

Abstract
Many advances in the understanding of meiosis have been made by measuring how often errors in chromosome segregation occur. This process of nondisjunction can be studied by counting experimental progeny, but direct measurement of nondisjunction rates is complicated by not all classes of nondisjunctional progeny being viable. For X chromosome nondisjunction in Drosophila female meiosis, all of the normal progeny survive, while nondisjunctional eggs produce viable progeny only if fertilized by sperm that carry the appropriate sex chromosome. The rate of nondisjunction has traditionally been estimated by assuming a binomial process and doubling the number of observed nondisjunctional progeny, to account for the inviable classes. However, the correct way to derive statistics (such as confidence intervals or hypothesis testing) by this approach is far from clear. Instead, we use the multinomial-Poisson hierarchy model and demonstrate that the old estimator is in fact the maximum-likelihood estimator (MLE). Under more general assumptions, we derive asymptotic normality of this estimator and construct confidence interval and hypothesis testing formulae. Confidence intervals under this framework are always larger than under the binomial framework, and application to published data shows that use of the multinomial approach can avoid an apparent type 1 error made by use of the binomial assumption. The current study provides guidance for researchers designing genetic experiments on nondisjunction and improves several methods for the analysis of genetic data.
MEIOSIS is a specialized cell division, where a diploid cell undergoes a single round of replication followed by two rounds of segregation to produce four haploid gametes. During this segregation, chromosomes must correctly separate (or disjoin) from their homologs at meiosis I, followed by sister chromatids disjoining at meiosis II. When chromosomes fail to disjoin from their partners, the resultant nondisjunction produces aneuploid gametes with the wrong number of chromosomes. The study of meiotic nondisjunction in Drosophila has a long and distinguished history of publication in genetics, with the inaugural article published in this journal being Calvin Bridges' use of nondisjunction to prove the chromosome theory of heredity (Bridges 1916). The first study that screened variants isolated from natural populations used nondisjunction to identify meiotic mutants (Sandler et al. 1968), as did the first EMS-induced mutant screen (Baker and Carpenter 1972). Subsequent screens using new mutagens or techniques have also relied on measuring nondisjunction to identify mutants of interest (Sekelsky et al. 1999). Indeed, much of the progress that has been made in the study of meiosis would not have been possible without the use of nondisjunction to identify new mutations that are defective at some step in chromosome segregation.
However, one difficulty in estimating nondisjunction rates is that in most instances the resulting aneuploid progeny cannot survive. Fortunately, in Drosophila it is possible to design crosses to recover them. Sex determination in flies is based on the number of X chromosomes, rather than a masculinizing Y chromosome as in mammals. This means that XO flies are viable (but sterile) males, while XXY flies are viable females. Therefore, it is possible to recover both normal and nondisjunctional progeny, as a nullo-X egg fertilized by an X-bearing sperm will survive as an XO male, while a diplo-X egg fertilized by a sperm lacking an X will be female (XXY). By using visible markers on the sex chromosomes, these exceptional progeny are straightforward to identify. However, if those eggs are fertilized by the other class of sperm, the resulting OY or XXX progeny are inviable. Therefore, the nondisjunction rate that occurs during meiosis is not equal to the proportion of nondisjunctional progeny, as only 50% of nondisjunctional eggs receive sperm compatible with viability, while all normal eggs are viable.
Given this experimental limitation, what is the correct method to calculate the error rate during meiosis? For this discussion, let N be the total number of progeny produced in an experiment, let X1 be the number of inviable nondisjunctional progeny (OY and XXX), let X2 be the number of viable nondisjunctional progeny (XO and XXY), and let X3 be the number of normal progeny (XY and XX), such that N = X1 + X2 + X3. If all progeny could be counted, then the nondisjunction rate 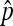 would simply be (X1 + X2)/N.
would simply be (X1 + X2)/N.
However, only flies that survive to adulthood can be counted, and therefore both X1 and N are unknown. As X- and Y-bearing sperm are produced in equal numbers, live and dead nondisjunctional progeny are also expected in equal numbers. Therefore, K.W. Cooper (Cooper 1948) proposed the widely used estimator for the X chromosome nondisjunction rate, where X2 is substituted for X1 in the above formula, giving the rate as:
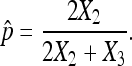 |
(1) |
While this estimator works, the statistical properties of this estimator are not clear. Instead of following the early literature to combine X1 and X2 and use a binomial distribution, we go back to the three original categories and model the process as a multinomial distribution with latent number of progeny N, considering all three possible phenotypes for each progeny (nondisjunctional dead, nondisjunctional living, and normal). Whether a nondisjunctional oocyte becomes a nondisjunctional dead or nondisjunctional living progeny depends on the sex chromosome content of the sperm that fertilized it. As X- and Y-bearing sperm are produced in equal numbers during male meiosis, the usual genetic expectation for the rates of nondisjunctional dead and living progeny will be 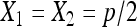 . However, even assuming that the rates of nondisjunctional dead and living progeny are different, with a Poisson assumption of N, we can derive the maximum-likelihood estimators (MLEs) for the nondisjunctional dead and nondisjunctional living rates. Under the usual genetic expectation of equality, the MLE of the nondisjunctional rate coincides with Cooper's estimator, and we furthermore derive the exact distribution of
. However, even assuming that the rates of nondisjunctional dead and living progeny are different, with a Poisson assumption of N, we can derive the maximum-likelihood estimators (MLEs) for the nondisjunctional dead and nondisjunctional living rates. Under the usual genetic expectation of equality, the MLE of the nondisjunctional rate coincides with Cooper's estimator, and we furthermore derive the exact distribution of 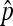 . Under another set of reasonable assumptions, we show the consistency and asymptotic normality of Cooper's estimator, and derive asymptotic results when comparing two nondisjunction rates. All these distributional results enable us to develop confidence interval and hypothesis testing related to p, or px − py in the case of comparing two nondisjunction rates from populations x and y.
. Under another set of reasonable assumptions, we show the consistency and asymptotic normality of Cooper's estimator, and derive asymptotic results when comparing two nondisjunction rates. All these distributional results enable us to develop confidence interval and hypothesis testing related to p, or px − py in the case of comparing two nondisjunction rates from populations x and y.
