

Abstract
DNA replication errors that escape polymerase proofreading and mismatch repair (MMR) can lead to base substitution and frameshift mutations. Such mutations can disrupt gene function, reduce fitness, and promote diseases such as cancer and are also the raw material of molecular evolution. To analyze with limited bias genomic features associated with DNA polymerase errors, we performed a genome-wide analysis of mutations that accumulate in MMR-deficient diploid lines of Saccharomyces cerevisiae. These lines were derived from a common ancestor and were grown for 160 generations, with bottlenecks reducing the population to one cell every 20 generations. We sequenced to between 8- and 20-fold coverage one wild-type and three mutator lines using Illumina Solexa 36-bp reads. Using an experimentally aware Bayesian genotype caller developed to pool experimental data across sequencing runs for all strains, we detected 28 heterozygous single-nucleotide polymorphisms (SNPs) and 48 single-nt insertion/deletions (indels) from the data set. This method was evaluated on simulated data sets and found to have a very low false-positive rate (∼6 × 10−5) and a false-negative rate of 0.08 within the unique mapping regions of the genome that contained at least sevenfold coverage. The heterozygous mutations identified by the Bayesian genotype caller were confirmed by Sanger sequencing. All of the mutations were unique to a given line, except for a single-nt deletion mutation which occurred independently in two lines. All 48 indels, composed of 46 deletions and two insertions, occurred in homopolymer (HP) tracts [i.e., 47 poly(A) or (T) tracts, 1 poly(G) or (C) tract] between 5 and 13 bp long. Our findings are of interest because HP tracts are present at high levels in the yeast genome (>77,400 for 5- to 20-nt HP tracts), and frameshift mutations in these regions are likely to disrupt gene function. In addition, they demonstrate that the mutation pattern seen previously in mismatch repair defective strains using a limited number of reporters holds true for the entire genome.
MUTATION rates in prokaryotic and eukaryotic organisms are typically determined by measuring reversion or forward mutation for specific marker alleles. These values are then extrapolated to obtain genome-wide estimates. Mutation rates in higher eukaryotes are also estimated by analyzing sequence divergence between different strains or species, followed by reconstructing the accumulation of mutations since divergence (reviewed in Nishant et al. 2009). These approaches suffer from two main limitations. First, recent studies have shown that mutation rate and repair efficiency vary across the genome and are affected by parameters that include base composition, local recombination rate, gene density, transcriptional activity, repair efficiency, chromatin structure, nucleosome position, and replication timing (Wolfe et al. 1989; Datta and Jinks-Robertson 1995; Matassi et al. 1999; Hardison et al. 2003; Arndt et al. 2005; Hawk et al. 2005; Teytelman et al. 2008; Washietl et al. 2008; Stamatoyannopoulos et al. 2009). Second, genomic comparisons can yield inaccurate rate measurements because DNA repair and subsequent purifying natural selection can bias the number and type of mutations that remain in the population, especially for mutations that occur in coding regions (reviewed in Nishant et al. 2009).
The DNA mismatch repair system improves the fidelity of DNA replication by about 1000-fold by excising DNA mismatches in the newly replicated strand that arise from polymerase misincorporation and slippage (reviewed in Modrich and Lahue 1996; Kunkel and Erie 2005; McCulloch and Kunkel 2008). Eukaryotes contain multiple MutS (MSH) and MutL (MLH) homologs (reviewed in Kunkel and Erie 2005). In Saccharomyces cerevisiae, two heterodimeric MutS homolog complexes, MSH2–MSH3 and MSH2–MSH6, act in mismatch recognition. MSH2–MSH6 is primarily involved in repairing base–base and small insertion/deletion loop mismatches. MSH2–MSH3 acts primarily on insertion/deletion loop mismatches up to 17 nt in length. In the presence of ATP, both MSH complexes interact primarily with MLH1–PMS1 to form a mismatch-MSH–MLH complex that interacts with downstream repair components. Recent work in humans and yeast suggests that MLH1–PMS1 contains an ATP–Mn2+-dependent latent endonuclease activity that acts near the mismatch and is essential for MMR, most likely in excision steps (Kadyrov et al. 2006, 2007). Null mutations in MSH2 and MLH1, the key partners in the MSH and MLH complexes, confer severe defects in MMR; reporter assays have shown that strains bearing these mutations display high rates of base substitutions and DNA slippages. For example, in an assay that measures frameshift mutations in homopolymeric runs, msh2Δ and mlh1Δ mutations confer mutation rates that are ∼10,000-fold higher than wild type (Marsischky et al. 1996; Tran et al. 1997, 2001; Gragg et al. 2002).
