F. Rollin, D. Desmecht, J. S. Agerholm et al., 2008. Highly effective SNP-based association mapping and management of recessive defects in livestock. Nat. Genet. 40 449–454. [DOI] [PubMed] [Google Scholar]
- Courdavault, V., V. Burlat, B. St-Pierre and N. Giglioli-Guivarc'h, 2009. Proteins prenylated by type I protein geranylgeranyltransferase act positively on the jasmonate signalling pathway triggering the biosynthesis of monoterpene indole alkaloids in Catharanthus roseus. Plant Cell Rep. 28 83–93. [DOI] [PubMed] [Google Scholar]
- Covert, S. F., A. Briley, M. M. Wallace, V. T. McKinney 1999. Partial MAT-2 gene structure and the influence of temperature on mating success in Gibberella circinata. Fungal Genet. Biol. 28 43–54. [DOI] [PubMed] [Google Scholar]
- D'Alfonso, S., M. Mellai, M. Giordano, A. Pastore, G. Malferrari et al., 2002. Identification of single nucleotide variations in the coding and regulatory regions of the myelin-associated glycoprotein gene and study of their association with multiple sclerosis. J. Neuroimmunol. 126 196–204. [DOI] [PubMed] [Google Scholar]
- Dangl, J. L., and J. D. G. Jones, 2001. Plant pathogens and integrated defence responses to infection. Nature 411 826–833. [DOI] [PubMed] [Google Scholar]
- Dwinell, L. D., J. B. Barrowsbroaddus and E. G. Kuhlman, 1985. Pitch canker: a disease complex of southern pines. Plant Dis. 69 270–276. [Google Scholar]
- Easton, D. F., K. A. Pooley, A. M. Dunning, P. D. Pharoah, D. Thompson et al., 2007. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447 1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, A. J., A. D. Bower, J. L. Wegrzyn, B. Pande, K. D. Jermstad et al., 2009. a Association genetics of coastal Douglas-fir (Pseudotsuga menziesii var. menziesii, Pinaceae) I. Cold-hardiness related traits. Genetics 182 1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, A. J., E. S. Ersoz, B. Pande, M. H. Wright, V. K. Rashbrook et al., 2009. b High-throughput genotyping and mapping of single nucleotide polymorphisms in loblolly pine (Pinus taeda L.). Tree Genet. Genome 5 225–234. [Google Scholar]
- Eckert, A. J., J. van Heerwaarden, J. L. Wegrzyn, C. D. Nelson, J. Ross-Ibarra et al., 2010. Patterns of population structure and environmental associations to aridity across the 4 ranges of loblolly pine (Pinus taeda L., Pinaceae). Genetics 185 969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enebak, S. A., and G. R. Stanosz, 2003. Responses of conifer species of the Great Lakes region of North America to inoculation with the pitch canker pathogen Fusarium circinatum. For. Pathol. 33 333–338. [Google Scholar]
- Flint, J., and T. F. Mackay, 2009. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res. 19 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint-Garcia, S. A., J. M. Thornsberry and E. S. Buckler, 2003. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54 357–374. [DOI] [PubMed] [Google Scholar]
- Flint-Garcia, S. A., A. C. Thuillet, J. M. Yu, G. Pressoir, S. M. Romero et al., 2005. Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J. 44 1054–1064. [DOI] [PubMed] [Google Scholar]
- Gilmour, A. R., B. J. Gogel, B. R. Cullis and R. Thompson, 2006. ASReml User Guide Release 2.0. VSN International Ltd, Hemel Hempstead, UK.
- Goddard, M. E., and B. J. Hayes, 2009. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10 381–391. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Martinez, S. C., N. C. Wheeler, E. Ersoz, C. D. Nelson and D. B. Neale, 2007. Association genetics in Pinus taeda L. I. Wood property traits. Genetics 175 399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Martinez, S. C., D. Huber, E. Ersoz, J. M. Davis and D. B. Neale, 2008. Association genetics in Pinus taeda L. II. Carbon isotope discrimination. Heredity 101 19–26. [DOI] [PubMed] [Google Scholar]
- Gopal, V., Z. Li and G. Casella, 2009. BAMD: Bayesian Association Model for genomic data with missing covariates. R package version 3.1. http://CRAN.R-project.org/package=BAMD.