FORMULATION OF THE PROBLEM
Suppose an experiment produces a total of N oocytes. There are three possible cases for each oocyte: nondisjunctional dead, nondisjunctional living, and normal. These classes have the corresponding probabilities p1, p2, and 1 − p1 − p2, where p1 (p2) is the nondisjunctional dead (living) rate. For the ith progeny, let Xi1 be the indicator of the i nondisjunctional dead defined as Xi1 = 1 if ith progeny is nondisjunctional dead, and Xi1 = 0, otherwise. Similarly, we define Xi2 and Xi3 as the indicators of the ith nondisjunctional living and regular progeny. Then, Xi1 + Xi2 + Xi3 = 1. For j = 1, 2, 3, 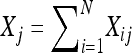 ,
,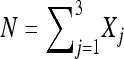 , and X1, X2, and X3 are the number of progeny in each of three categories.
, and X1, X2, and X3 are the number of progeny in each of three categories.
Given N = n, the conditional distribution of (X1, X2, X3) is a multinomial distribution with (p1, p2, 1 − p1 − p2). The probability mass function (p.m.f.) is
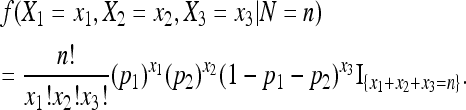 |
(2) |
THE EXACT DISTRIBUTION OF 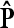 UNDER POISSON ASSUMPTION
UNDER POISSON ASSUMPTION
First, we make a Poisson assumption for N, which naturally comes from the most classical hierarchical model, known as binomial-Poisson hierarchy (see Casella and Berger 2001, Examples 4.4.1 and 4.4.2). We then derive 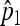 and
and 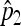 , the maximum-likelihood estimators for p1 and p2. Under the usual genetic expectation that X- and Y-bearing sperm are produced in equal numbers (and therefore p1 = p2), and ignoring all other causes of mortality, we show that
, the maximum-likelihood estimators for p1 and p2. Under the usual genetic expectation that X- and Y-bearing sperm are produced in equal numbers (and therefore p1 = p2), and ignoring all other causes of mortality, we show that 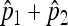 is equal to Cooper's estimator of
is equal to Cooper's estimator of 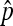 , and we further derive its exact distribution.
, and we further derive its exact distribution.
The likelihood function:
To specify the likelihood function of the observed (X2, X3), we assume that the number of progeny, N, has a Poisson probability distribution: 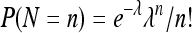 . Then, the joint p.m.f. can be written as
. Then, the joint p.m.f. can be written as
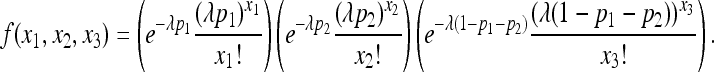 |
(3) |
This implies that under the Poisson progeny assumption, X1, X2, and X3 are independent Poisson random variables with parameters λp1, λp2, and λ(1 − p1 − p2), respectively. This desirable property with the observation that 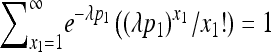 helps to obtain a simple likelihood of (p1, p2) by summing over x1 as follows:
helps to obtain a simple likelihood of (p1, p2) by summing over x1 as follows:
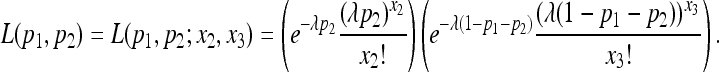 |
(4) |
Let l(p1, p2) = log L(p1, p2) be the log likelihood.
The maximum-likelihood estimators:
Setting 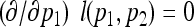 and
and 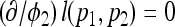 , we obtain
, we obtain
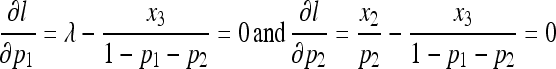 |
with roots:
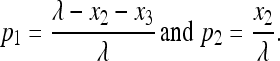 |
It can be checked that the second-order Jacobian matrix is nonpositive definite, ensuring that 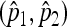 is the maximizer.
is the maximizer.
To realize the estimators of p1 and p2, we need to estimate λ. However, without further constraint on p1 and p2, λ can be any positive number larger than x2 + x3 because the given observations of x2 and x3 allow us to only estimate the ratio of p2 and p3. Further restricting p1 = kp2 for a positive k, a reasonable estimate for λ is λ = (1/k + 1)x2 + x3. and then MLEs for p1 and p2 are
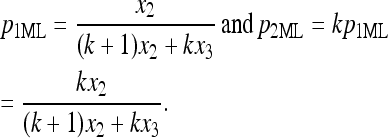 |
Of course, the usual genetic case is k = 1. In such a case, we obtain λ = 2x2 + x3 and the nondisjunctional rate p = p1 + p2. The invariance property of maximum-likelihood estimators implies that 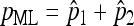 and interestingly, pML turns out to be
and interestingly, pML turns out to be
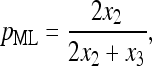 |
(5) |
which is exactly Cooper's estimator, 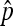 in (1).
in (1).
The exact distribution of 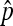 :
:
Focusing on the case p1 = p2 and letting p = p1 + p2, we can rewrite (4) as
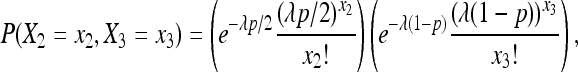 |
(6) |
with λ = 2x2 + x3. By defining a transformation as y2 = 2x2 + x3 and 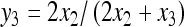 , we can derive the joint p.m.f. of (Y2, Y3) using (6), and then get the marginal exact p.m.f. of Y3
, we can derive the joint p.m.f. of (Y2, Y3) using (6), and then get the marginal exact p.m.f. of Y3
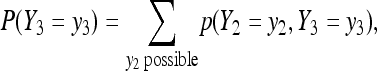 |
(7) |
which is the p.m.f. of 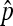 . This distribution could be obtained numerically and an R script is available upon request.
. This distribution could be obtained numerically and an R script is available upon request.
ASYMPTOTIC RESULTS WITHOUT POISSON ASSUMPTION
For the asymptotic properties of 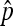 , if N = n is known (equivalently, X1 is observed), it is the classical parameter estimation problem of multinomial distribution. It is well known that
, if N = n is known (equivalently, X1 is observed), it is the classical parameter estimation problem of multinomial distribution. It is well known that 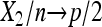 in probability, and
in probability, and 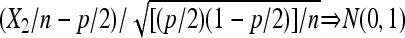 , where the ⇒ means convergence in distribution. However, in this framework X1 is not observed and N is unknown. Hence, we cannot apply the existing results.