Our goal in this study was to analyze with limited bias the rate at which mutations occur in MMR-defective lines due to DNA polymerase errors during DNA replication, and to identify novel genomic features associated with these errors. The baker's yeast S. cerevisiae is an ideal model system in which to perform these studies because genetic analysis of many of the key MMR factors has been performed; more importantly the effect of null mutations in these factors has been extensively characterized using a variety of mutator assays (Kunkel and Erie 2005). Previously, one of our groups (Heck et al. 2006) grew wild-type and conditional mlh1 (mlh1-7ts) diploid strains of S. cerevisiae for 160 generations with bottlenecks that reduced the population size to one cell every 20 generations. These lines were grown at 35°, the nonpermissive temperature for mlh1-7ts. A conditional mlh1 allele was chosen instead of a null so that mutation accumulation in the absence of MMR could be limited to 160 generations by shifting cells at generation 160 to the permissive temperature for MMR function. The mlh1-7ts mutation contains two mutations within the ATP-binding domain of MLH1 (K67A, D69A). Unlike mlh1Δ strains that display poor spore viability due to defects in meiotic crossing over, mlh1-7ts lines display wild-type spore viability at the permissive temperature. Such a phenotype allowed us to easily identify recessive lethal mutations (Heck et al. 2006). At the nonpermissive temperature, the mlh1-7ts mutation conferred a phenotype similar to the null in the canavanine resistance mutation assay and a mutator phenotype in the lys2-A14 reversion assay that was 1000-fold higher than MLH1 but 4-fold lower than the null (Heck et al. 2006); J. Heck and E. Alani, unpublished observations).
Tetrad analysis showed that the mlh1-7ts bottleneck lines would be ideally suited for a high-throughput DNA sequencing approach that would identify mutagenesis patterns. First, the wild-type lines maintained high spore viability (∼94%) at generation 160. In contrast, mlh1-7ts lines displayed spore viabilities that ranged from 1.1 to 77%, demonstrating that the lines had accumulated recessive lethal mutations. Second, comparative genome hybridization (CGH) and pulse-field (PFGE) analyses of the mlh1-7ts strains indicated that they did not undergo major genome rearrangements (Heck et al. 2006). Third, because the lines were grown as diploids for a limited number of generations, secondary mutations, dominant or recessive, that alter the rate or type of mutagenesis should rarely occur. Also, because there is no sexual reproduction and mutations should clonally propagate after escaping the initial bottleneck, newly arising mutations should appear as heterozygous sites. Finally, the above strategy should limit biases in mutation accumulation because the diploid cells were grown in rich media under minimal selection pressure where deleterious mutations could accumulate (Heck et al. 2006).
As described below, a Bayesian method was developed to detect heterozygous mutations in one wild-type and three mlh1-7ts lines using whole-genome sequencing. We detected 28 heterozygous single-nucleotide polymorphisms (SNPs) and 48 single-nt insertion/deletion (indels) in the mutator lines, all of which mapped to homopolymeric runs of nucleotides (HP tracts). The mutation spectra match closely with that seen in MMR defective strains using different reporter constructs (Marsischky et al. 1996; Tran et al. 1997, 2001). This demonstrates that the mutation pattern seen previously using a limited number of reporters holds true for the entire genome. In addition, we were able to correlate genotype to phenotype for one locus in one mutator line. Together this work provides new insights into how mismatch repair can shape genome stability and dynamics, mutation mechanisms, and evolution.