- Goring, H. H. H., J. D. Terwilliger and J. Blangero, 2001. Large upward bias in estimation of locus-specific effects from genomewide scans. Am. J. Hum. Genet. 69 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goritschnig, S., T. Weihmann, Y. L. Zhang, P. Fobert, P. McCourt et al., 2008. A novel role for protein farnesylation in plant innate immunity. Plant Physiol. 148 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbers, K., G. Monke, R. Badur and U. Sonnewald, 1995. A simplified procedure for the subtractive cDNA cloning of photoassimilate-responding genes: isolation of cDNAs encoding a new class of pathogenesis-related proteins. Plant Mol. Biol. 29 1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbers, K., P. Meuwly, W. B. Frommer, J. P. Metraux and U. Sonnewald, 1996. Systemic acquired resistance mediated by the ectopic expression of invertase: possible hexose sensing in the secretory pathway. Plant Cell 8 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge, G. R., and W. S. Dvorak, 2000. Differential responses of Central American and Mexican pine species and Pinus radiata to infection by the pitch canker fungus. New Forests 19 241–258. [Google Scholar]
- Hodge, G. R., and W. S. Dvorak, 2007. Variation in pitch canker resistance among provenances of Pinus patula and Pinus tecunumanii from Mexico and Central America. New Forests 33 193–206. [Google Scholar]
- Ingvarsson, P. K., M. V. Garcia, V. Luquez, D. Hall and S. Jansson, 2008. Nucleotide polymorphism and phenotypic associations within and around the phytochrome B2 locus in European aspen (Populus tremula, Salicaceae). Genetics 178 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inostroza, L., A. del Pozo, I. Matus, D. Castillo, P. Hayes et al., 2009. Association mapping of plant height, yield, and yield stability in recombinant chromosome substitution lines (RCSLs) using Hordeum vulgare subsp spontaneum as a source of donor alleles in a Hordeum vulgare subsp vulgare background. Mol. Breed. 23 365–376. [DOI] [PubMed] [Google Scholar]
- Jiang, N., E. Emberly, O. Cuvier and C. M. Hart, 2009. Genome-wide mapping of boundary element-associated factor (BEAF) binding sites in Drosophila melanogaster links BEAF to transcription. Mol. Cell. Biol. 29 3556–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johal, G. S., and S. P. Briggs, 1992. Reductase-activity encoded by the Hm1 disease resistance gene in maize. Science 258 985–987. [DOI] [PubMed] [Google Scholar]
- Jones, J. D. G., and J. L. Dangl, 2006. The plant immune system. Nature 444 323–329. [DOI] [PubMed] [Google Scholar]
- Kanzaki, H., H. Saitoh, Y. Takahashi, T. Berberich, A. Ito et al., 2008. NbLRK1, a lectin-like receptor kinase protein of Nicotiana benthamiana, interacts with Phytophthora infestans INF1 elicitin and mediates INF1-induced cell death. Planta 228 977–987. [DOI] [PubMed] [Google Scholar]
- Kayihan, G. C., D. A. Huber, A. M. Morse, T. L. White and J. M. Davis, 2005. Genetic dissection of fusiform rust and pitch canker disease traits in loblolly pine. Theor. Appl. Genet. 110 948–958. [DOI] [PubMed] [Google Scholar]
- Kennington, W. J., A. A. Hoffmann and L. Partridge, 2007. Mapping regions within cosmopolitan inversion In(3R)Payne associated with natural variation in body size in Drosophila melanogaster. Genetics 177 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebude, A. V., B. Goldfarb, F. A. Blazich, F. C. Wise and J. Frampton, 2004. Mist, substrate water potential and cutting water potential influence rooting of stem cuttings of loblolly pine. Tree Physiol. 24 823–831. [DOI] [PubMed] [Google Scholar]
- Lee, J. H., R. Cheng, N. Schupf, J. Manly, R. Lantigua et al., 2007. The association between genetic variants in SORL1 and Alzheimer disease in an urban, multiethnic, community-based cohort. Arc. Neurol. 64 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., 2008. Bayesian methodologies for genomic data with missing covariates. Ph.D. Dissertation, University of Florida, Gainesville, FL.
- Llorente, F., P. Muskett, A. Sanchez-Vallet, G. Lopez, B. Ramos et al., 2008. Repression of the auxin response pathway increases Arabidopsis susceptibility to necrotrophic fungi. Mol. Plant 1 496–509. [DOI] [PubMed] [Google Scholar]
- Loopstra, C. A., and R. R. Sederoff, 1995. Xylem-specific gene-expression in loblolly-pine. Plant Mol. Biol. 27 277–291. [DOI] [PubMed] [Google Scholar]
- Ma, L-J., H. C. van der Does, K. A. Borkovich, J. J. Coleman, M-J. Daboussi et al., 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malosetti, M., C. G. van der Linden, B. Vosman and F. A. van Eeuwijk, 2007. A mixed-model approach to association mapping using pedigree information with an illustration of resistance to Phytophthora infestans in potato. Genetics 175 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, M. L., M. A. Machado, C. S. Nascimento, M. V. Silva, R. L. Teodoro et al., 2006. Association of BoLA-DRB3.2 alleles with tick (Boophilus microplus) resistance in cattle. Genet. Mol. Res. 5 513–524. [PubMed] [Google Scholar]
- McGuffin, P., K. Tandon and A. Corsico, 2003. Linkage and association studies of schizophrenia. Curr. Psychiatr. Rep. 5 121–127. [DOI] [PubMed] [Google Scholar]
- Morse, A. M., C. D. Nelson, S. F. Covert, A. G. Holliday, K. E. Smith et al., 2004. Pine genes regulated by the necrotrophic pathogen Fusarium circinatum. Theor. Appl. Genet. 109 922–932. [DOI] [PubMed] [Google Scholar]
- Murray, S. C., W. L. Rooney, M. T. Hamblin, S. E. Mitchell and S. Kresovich, 2009. Sweet sorghum genetic diversity and association mapping for brix and height. Plant Genome 2 48–62. [Google Scholar]
- Navarro, L., R. Bari, P. Achard, P. Lison, A. Nemri et al., 2008. a DELLAs control plant immune responses by modulating the balance and salicylic acid signaling. Curr. Biol. 18 650–655. [DOI] [PubMed] [Google Scholar]
- Navarro, L., R. Bari, A. Seilaniantz, A. Nemri and J. D. G. Jones, 2008. b Roles of Plant Hormones in Plant Resistance and Susceptibility to Pathogens. Springer-Verlag, New York.