, where the ⇒ means convergence in distribution. However, in this framework X1 is not observed and N is unknown. Hence, we cannot apply the existing results.
We study the asymptotic properties of 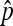 with more general assumptions, and the asymptotic properties of
with more general assumptions, and the asymptotic properties of 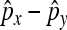 , which allow the testing of differences between two nondisjunctional rates.
, which allow the testing of differences between two nondisjunctional rates.
One nondisjunction rate:
Let the number of progeny produced in an experiment, Nn, be a random variable taking only nonnegative integer values with a probability distribution P(Nn = k). Each individual progeny can only have three possible outcomes (nondisjunctional dead, nondisjunctional living, and normal), and progeny are independent of each other. Let the probabilities of a progeny being in the three categories be (p/2, p/2, 1−p). If Xi denotes the number of progeny resulting in outcome i(i = 1, 2, 3), then the joint p.m.f. of (X1, X2, X3) given Nn = k is the multinomial distribution M(p/2, p/2, 1 − p;k), whose p.m.f. is given by Equation 2.
Theorem 1. Assume that {Nn} is a sequence of random variables such that E(Nn) = cn and 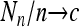 in probability for a constant c. Moreover, assume that as
in probability for a constant c. Moreover, assume that as 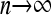 ,
, 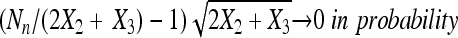 . Then, Cooper's estimator
. Then, Cooper's estimator 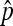 has the following property: (1)
has the following property: (1) 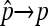 in probability, and (2)
in probability, and (2) 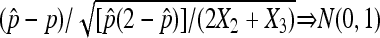 .
.
Remark 1. The assumptions of Theorem 1 are necessarily met by a Poisson distribution for N.
The proof of this remark as well as all the theorems are provided in the Appendix.
Similar to the usual normal approximation to the binomial, we require that 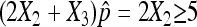 and
and 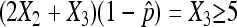 to ensure a good approximation as our simulation demonstrates. On the basis of the above theorem, we can easily obtain the (1 − α) 100% confidence interval for p as
to ensure a good approximation as our simulation demonstrates. On the basis of the above theorem, we can easily obtain the (1 − α) 100% confidence interval for p as 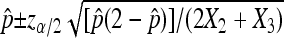 . For hypothesis testing with H0: p = p0 vs. H1: p > p0 (for example), let
. For hypothesis testing with H0: p = p0 vs. H1: p > p0 (for example), let 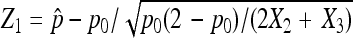 . Then, the decision rule at significance level α is to reject H0 if Z1 > zα.
. Then, the decision rule at significance level α is to reject H0 if Z1 > zα.
The difference of two nondisjunction rates:
Suppose that there are two progeny populations X and Y. We observed X2, Y2, X3, Y3 as the number of nondisjunctional living and regular normal progeny for both populations. We would like to assess whether the nondisjunction rates of two populations are statistically different from each other. Specifically, we are interested in testing: H0: px − py = δ0 vs. H1: px − py ≠ δ0, for example, or in constructing the confidence interval of px − py. Similarly, let the number of progeny from the X population be Nn, and the number of progeny from the Y population be Mm, where both Nn and Mm are random variables. Let the probabilities of a progeny's outcome being in the three categories (X1, X2, X3) be (px/2, px/2, 1 − px) in the X population, and the probabilities of a progeny's outcome being in the three categories (Y1, Y2, Y3) be (py/2, py/2, 1 − py) in the Y population. We define
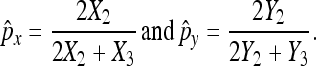 |
Theorem 2. Assume that {Nn} is a sequence of random variables such that E(Nn) = c1n and 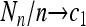 in probability for a constant c1. Assume that {Mm} is a sequence of random variables such that E(Mm) = c2m and
in probability for a constant c1. Assume that {Mm} is a sequence of random variables such that E(Mm) = c2m and 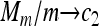 in probability for a constant c2. Moreover, assume that as
in probability for a constant c2. Moreover, assume that as
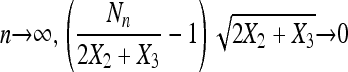 |
in probability, and as
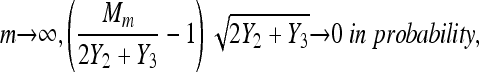 |
then: (1) 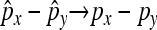 in probability, and (2)
in probability, and (2)
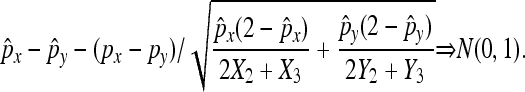 |
Similarly, the Poisson assumptions of Nn and Mm satisfy the assumptions of Theorem 2.
Again, we require that 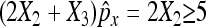 and
and 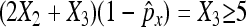 as well as
as well as 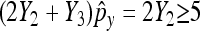 and
and 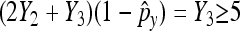 to ensure a good approximation. On the basis of the above theorem, we can easily obtain the (1 − α)100% confidence interval for px − py as
to ensure a good approximation. On the basis of the above theorem, we can easily obtain the (1 − α)100% confidence interval for px − py as
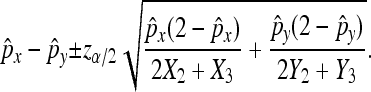 |
For hypothesis testing with H0: px − py = δ0 vs. H1: px − py ≠ δ0 (for example), let
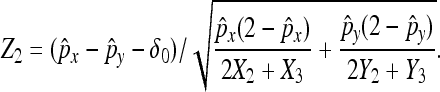 |
Then, the decision rule at significance level α is to reject H0 if |Z2| > zα/2. Finally, for the future experiment with the expected difference as δ0, the sample size can be calculated as 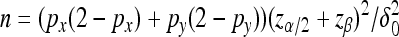 with power 1 − β and probability of type I error as α. For readers not interested in the derivation, the final equations are summarized in File S1.
with power 1 − β and probability of type I error as α. For readers not interested in the derivation, the final equations are summarized in File S1.