MATERIALS AND METHODS
Whole-genome sequencing analysis of Mut lines:
Bottleneck experiments involving 10 independent wild-type (MATa/MATα, his3/HIS3, LEU2/leu2, cyhr/cyhs, ade2/ADE2, ura3/ura3, trp1/trp1) and mlh1-7ts (MATa/MATα, mlh1-7∷KanMX4/mlh1-7∷KanMX4, his3/HIS3, LEU2/leu2, cyhr/cyhs, ade2/ADE2, ura3/ura3, trp1/trp1) lines were performed previously (Heck et al. 2006). Three of the 10 mlh1-7ts lines at generation 160 were analyzed by whole-genome sequencing. These lines were chosen to ensure a reasonable sample set of mutations and displayed a lower range of spore viabilities (2.5–15.6%) following tetrad dissection compared to the entire set (1.1–77%).
Whole-genome sequencing was performed at the Cornell University Life Sciences Core Laboratory Center (CLC) using an Illumina Genome Analyzer (http://www.illumina.com). Yeast genomic DNA for whole genome sequencing was prepared using a Qiagen genomic DNA preparation kit (http://www.qiagen.com). Sequencing was performed using the Illumina pipeline for 36-bp single-end reads. Reads were aligned onto the S288c genome (http://genome.ucsc.edu/cgi-bin/hgGateway) using Novoalign (http://www.novocraft.com), a program that performs a gapped alignment with high specificity and sensitivity.
Detection of DNA sequence heterozygosity using a Bayesian approach:
We analyzed five diploid strains in this study: a wild-type strain at generations 0 and 160 (Wt0, Wt160) and three derived mlh1-7ts mutator lines grown vegetatively (i.e., no meiosis) and bottlenecked to one cell every 20 generations until generation 160 (Mut2, Mut3, Mut4). Several aspects of the experiment required us to develop a novel approach for calling genotypes from the sequencing data. First, the initial wild-type strain (Wt0) likely contained SNPs and indels that distinguish it from the reference yeast genome. Because all lines were grown vegetatively, they were all expected to have these “propagated” SNPs and indels. Thus reads from the five sequenced lines were used to identify these variants. Furthermore, we expect new mutations (as this those occurring in Wt160, Mut2, Mut3, or Mut4 during generations 1–160) to be heterozygous at the end of the experiment and few, if any, variants are expected to be shared (as this would require independent hits in replicate lines). Finally, the sequencing depth (∼8–20×) suggests moderate but not exceptional power to detect heterozygous mutations from the sequence of a single line on its own. Therefore, we developed a Bayesian SNP caller that (1) aligns all reads to the genome and (2) uses read depth and quality scores at a given position to call genotypes for all five lines simultaneously.
Importantly, our Bayesian model allows us to distinguish between a propogated mutation, (defined as a variant seen in all five strains in either heterozygous or homozygous state from Wt0) and a derived mutation, defined as a DNA sequence variant that arose in only a single line. First, we indexed the five diploid strains as s = 1, 2, 3, 4, 5 for Wt0, Wt160, Mut2, Mut3, and Mut4, respectively. We set the prior probability of strain s being heterozygous as Priors = 10−7, 10−8, 10−5, 10−5, 10−5 for s = 1, 2, 3, 4, and 5, respectively, according to mutation rates previously determined in wild-type and mismatch-repair defective organisms (Denver et al. 2005; Iyer et al. 2006; Nishant et al. 2009). It is important to note that Wt160 was assigned a lower prior probability of being heterozygous relative to Wt0. This is because a heterozygosity in Wt0 is defined as the difference between the Wt0 strain (Heck et al. 2006) and the S288c reference genome (http://genome.ucsc.edu/cgi-bin/hgGateway). There were a significant number of differences between the two strains. On the other hand, a heterozygosity in Wt160 was defined as one that occurred during the bottleneck experiment (propogated). Because there were only 160 generations between Wt0 and Wt160, we expected the number of differences between the lines to be small; in fact, none were detected.
At a given locus, let A and a be the major and minor allele types, respectively, based on the allele counts from all the strains. Let Ns be the total number of alleles observed for strain s; let Aj,s be the type of the jth allele copy among these Ns alleles, j = 0, 1, …, Ns. Let ej be the probability that the jth allele has been assigned the wrong allele type. We estimated ej from the error rates given by Dohm et al. (2008) for 36-bp Solexa reads as a function of read position.