- Neale, D. B., and O. Savolainen, 2004. Association genetics of complex traits in conifers. Trends Plant. Sci. 9 325–330. [DOI] [PubMed] [Google Scholar]
- Nersissian, A. M., C. Immoos, M. G. Hill, P. J. Hart, G. Williams et al., 1998. Uclacyanins, stellacyanins, and plantacyanins are distinct subfamilies of phytocyanins: plant-specific mononuclear blue copper proteins. Prot. Sci. 7 1915–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norry, F. M., F. H. Gomez and V. Loeschcke, 2007. Knockdown resistance to heat stress and slow recovery from chill coma are genetically associated in a quantitative trait locus region of chromosome 2 in Drosophila melanogaster. Mol. Ecol. 16 3274–3284. [DOI] [PubMed] [Google Scholar]
- Poland, J. A., P. J. Balint-Kurti, R. J. Wisser, R. C. Pratt and R. J. Nelson, 2009. Shades of gray: the world of quantitative disease resistance. Trends Plant Sci. 14 21–29. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team, 2005. R: A language and environment for statistical computing, reference index version 3.3.1. R Foundation for Statistical. Computing, Vienna, Austria, http://www.R-project.org
- Sakamoto, J. M., and T. R. Gordon, 2006. Factors influencing infection of mechanical wounds by Fusarium circinatum on Monterey pines (Pinus radiata). Plant Pathol. 55 130–136. [Google Scholar]
- Schmidtling, R. C., E. Carroll and T. LaFarge, 1999. Allozyme diversity of selected and natural loblolly pine populations. Silvae Genetica 48 35–45. [Google Scholar]
- Stich, B., H. P. Piepho, B. Schulz and A. E. Melchinger, 2008. Multi-trait association mapping in sugar beet (Beta vulgaris L.). Theor. Appl. Genet. 117 947–954. [DOI] [PubMed] [Google Scholar]
- Stracke, S., G. Haseneyer, J.-B. Veyrieras, H. H. Geiger, S. Sauer et al., 2009. Association mapping reveals gene action and interactions in the determination of flowering time in barley. Theor. Appl. Genet. 118 259–273. [DOI] [PubMed] [Google Scholar]
- Thumma, B. R., M. F. Nolan, R. Evans and G. F. Moran, 2005. Polymorphisms in cinnamoyl CoA reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics 171 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., P. E. McClean, R. Lee, R. J. Goos and T. Helms, 2008. Association mapping of iron deficiency chlorosis loci in soybean (Glycine max L. Merr.) advanced breeding lines. Theor. Appl. Genet. 116 777–787. [DOI] [PubMed] [Google Scholar]
- Wei, X. M., P. A. Jackson, C. L. McIntyre, K. S. Aitken and B. Croft, 2006. Associations between DNA markers and resistance to diseases in sugarcane and effects of population substructure. Theor. Appl. Genet. 114 155–164. [DOI] [PubMed] [Google Scholar]
- Yahiaoui, S., A. M. Casas, M. A. Moralejo, B. Medina, M. P. Gracia et al., 2008. Genome-wide association mapping in barley. Options Mediterraneennes. Series A, Seminaires Mediterraneens: 123–126.
- Yoshida, S., M. Ito, I. Nishida and A. Watanabe, 2002. Identification of a novel gene HYS1/CPR5 that has a repressive role in the induction of leaf senescence and pathogen-defence responses in Arabidopsis thaliana. Plant J. 29 427–437. [DOI] [PubMed] [Google Scholar]
- Young, C. H., B. S. Minton and J. J. Bronson, 2006. Resistance Screening Center Procedures Manual: A Guide Used in the Operational Screening of Pines for Resistance to Pitch Canker Disease. United States Department of Agriculture, Southern Region, Forest Health Protection.
- Yu, J. M., and E. S. Buckler, 2006. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 17 155–160. [DOI] [PubMed] [Google Scholar]
- Zhao, J., M. J. Paulo, D. Jamar, P. Lou, F. van Eeuwijk et al., 2007. Association mapping of leaf traits, flowering time, and phytate content in Brassica rapa. Genome 50 963–973. [DOI] [PubMed] [Google Scholar]