COMPARISON OF THE EXACT AND THE ASYMPTOTIC DISTRIBUTIONS
In this study, we present two ways of getting the distribution of nondisjunction rate estimator 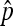 . The exact distribution of
. The exact distribution of 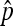 is derived with stronger assumptions, namely, the Poisson distribution for the total number of progeny (N) with its mean equal to 2x2 + x3. The asymptotic results are derived with weaker assumptions and are applicable as long as N satisfies conditions in Theorem 1. The Poisson assumption of N is one special case where Theorem 1 can be applied. When the number of nondisjunctional living progeny (x2) is not too small, usually x2 ≥ 5, the approximation is good. We demonstrate this by comparing the two distributions assuming there is a total of 1000 progenies for three cases: (1) X2 = 25, X3 = 950, then p = 0.05; (2) X2 = 5, X3 = 990, then p = 0.01; and (3) X2 = 2, X3 = 996, then p = 0.004.
is derived with stronger assumptions, namely, the Poisson distribution for the total number of progeny (N) with its mean equal to 2x2 + x3. The asymptotic results are derived with weaker assumptions and are applicable as long as N satisfies conditions in Theorem 1. The Poisson assumption of N is one special case where Theorem 1 can be applied. When the number of nondisjunctional living progeny (x2) is not too small, usually x2 ≥ 5, the approximation is good. We demonstrate this by comparing the two distributions assuming there is a total of 1000 progenies for three cases: (1) X2 = 25, X3 = 950, then p = 0.05; (2) X2 = 5, X3 = 990, then p = 0.01; and (3) X2 = 2, X3 = 996, then p = 0.004.
We further generate the empirical distributions of p′s under the three cases by simulations to see how our derived distributions matched the simulated ones. The detailed procedures are to first, simulate a N from a Poisson distribution with mean being 1000; second, simulate x1, x2, x3 from a multinomial distribution with (p/2, p/2, 1 − p); and third, calculate 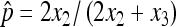 . The procedure is repeated 50, 000 times each, with p set to 0.05, 0.01, or 0.004, respectively, as shown in Figure 1. When X2 is large, the assumptions for asymptotic results are well met and the three distributions (exact, asymptotic, and empirical) are almost identical (case 1 and 2). When X2 is small (2X2 < 5 and p is close to 0) (case 3), the asymptotic density deviates more from the exact distribution, but still in good agreement. These results show that the asymptotic normal distribution is a very good approximation of the exact distribution. In the extreme case that the data are not well modeled by the Poisson distribution, the asymptotic results are still valid. We suggest using the asymptotic results for constructing confidence intervals and doing hypothesis tests unless either 2X2 or X3 is small (<5). As nondisjunction assays in Drosophila usually have sample sizes of at least several hundred, this condition is most likely to be violated in cases where the value of p is close to 1/N.
. The procedure is repeated 50, 000 times each, with p set to 0.05, 0.01, or 0.004, respectively, as shown in Figure 1. When X2 is large, the assumptions for asymptotic results are well met and the three distributions (exact, asymptotic, and empirical) are almost identical (case 1 and 2). When X2 is small (2X2 < 5 and p is close to 0) (case 3), the asymptotic density deviates more from the exact distribution, but still in good agreement. These results show that the asymptotic normal distribution is a very good approximation of the exact distribution. In the extreme case that the data are not well modeled by the Poisson distribution, the asymptotic results are still valid. We suggest using the asymptotic results for constructing confidence intervals and doing hypothesis tests unless either 2X2 or X3 is small (<5). As nondisjunction assays in Drosophila usually have sample sizes of at least several hundred, this condition is most likely to be violated in cases where the value of p is close to 1/N.
Figure 1.—

Comparison of the exact distribution and the asymptotic distribution of the nondisjunction rate. Three cases are considered, the nondisjunction rates being 0.05, 0.01, 0.004, to show the difference of two distributions. The green curve is the empirical distribution of p with 50,000 simulations.
ANALYSIS USING REAL DATA
Case study I:
The common objectives for doing a nondisjunction assay include estimating the nondisjunction rate and testing if two genotypes have rates that are statistically significantly different. In the first example, we compare results of point estimation and hypothesis tests between the asymptotic results derived in this study and the asymptotic results assuming the traditional binomial distribution. As we discussed, most published literature has used the binomial distribution to model the nondisjunctional event as Binomial (N, p) assuming that N is observed and N = 2X2 + X3. With this assumption, the estimator turns out to be the same as one in this study, 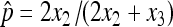 , but the standard deviation is calculated as
, but the standard deviation is calculated as 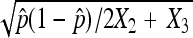 . This approximation ignores the fact that the number of nondisjunctional dead progeny is an unobserved random number. When this randomness is accounted for, as we do in this study, the standard deviation is calculated as
. This approximation ignores the fact that the number of nondisjunctional dead progeny is an unobserved random number. When this randomness is accounted for, as we do in this study, the standard deviation is calculated as 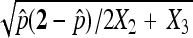 , which is at least 1.414 times as large as the one calculated with the binomial distribution (Figure 2). Unlike the binomial assumption that the standard deviation reaches to the largest when p = 0.5, under the multinomial assumption, the standard deviation of p increases as p increases. Therefore, as p gets larger, the ratio between these two standard deviations gets larger. We illustrate this using a published data set (Zhang and Hawley 1990). This study tested nondisjunction rates from a number of different mutant alleles of the gene nod. The estimated X nondisjunctional rate for these mutants is around 0.5 (Table 1). The standard deviation calculated using our asymptotic results is always larger (1.74–1.83 times as large) and the difference tends to increase as p gets larger.
, which is at least 1.414 times as large as the one calculated with the binomial distribution (Figure 2). Unlike the binomial assumption that the standard deviation reaches to the largest when p = 0.5, under the multinomial assumption, the standard deviation of p increases as p increases. Therefore, as p gets larger, the ratio between these two standard deviations gets larger. We illustrate this using a published data set (Zhang and Hawley 1990). This study tested nondisjunction rates from a number of different mutant alleles of the gene nod. The estimated X nondisjunctional rate for these mutants is around 0.5 (Table 1). The standard deviation calculated using our asymptotic results is always larger (1.74–1.83 times as large) and the difference tends to increase as p gets larger.
Figure 2.—

Comparison of standard deviations calculated using asymptotic results assuming a binomial distribution 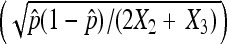 and asymptotic results assuming a multinomial distribution as proposed in this study
and asymptotic results assuming a multinomial distribution as proposed in this study 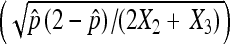 . In the plot, we use 2X2 + X3 = 1000.