To call SNPs and indels in Wt0, we used the allele count data from Wt0 along with that from the other four strains. The posterior probabilities of a given genomic position being homozygous or heterozygous in Wt0 are
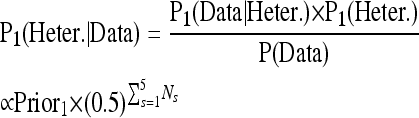 |
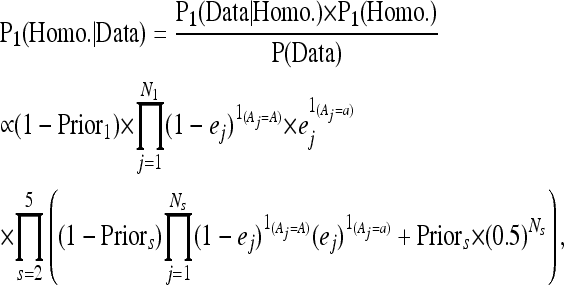 |
where Ps(·) denotes the probability in the context of strain s. On the basis of the posterior probabilities above, we classified each locus as homozygous or heterozygous for Wt0. If a locus was classified as heterozygous for Wt0, then it was assumed to have a propagated mutation in the rest of the strains. To call derived mutation in strains s = 2, 3, 4, 5, we use similar logic:
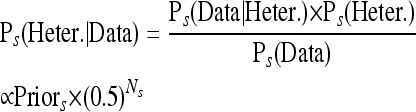 |
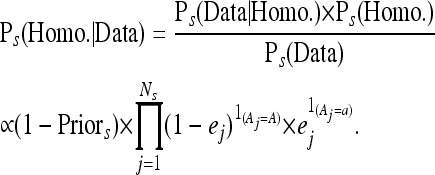 |
We use the posterior probabilities calculated above, to make a decision as to whether a site is called as heterozygous for a new mutation, heterozygous for a propagated mutation, or invariant for the four evolved strains: s = 2, 3, 4, 5. Specifically, if the posterior probability of heterozygosity was greater than 50% at a given position, then we classified the site as containing a SNP or indel. Visual inspection of the alignments for some of the inferred indel positions revealed that pairwise alignment of reads could induce false positives across multiple lines due to variations on how the alignment software interprets the alignment of different reads around a given position. These are characterized by one allele count being much smaller (but nonzero) compared to the other, across multiple strains. To bioinformatically cull such sites from our data set, we carried out an additional likelihood-ratio test for the allele frequencies to be equal (i.e., a propagated SNP had to have statistical support for the model of 50% frequency across Wt0, Wt160, Mut2, Mut3, and Mut4; a derived SNP had to have statistical support for 50% in one of the evolved lines, and 0% in all the others). If the hypothesis of equality was rejected for an indel, we flagged it as low confidence (Figure 1).
Figure 1.—

Flow chart describing bioinformatic methods used to identify heterozygous mutations from Illumina GA whole-genome sequencing. See text for details.
We expected, on the basis of previous estimates of mutation rate in MMR defective strains, to find ∼125 mutations for each of the MMR deficient strains (approximately one mutation per line generation). This corresponds to a prior mutation rate of 10−5 mutations/site/generation. However, we detected 12, 24, 40 mutations for each of the MMR-deficient strains, which yield mutation rates of 1 × 10−6, 2 × 10−6, and 3 × 10−6 in each line, respectively. Although our estimated prior values differ somewhat from the real data, the alignment analysis allowed us to calculate very accurate posterior subjective probabilities. This accuracy is due to the large number of observations and has in practice made the influence of the prior negligible. Thus given the high coverage for the Mut lines, the difference in our prior estimates does not influence our analysis. Even with low coverage data where accurate estimates of prior are critical, a higher prior value would yield a larger number of false positives. The majority of mutations (and all low confidence mutations) were verified by Sanger sequencing, suggesting that false positives were rare, but we may have false negatives (i.e., missed variants) due to the medium coverage (∼8–20×) of the lines.