. In the plot, we use 2X2 + X3 = 1000.
TABLE 1.
Comparisons of standard deviation calculation between the asymptotic results derived in this study and the asymptotic results assuming the binomial distribution
| FM7a, nodb27/noda | FM7a, nodb34/noda | FM7a, nodb9/noda | FM7a, nodb1/noda | FM7a, nodb17/noda | FM7a, nodb29/noda | FM7a, nodbd/noda | |
|---|---|---|---|---|---|---|---|
| Regular | 1167 | 639 | 844 | 897 | 2566 | 598 | 639 |
| X NDJ | 661 | 323 | 527 | 573 | 1319 | 400 | 378 |
| Total | 1828 | 962 | 1371 | 1470 | 3885 | 998 | 1017 |
| Adj.total | 2489 | 1285 | 1898 | 2043 | 5204 | 1398 | 1395 |
| X NDJ rate | 0.5311 | 0.5027 | 0.5553 | 0.5609 | 0.5069 | 0.5722 | 0.5419 |
| std1 (asymp.normal) | 0.0177 | 0.0242 | 0.0206 | 0.0199 | 0.0121 | 0.0242 | 0.0238 |
| std2 (binomial) | 0.0100 | 0.0139 | 0.0114 | 0.0110 | 0.0069 | 0.0132 | 0.0133 |
| Ratio (std1/std2) | 1.77 | 1.74 | 1.80 | 1.81 | 1.74 | 1.83 | 1.78 |
The data are taken from Zhang and Hawley (1990), which studied nondisjunction rates from a number of different mutant alleles of the gene nod.
Taking this randomness into consideration also has a large effect in terms of hypothesis tests. For comparing two nondisjunction rates px and py, our results show that
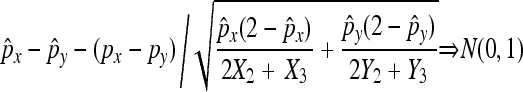 |
under the null hypothesis, which is different from
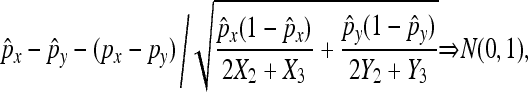 |
when N is assumed to be observed. When we test if all seven mutants have the same nondisjunction rates by pairwise comparison (Table 1), we found that there are no statistically significant differences among them with the family-wise error rate ≤0.05 (Bonferroni multitest correction). This is consistent with the genetic analysis of these alleles, which appear to act as complete nulls that have lost all gene function. In contrast, using the same multitest correction method with asymptotic results derived from the traditional binomial distribution, the b34 and b17 alleles appear to be significantly different from b9, b1, and b29 (Table 2). This suggests that the genetic analysis is wrong and that these alleles retain some residual function. However, in light of our current analysis, the traditional binomial method would appear to yield false-positive results caused by ignoring the randomness in the number of nondisjunctional dead progeny.
TABLE 2.
Comparisons of Bonferroni-adjusted P-values from the hypothesis tests between the asymptotic results derived in this study (conditional multinomial) and the asymptotic results assuming the binomial distribution using data in Table 1
| genotype1 | genotype2 | adjp(Multinomial) | adjp(Binomial) |
|---|---|---|---|
| nodb34 | nodb9 | 1 | 0.0737 |
| nodb34 | nodb1 | 1 | 0.0218 |
| nodb34 | nodb29 | 0.8879 | 0.0063 |
| nodb17 | nodb9 | 0.8879 | 0.0060 |
| nodb17 | nodb1 | 0.4232 | 0.0007 |
| nodb17 | nodb29 | 0.3276 | 0.0003 |
Case study II:
In the second data set, a collection of fly lines isolated from nature that had been used in a population genetics sequencing project for meiotic genes (Anderson et al. 2009) was assayed for their X nondisjunction rates. The nondisjunction rates observed among these lines were small (ranging from p = 0 to p = 0.014; Table 3). After multitest correction to control the FDR 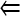 0.05 (Benjamini and Hochberg 1995), the line MW9X showed a significant difference with several other lines (marked with * in Table 3, P-values = 0.05) with changes ranging between 6- and 20-fold. This result shows that while these lines do not carry alleles of large effect, such as those isolated by a screen of natural variation (Sandler et al. 1968), these assays have nonetheless successfully identified naturally occurring phenotypic variation in the trait of meiotic segregation. This is consistent with the genotypic variation identified in these same natural populations having phenotypic consequences as well. While these phenotypic differences are only just statistically significant at these sample sizes, at the population level these differences should clearly be subject to natural selection. This result also raises several experimental design considerations, such as when designing an assay to compare the nondisjunction rate for alleles of small effect, what sample size would be needed to reject H0: px − py = 0 with 80% power? For example, if the values of p for two lines differ by 1% (e.g., px = 0.005, py = 0.015), a sample size of 2338 per group is required to achieve a power of at least 0.8 with a two-sided significance level of 0.05. In Table 4, we list the sample size required for pairwise comparisons of a list of nondisjunction rates, ranging from 0.01 to 0.31. This table indicates that if the expected difference in rates is quite large (e.g., 20% vs. 1%, as might be seen in comparing a mutant to a mutant plus rescue construct) then sample sizes of only a few hundred would be more than sufficient. Conversely, as the real rates under consideration become closer, the needed sample size becomes much larger and quickly becomes experimentally intractable. This indicates that any experimental outcome that hinges on nondisjunction rates being different by only 1% or 2% should be viewed with great skepticism.
0.05 (Benjamini and Hochberg 1995), the line MW9X showed a significant difference with several other lines (marked with * in Table 3, P-values = 0.05) with changes ranging between 6- and 20-fold. This result shows that while these lines do not carry alleles of large effect, such as those isolated by a screen of natural variation (Sandler et al. 1968), these assays have nonetheless successfully identified naturally occurring phenotypic variation in the trait of meiotic segregation. This is consistent with the genotypic variation identified in these same natural populations having phenotypic consequences as well. While these phenotypic differences are only just statistically significant at these sample sizes, at the population level these differences should clearly be subject to natural selection. This result also raises several experimental design considerations, such as when designing an assay to compare the nondisjunction rate for alleles of small effect, what sample size would be needed to reject H0: px − py = 0 with 80% power? For example, if the values of p for two lines differ by 1% (e.g., px = 0.005, py = 0.015), a sample size of 2338 per group is required to achieve a power of at least 0.8 with a two-sided significance level of 0.05. In Table 4, we list the sample size required for pairwise comparisons of a list of nondisjunction rates, ranging from 0.01 to 0.31. This table indicates that if the expected difference in rates is quite large (e.g., 20% vs. 1%, as might be seen in comparing a mutant to a mutant plus rescue construct) then sample sizes of only a few hundred would be more than sufficient. Conversely, as the real rates under consideration become closer, the needed sample size becomes much larger and quickly becomes experimentally intractable. This indicates that any experimental outcome that hinges on nondisjunction rates being different by only 1% or 2% should be viewed with great skepticism.
TABLE 3.
X nondisjunctional rate estimates for lines from two natural populations (North American and Africa)
| Line | NonX | Normal | X nondis rate (std) |
|---|---|---|---|
| 301 | 0 | 177 | 0 (−) |
| 303 | 3 | 1905 | 0.0031(0.0018) |
| 304* | 2 | 3818 | 0.0010(0.0007) |
| 306 | 0 | 1601 | 0 (−) |
| 319 | 2 | 2295 | 0.0017(0.0012) |
| 322 | 4 | 3826 | 0.0021(0.0010) |
| 335 | 3 | 2784 | 0.0022(0.0012) |
| 336 | 7 | 3168 | 0.0044(0.0017) |
| 350 | 7 | 3843 | 0.0036(0.0014) |
| 357 | 3 | 2658 | 0.0023(0.0013) |
| 358 | 6 | 2908 | 0.0041(0.0017) |
| 359 | 2 | 525 | 0.0076(0.0053) |
| 361 | 3 | 3651 | 0.0016(0.0009) |
| 375 | 6 | 2650 | 0.0045(0.0018) |
| 390 | 5 | 1122 | 0.0088(0.0039) |
| 397 | 0 | 664 | 0 (−) |
| 399* | 1 | 3053 | 0.0007(0.0007) |
| 732 | 4 | 1845 | 0.00439(0.0022) |
| 740* | 1 | 2691 | 0.0007(0.0007) |
| 774 | 2 | 1222 | 0.0033(0.0023) |
| MW11-3 | 0 | 218 | 0 (−) |
| MW25X | 2 | 2909 | 0.0014(0.0010) |
| MW27X* | 1 | 1937 | 0.0010(0.0010) |
| MW28X | 0 | 159 | 0 (−) |
| MW28-5 | 0 | 148 | 0 (−) |
| MW38X | 6 | 2155 | 0.0055(0.0023) |
| MW46-1 | 0 | 499 | 0 (−) |
| MW6-3II | 1 | 1482 | 0.0013(0.0013) |
| MW6X | 3 | 1526 | 0.0039(0.0023) |
| MW9-2 | 1 | 919 | 0.0022(0.0022) |
| MW9-4* | 1 | 2162 | 0.0009(0.0009) |
| MW9X | 14 | 2024 | 0.0136(0.0036) |
Nondisjunction rates were measured by crossing wild-type females to y cv v f car/BSY males under standard conditions, which allowed identification of nondisjunctional progeny as multiply-marked males (XO) or BS females (XXY). The numbers in parentheses are the standard deviations of the X nondisjunction rates. The lines marked with * have significantly different nondisjunction rates when compared to MW9X.
TABLE 4.
The expected sample size needed from each of two populations with the given nondisjunction rates (px and py) to declare a statistically significant difference with probability of type I error of α = 0.05 and power of β = 0.90
|
py |
||||||
|---|---|---|---|---|---|---|
| px | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 |
| 0.01 | 771 | 273 | 160 | 111 | 84 | 67 |
| 0.06 | 22,476 | 2,013 | 511 | 256 | 162 | 115 |
| 0.11 | 892 | 41,810 | 3,188 | 737 | 346 | 209 |
| 0.16 | 341 | 1,414 | 60,092 | 4,298 | 950 | 432 |
| 0.21 | 195 | 492 | 1,908 | 77,325 | 5,342 | 1,150 |
| 0.26 | 132 | 264 | 634 | 2,372 | 93,506 | 6,321 |
| 0.31 | 97 | 171 | 329 | 768 | 2,807 | 108,637 |
DISCUSSION
The nondisjunction rate is an important parameter in the study of meiosis. We have studied the statistical properties of the currently widely used Cooper's estimator 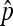 , which is
, which is 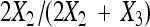 . Under stringent assumptions, the estimator turns out the be the MLE and the exact distribution of
. Under stringent assumptions, the estimator turns out the be the MLE and the exact distribution of 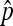 could be obtained numerically. When p is not too close to 0 and the observed nondisjunctional progeny (X2) is not too small (2X2 ≤ 5),
could be obtained numerically. When p is not too close to 0 and the observed nondisjunctional progeny (X2) is not too small (2X2 ≤ 5), 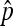 is shown to have an asymptotic normal distribution (Theorem 1), and the asymptotic distribution approximates the exact distribution well when p is large. In the real data analysis, we suggest use of asymptotic results whenever possible because it requires no specific distribution on N. Unless both 2X2 and X3 are small (<5), the asymptotic normal distribution is a good approximation of the exact distribution as shown in our simulation study. The use of the normal approximation also enables us to apply classical statistical tools to this problem. For example (as shown in Table 4), the power/sample size calculation can be carried out and this can provide experimental guidelines for designing nondisjunction assays. Statistical significance tests (P-value calculation) also can be carried out on the basis of Theorem 2. We provide a MS EXCEL file to do these calculations as supporting information material in File S2.
is shown to have an asymptotic normal distribution (Theorem 1), and the asymptotic distribution approximates the exact distribution well when p is large. In the real data analysis, we suggest use of asymptotic results whenever possible because it requires no specific distribution on N. Unless both 2X2 and X3 are small (<5), the asymptotic normal distribution is a good approximation of the exact distribution as shown in our simulation study. The use of the normal approximation also enables us to apply classical statistical tools to this problem. For example (as shown in Table 4), the power/sample size calculation can be carried out and this can provide experimental guidelines for designing nondisjunction assays. Statistical significance tests (P-value calculation) also can be carried out on the basis of Theorem 2. We provide a MS EXCEL file to do these calculations as supporting information material in File S2.