Simulation study:
To estimate the false-positive and false-negative rates, as well as to check our bioinformatics and SNP/indel calling pipelines, we set up a simulation to test the accuracy of our Bayesian approach. We started with a complete genome of a yeast S288c strain (http://genome.ucsc.edu/cgi-bin/hgGateway; June 2008 assembly from the Saccharomyces Genome Database (SGD, http://www.yeastgenome.org/) and introduced SNPs and indels to simulate five strains: Wt0, Wt160, Mut2, Mut3, and Mut4. To simulate Wt0, we duplicated the S288c genome to create a diploid. We then randomly selected nμ and nd positions for SNPs and indels respectively. (nμ = 2, nd = 8; the values of nμ and nd were chosen to mimic changes between S288c and the Wt0 strain used in the bottleneck experiment). One of the two copies of S288c was randomly selected to incur each SNP or indel. For an indel mutation, the nt in that copy was deleted, or a new randomly chosen allele was inserted after it. For a SNP position, the nt was randomly changed to another nt. The resulting two copies of the genome were defined as the Wt0 diploid. The other four strains were all simulated directly from Wt0 by introducing SNPs and indels in the two copies of Wt0. The mechanism of adding SNPs and indels was exactly as described above. The values of nμ and nd for each of the simulations are given below. These values mimic the number of mutations that were expected in the bottleneck experiments. One distinction between the simulations and the real data is that the SNPs and indels in the simulations were not introduced into HP tracts. As described below, we believe that our ability to detect indels in HP tracts is lower because indels in HP tracts can be identified only if the entire tract and sequence flanking both sides are present in a 36-nt read.
Table 4.
| 0 → Wt0 | Wt0 → Wt160 | Wt0 → Mut2 | Wt0 → Mut3 | Wt0 → Mut4 | |
|---|---|---|---|---|---|
| nμ | 2 | 1 | 25 | 25 | 25 |
| nd | 1 | 1 | 100 | 100 | 100 |
Next, we simulated 32-nt Illumina GA reads from each of the five strains by randomly choosing read-start positions and copying 32 nt of strain s starting from that position. For each strain, the number of reads simulated matches the coverage achieved in the real sequencing experiment. We also simulated a quality score for each position of each read, following the error rate distribution given in Dohm et al. (2008). The reads were aligned with S288c using Novoalign (http://www.novocraft.com). Based on the alignment, we listed the allele counts and associated quality scores in each of the variable, potentially heterozygous, positions. We used this list as the input to a computer program created on the basis of our method of heterozygosity detection, which went through all the steps described in the last section. The rates of false positives and negatives (based on the output of the program) are given in Table 1. We believe that these rates are similar to those seen in the bottleneck experiment.
TABLE 1.
False-positive and -negative rates based on the simulation analysis
| False-positive rate (in units of no. of SNP calls) | False-negative rate (in units of no. of SNP calls) | |
|---|---|---|
| Mutant | 6 × 10−5 | 0.030 |
| Indel | 0 | 0.089 |
| Total | 6 ×10−5 | 0.078 |
| Propagated | 0 | 0 |
| Derived | 6 ×10−5 | 0.091 |
| Total | 6 ×10−5 | 0.078 |
See materials and methods for details.
Verifying mutations identified using Bayesian method:
Our method for heterozygous mutation calling from the whole-genome sequencing data yielded both low- and high-confidence predictions (see above). All low-confidence predictions (10 in total) were verified and either validated (n = 4) or disproved (n = 6) using Sanger sequencing. Briefly, to assay heterozygous mutations predicted from the whole-genome sequence data, genomic DNA was prepared from wild-type generation zero, and mutation accumulation lines Mut2, Mut3, and Mut4 using standard techniques. Approximately 400 bp of DNA flanking the predicted mutated site was amplified in all lines using PCR and Sanger sequenced at the Cornell CLC using an Applied Biosystems Automated 3730 DNA analyzer. The sequencing traces were all analyzed visually. A heterozygous base change mutation was confirmed if a doublet representing both alleles was observed only in the sequencing trace of the predicted Mut line, but all other lines showed only a singlet representing the parental allele. A heterozygous indel mutation was confirmed if the sequencing reaction failed (i.e., tall singlet peaks fall to small doublet peaks or random noise) at the predicted location only in the predicted Mut line, but the sequencing reactions in all other lines were able to successfully sequence past the site.