The analysis of nondisjunction data using this framework suggests several important conclusions. The first is that as nondisjunction rates approach zero, the number of nondisjunctional progeny expected approaches zero. It is in this region that the random number of progeny surviving fertilization has its greatest effect on the estimated rate. Second, even for cases where p is far from zero, the variance of this process is greater than that of a binomial. The practical impact of this is clearly seen in our analysis of the published nod nondisjunction data (Zhang and Hawley 1990). While the genetic analysis indicated that the nod alleles were complete nulls, the binomial approach finds that their nondisjunction rates are statistically significantly different from one another, suggesting that these alleles retain at least some residual function. When the increased variance due to lethal aneuploidy after fertilization is accounted for, the differences are no longer significant, which is consistent with the genetic analysis. This avoidance of an apparent false-positive result is a clear benefit to using the multinomial approach. Third, this suggests that differences in the nondisjunction rate of less than around 2% may simply not be amenable to direct experimental analysis, even with sample sizes of several thousand. This is a point of concern for population genetics, as variants that reduced nondisjunction by even a fraction of a percent should be advantageous and undergo positive selection in species as numerous as Drosophila. Our results suggest that any experimental program working with alleles of small effect should consider the use of sensitized assays, where the genetic background is weakened so that small genotypic differences are magnified to an experimentally tractable level (Zwick et al. 1999). Finally, while increasing sample sizes does decrease confidence intervals, sample size increases rapidly experience diminishing returns. As a rule of thumb, Table 4 appears to show that reasonable statistical payoffs (such as reduction of sizes of confidence intervals) in increasing sample sizes from ∼100 to ∼1000, but very little improvement in increasing sample sizes from ∼3000 to > 10,000. The exact sample sizes aimed for in an experiment should be considered in light of the data's intended purpose to meet research goals without wasted efforts.
In the current work, we have considered only estimating the rate of X nondisjunction in female meiosis. The small 4 chromosome can also be used in nondisjunction assays, as triplo-4 progeny are viable and can therefore be observed. By mating experimental females to males bearing a compound-4, both normal and nondisjunctional oocytes have the same 50% chance of being fertilized by the type of sperm that results in viable progeny. This means that the rate of nondisjunction is expected to be equal to the proportion of nondisjunctional progeny observed, without the doubling used in Cooper's estimator for X chromosome nondisjunction. In light of our current results, it is clear that the use of a binomial model for 4 nondisjunction would also underestimate the true size of the confidence intervals. A preliminary examination of this process suggests that as random survival is applied to all progeny, instead of solely to the nondisjunctional classes, the increase in variance of estimates of 4 nondisjunction rates due to sperm chromosome content may be even greater than that for the X. This appears to be because in the X-only case the 50% chance of dying from fertilization by the wrong sperm is applied solely to nondisjunctional progeny, while all of the normal progeny are assumed to survive. In the 4-only case, the same 50% chance of dying is applied to both nondisjunctional and normal progeny. Therefore, while the value of 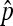 is equal to the observed proportion of nondisjunctional progeny observed, the variance of 4-only nondisjunction should be greater than that of the X-only case. Furthermore, in practice nondisjunction for the X and 4 are often scored simultaneously. This practice is biologically relevant, as it has revealed the intriguing observation that rates of X and 4 nondisjunction are often found in a 2:1 ratio across certain classes of mutants (Zitron and Hawley 1989; Sekelsky et al. 1999). In this case, as X nondisjunctional oocytes have only a 25% chance of being viable after fertilization, this should result in an even larger increase in the variance than that of the X-only case. Therefore, researchers should be aware that when compound-4 is used to simultaneously measure X and 4 nondisjunction, our method for calculating confidence intervals for X nondisjunction rates will be an underestimate of the true interval. We are continuing to study the process of X and 4 nondisjunction and hope to be able to develop similar multinomial results for the 4-only and X/4 simultaneous cases in the future.
is equal to the observed proportion of nondisjunctional progeny observed, the variance of 4-only nondisjunction should be greater than that of the X-only case. Furthermore, in practice nondisjunction for the X and 4 are often scored simultaneously. This practice is biologically relevant, as it has revealed the intriguing observation that rates of X and 4 nondisjunction are often found in a 2:1 ratio across certain classes of mutants (Zitron and Hawley 1989; Sekelsky et al. 1999). In this case, as X nondisjunctional oocytes have only a 25% chance of being viable after fertilization, this should result in an even larger increase in the variance than that of the X-only case. Therefore, researchers should be aware that when compound-4 is used to simultaneously measure X and 4 nondisjunction, our method for calculating confidence intervals for X nondisjunction rates will be an underestimate of the true interval. We are continuing to study the process of X and 4 nondisjunction and hope to be able to develop similar multinomial results for the 4-only and X/4 simultaneous cases in the future.
Acknowledgments
The authors thank Boris Rubinstein and Arcady Mushegian for helpful discussion and comments, the editor, and two anonymous reviewers for their helpful suggestions to improve the manuscript. This work was supported by a Stowers Summer Scholarship to N.M.S., an American Cancer Society Research Professorship to R.S.H., and an American Cancer Society Postdoctoral Fellowship to W.D.G.
APPENDIX
Proof of Theorem 1:
The key result to obtain the asymptotic properties of 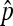 is the following Chung's lemma, which is Theorem 7.3.2 (Chung 1974).
is the following Chung's lemma, which is Theorem 7.3.2 (Chung 1974).
Lemma 1. Suppose that {Xi, i ≥ 1} is a sequence of i.i.d. random variables with mean 0 and variance 1. Define 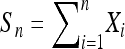 . Let {γn,n ≥ 1} be a sequence of random variables taking only strictly positive integer values (can be relaxed to “taking only nonnegative integers”) such that
. Let {γn,n ≥ 1} be a sequence of random variables taking only strictly positive integer values (can be relaxed to “taking only nonnegative integers”) such that 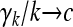 in probability, where c is a positive constant. Then,
in probability, where c is a positive constant. Then, 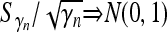 .
.
The proof of Theorem 1 also relies on the two lemmas below (their proofs are available upon request).
Lemma 2.
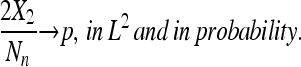 |
Lemma 3.