For the high confidence predictions, 31 (of 65) were sequenced and verified using the methods described above. Of those 31 mutations, 10 were further verified by genotyping the haploid progeny of the diploid containing the heterozygous mutation via Sanger sequencing. Both alleles comprising the heterozygote were observed in the haploid progeny with the exception of the frameshift mutation in the essential MDN1 gene. Six additional high-confidence predictions were also verified by genotyping the haploid progeny of the heterozygous diploid.
By Sanger sequencing of the diploid lines (see above) we also found and verified four heterozygous mutations that were detected in earlier, less accurate prediction protocols that were not found using the final more stringent prediction method.
RESULTS AND DISCUSSION
Identification of mutations in diploid bottleneck lines using maximum-likelihood and Bayesian methods:
One wild-type and three mlh1-7ts lines (Mut2, Mut3, and Mut4) allowed to accumulate mutations for 160 generations were sequenced using the Illumina genome analyzer technology (materials and methods; http://www.illumina.com). The wild-type progenitor of all the strains was also sequenced. The analysis was performed with three independent mlh1-7ts lines to control for chance associations within an individual line and for mutations that could alter the mutation rate of a given line. The Mut2, Mut3, and Mut4 lines at generation 160 displayed 15.6, 7.1, and 2.5% spore viability, respectively (Heck et al. 2006). As shown below and in Tables 2 and 3, our data analysis indicated that the mutation spectra and rates in the three mlh1-7ts lines were indistinguishable. In total, 25 million, out of 35 million sequenced, 36 nt sequence reads were uniquely mapped to the yeast genome, allowing up to two mismatches per read (materials and methods). The wild-type and Mut2 generation 160 strains were sequenced to 9× and 8× average genome coverage depth, respectively. Mut3 (160) and Mut4 (160) were sequenced to average depths of 18× and 22×, respectively. We then developed and employed an “experiment aware” probabilistic framework using maximum-likelihood and Bayesian methods that utilized sequence coverage of the entire data set (∼70-fold; Figure 1; materials and methods; Dohm et al. 2008). Briefly, the approach classifies each site in the yeast genome with uniquely mapping reads into one of three categories: (1) invariant across all strains, (2) heterozygous in the wild-type (and all derived strains), which we term “propagated” SNPs or indels, or (3) heterozygous in one of the mutant strains, which we term “derived” SNPs or indels. As described below, this method allowed us to pool experimental data across sequencing runs for all strains and detect with high reliability heterozygous SNPs (28 identified) and single-nt indels (48 identified) from the 36-nt read data set. This method was evaluated on simulated data sets and found to have a very low false-positive rate (∼6 × 10−5) and a false-negative rate of 0.08 within the unique mapping regions of the genome that contained at least sevenfold coverage (Table 1). The low false-positive rate was verified by PCR amplifying genomic fragments covering a specific mutation site and then confirming the presence of a heterozygous mutation by Sanger sequencing the fragment (materials and methods). On the basis of simulations, we estimated that the method, as applied to regions with at least sevenfold sequencing coverage, allowed us to detect heterozygous mutations in 60, 41, 69, and 84% of the total genome for the generation 160 wild-type, Mut2, Mut3, and Mut4 lines, respectively.
TABLE 2.
Genome location of mutations detected in the Mut2, 3, and 4 lines
| Amino acid change | Distribution of sequence reads |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | SGD Position | HP tract | Strain | Gene | Mutation | A | G | T | C | Indel | |
| 1 | 139,349–139,358 | 10 | 4 | del | 1 | 11 | 8 | ||||
| 2 | 275,549–275,557 | 9 | 2 | del | 3 | 3 | |||||
| 2 | 92,271–92,279 | 9 | 3 | del | 1 | 10 | 8 | ||||
| 2 | 92,271–92,279 | 9 | 2 | del | 3 | 0 | |||||
| 2 | 423,462–423,469 | 8 | 3 | ins | 8 | 16 | |||||
| 2 | 662,560–662,569 | 10 | 4 | YBR219C | fs; 103–106 | del | 6 | 5 | |||
| 2 | 653,035–653,045 | 11 | 4 | del | 13 | 9 | |||||
| 3 | 212,451–212,457 | 7C | 3 | YCR048W/ARE1 | fs; 176–178 | del | 15 | 11 | |||
| 3 | 275,289 | NA | 4 | YCR091W/KIN82 | 297; V to I | G to A | 12 | 15 | |||
| 4 | 512,796 | NA | 2 | A to T | 8 | 7 | |||||
| 4 | 814,336 | NA | 2 | G to A | 10 | 6 | |||||
| 4 | 929,182–929,193 | 12 | 3 | del | 1 | 5 | 4 | ||||
| 4 | 963,768 | NA | 3 | YDR252W/BTT1 | 120: G to V | T to G | 12 | 18 | |||
| 4 | 231,908–231,914 | 7 | 4 | del | 12 | 12 | |||||
| 4 | 1,386,657–1,386,664 | 8 | 4 | del | 17 | 11 | |||||
| 4 | 470,576–470,584 | 9 | 4 | del | 19 | 12 | |||||
| 4 | 832,716–832,726 | 11 | 4 | del | 15 | 7 | |||||
| 4 | 50,592–50,603 | 12 | 4 | del | 10 | 9 | |||||
| 1,054,759 | NA | 4 | A to T | 9 | 11 | ||||||
| 5 | 305,972 | NA | 2 | C to A | 5 | 5 | |||||
| 5 | 479,369 | NA | 2 | YER155C/BEM2 | 1159; S to C | T to A | 8 | 3 | |||
| 5 | 225,319–225,327 | 9 | 3 | Del | 9 | 7 | |||||
| 5 | 403,576 | NA | 3 | YER122C/GLO3 | 258; A to E | G to T | 8 | 14 | |||
| 5 | 34,325–34,333 | 9 | 4 | del | 11 | 7 | |||||
| 5 | 402,832–402,843 | 12 | 4 | del | 13 | 7 | |||||
| 6 | 223,108–223,118 | 11 | 3 | del | 10 | 7 | |||||
| 6 | 114,200–114,210 | 11 | 4 | del | 6 | 7 | |||||
| 6 | 88,832 | NA | 4 | YFL024C/EPL1 | 504; D to E | G to T | 15 | 13 | |||
| 6 | 225,229 | NA | 4 | YFR034C/PHO4 | 240; R to G | G to C | 9 | 8 | |||
| 7 | 194,092–194,098 | 7 | 3 | YGL163C/RAD54 | fs; 771–773 | del | 13 | 13 | |||
| 7 | 878,690–878,701 | 12 | 3 | del | 6 | 4 | |||||
| 7 | 653,363–653,369 | 7 | 4 | del | 14 | 10 | |||||
| 7 | 882,549–882,558 | 10 | 4 | del | 10 | 6 | |||||
| 7 | 20,017–20,027 | 11 | 4 | del | 10 | 5 | |||||
| 7 | 678,172–678,182 | 11 | 4 | del | 11 | 9 | |||||
| 8 | 150,380–150,386 | 7 | 3 | del | 11 | 1 | 12 | ||||
| 8 | 472,612–472,624 | 13 | 3 | del | 10 | 8 | |||||
| 8 | 288,299 | NA | 3 | YHR092C/HXT4 | 172; K to K | C to T | 5 | 6 | 1 | ||
| 8 | 370,253 | NA | 3 | YHR132W-A/IGO2 | 46: Y to F | A to T | 15 | 12 | 1 | 1 | |
| 9 | 270,327 | NA | 2 | YIL046W/MET30 | 560; A to S | G to T | 5 | 4 | |||
| 9 | 375,856 | NA | 3 | YIR010W/DSN1 | 143; M to I | G to A | 7 | 10 | |||