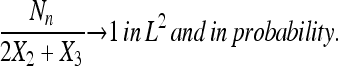 |
Lemma 4.
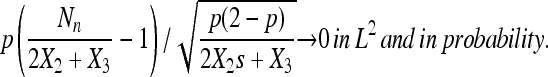 |
Proof. Since 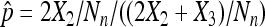 , Lemmas 2 and 3 imply the consistency of
, Lemmas 2 and 3 imply the consistency of 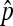 , namely,
, namely, 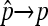 in probability.
in probability.
Observe that 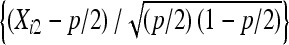 is a sequence of i.i.d. random variables with mean 0 and variance 1. Then,
is a sequence of i.i.d. random variables with mean 0 and variance 1. Then,
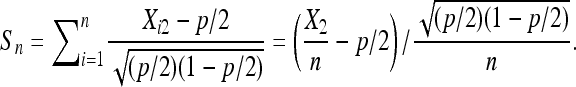 |
So, Chung's lemma and the assumption imply
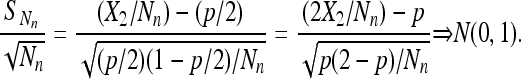 |
(8) |
Next, consider
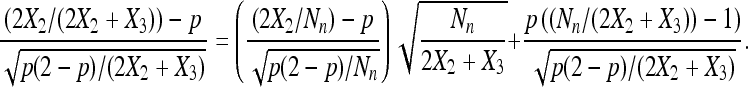 |
(9) |
Slutsky's theorem with (8) and Lemmas 3 and 4 imply
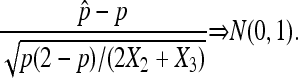 |
(10) |
Finally, observe that 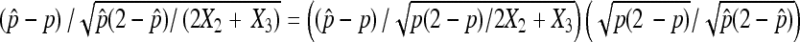 . The consistency of
. The consistency of  implies
implies 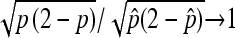 /
/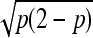 in probability. Together with (10), Slutsky's theorem gives the desired asymptotic normal result. ▪
in probability. Together with (10), Slutsky's theorem gives the desired asymptotic normal result. ▪
Proof of Remark 1:
With the Poisson assumption, Nn becomes Nλ having a Poisson distribution with parameter λ. It is well known that E(Nλ) = λ and 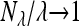 in probability. The first assumption is satisfied. To check the second assumption, it suffices to show the L1 convergence by Markov inequality. Observe that E(2X2 + X3) = E(Nλ) = λ. Applying Cauchy–Schwartz inequality, we have
in probability. The first assumption is satisfied. To check the second assumption, it suffices to show the L1 convergence by Markov inequality. Observe that E(2X2 + X3) = E(Nλ) = λ. Applying Cauchy–Schwartz inequality, we have
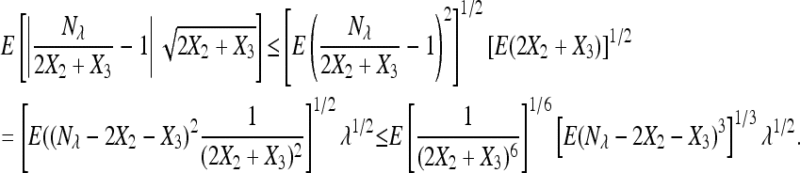 |
The last inequality comes by applying Hölder's inequality with p = 3 and q = 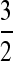 . It goes to zero because it can be shown that given the first assumption of Theorem 1, [E(2X2 + X3)−6]1/6 = O(λ−1) and [E(Nλ − 2X2 − X3)3]1/3 = O(λ1/3).
. It goes to zero because it can be shown that given the first assumption of Theorem 1, [E(2X2 + X3)−6]1/6 = O(λ−1) and [E(Nλ − 2X2 − X3)3]1/3 = O(λ1/3).
Proof of Theorem 2:
Proof. Observe that the two samples are independent. The consistency follows immediately. With the assumptions, we can apply Theorem 1 to each sample and obtain
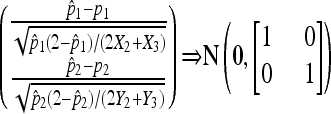 |
(11) |
Let g(x, y) = x − y and 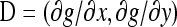 . Observe that
. Observe that 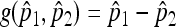 and D = (1, −1). Then, the asymptotic normality comes from applying the multivariate δ methods.
and D = (1, −1). Then, the asymptotic normality comes from applying the multivariate δ methods.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.118778/DC1.
References
- Anderson, J. A., W. D. Gilliland and C. H. Langley, 2009. Molecular population genetics and evolution of Drosophila meiosis genes. Genetics 181 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, B. S., and A. T. Carpenter, 1972. Genetic analysis of sex chromosomal meiotic mutants in Drosophila melanogaster. Genetics 71 255–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B Methodol. 57 289–300. [Google Scholar]
- Bridges, C. B., 1916. Non-disjunction as proof of the chromosome theory of heredity. Genetics 1 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella, G., and L. B. Berger, 2001. Statistical Inference, Ed. 2. Duxbury Press, Pacific Grove, CA.
- Chung, K. L., 1974. A Course in Probability Theory, Ed. 2. Academic Press, New York.
- Cooper, K. W., 1948. A new theory of secondary non-disjunction in female Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 34 179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler, L., D. L. Lindsley, B. Nicoletti and G. Trippa, 1968. Mutants affecting meiosis in natural populations of Drosophila melanogaster. Genetics 60 525–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekelsky, J. J., K. S. McKim, L. Messina, R. L. French, W. D. Hurley et al., 1999. Identification of novel Drosophila meiotic genes recovered in a P-element screen. Genetics 152 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P., and R. S. Hawley, 1990. The genetic analysis of distributive segregation in Drosophila melanogaster. II. Further genetic analysis of the nod locus. Genetics 125 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitron, A. E., and R. S. Hawley, 1989. The genetic analysis of distributive segregation in Drosophila melanogaster. I. Isolation and characterization of Aberrant X segregation (AXS), a mutation defective in chromosome partner choice. Genetics 122 801–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick, M. E., J. L. Salstrom and C. H. Langley, 1999. Genetic variation in rates of nondisjunction: association of two naturally occurring polymorphisms in the chromokinesin nod with increased rates of nondisjunction in Drosophila melanogaster. Genetics 152 1605–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]