| 9 | 199,995 | NA | 4 | YIL087C/AIM19 | 41; T to I | G to A | 12 | 18 | |||
| 10 | 445,012–445,020 | 9 | 3 | del | 10 | 6 | |||||
| 10 | 131,051–131,059 | 9 | 4 | del | 11 | 15 | |||||
| 10 | 469,684–469,694 | 11 | 4 | del | 19 | 6 | |||||
| 11 | 162,688–162,695 | 8 | 4 | del | 9 | 1 | 7 | ||||
| 11 | 403,466 | NA | 4 | YKL018C-A | 19; S to S | C to T | 13 | 13 | |||
| 12 | 405,712–405,719 | 8 | 2 | YLR131C/ACE2 | fs; 369–371 | del | 4 | 3 | |||
| 12 | 32,320-32,330 | 11 | 3 | del | 9 | 5 | |||||
| 12 | 964,065 | NA | 3 | YLR420W/URA4 | 95: R to H | G to A | 11 | 12 | |||
| 12 | 1,009,007 | NA | 3 | YLR436C/ECM30 | 746; I to V | T to C | 12 | 12 | |||
| 12 | 363,531-363,537 | 7 | 4 | YLR106C/MDN1 | fs; 68–70 | del | 11 | 13 | |||
| 12 | 201,846-201,856 | 11 | 4 | del | 14 | 9 | |||||
| 12 | 1,047,741 | NA | 4 | YLR454W/FMP27 | 1249: D to G | A to G | 10 | 12 | 1 | ||
| 13 | 763,010-763,016 | 7 | 2 | SNR86 (small nucleolar RNA) | ins | 5 | 5 | ||||
| 13 | 241,855-241,867 | 13 | 3 | del | 12 | 6 | |||||
| 13 | 311,843 | NA | 3 | C to T | 15 | 25 | |||||
| 13 | 139,705-139,709 | 5 | 4 | YML067C/ERV41 | fs; 138–139 | del | 15 | 13 | |||
| 13 | 816,457-816,463 | 7 | 4 | YMR275C/BUL1 | fs; 706–708 | del | 11 | 9 | |||
| 14 | 761,792 | NA | 2 | YNR069C/BSC5 | 267; V to V | C to T | 8 | 6 | 1 | ||
| 14 | 222,733 | NA | 3 | YNL225C/CMN67 | 580; V to M | C to T | 1 | 17 | 10 | ||
| 14 | 435,595-435,601 | 7 | 4 | YNL101W/AVT4 | fs; 199–201 | del | 9 | 10 | |||
| 14 | 481,123-481,129 | 7 | 4 | del | 15 | 8 | |||||
| 14 | 685,574-685,582 | 9 | 4 | del | 12 | 1 | 9 | ||||
| 14 | 575,616-575,626 | 11 | 4 | del | 12 | 8 | |||||
| 14 | 400,002 | NA | 4 | YNL121C/TOM70 | 180; G to STOP | C to A | 15 | 15 | |||
| 14 | 734,521 | NA | 4 | YNR058W/BIO3 | 77; L to L | A to G | 11 | 10 | |||
| 15 | 854,146-854,153 | 8 | 2 | del | 4 | 5 | |||||
| 15 | 874,052-874,057 | 6 | 3 | YOR296W | fs; 1284–1286 | del | 10 | 7 | |||
| 15 | 767,667-767,673 | 7 | 3 | YOR228C | fs; 36–38 | del | 10 | 7 | |||
| 15 | 822,829-822,835 | 7 | 3 | YOR267C/HRK1 | fs; 678–680 | del | 11 | 8 | |||
| 16 | 146,421-146,427 | 7 | 2 | YPL216W | fs; 868–870 | del | 7 | 5 | |||
| 16 | 22,677 | NA | 4 | C to T | 20 | 14 | |||||
| 16 | 131,583 | NA | 4 | YPL222W/FMP40 | 475; A to T | G to A | 11 | 15 | 1 | ||
| 16 | 509,632 | NA | 4 | YPL022W/RAD1 | 980; A to S | G to T | 19 | 18 | 1 | ||
| 16 | 570,131 | NA | 4 | YPR007C/REC8 | 415; S to M | G to A | 11 | 11 | |||
The type of mutation [base substitution, single-nt insertion (ins), single-nt deletion (del)] is shown, as well as the length of the HP tract that contains an indel. The specific Mut line (2, 3, or 4) is indicated under “strain.” All HP tracts were poly(A) or poly(T) except for the mutation in chromosome 3 at 212,451–212,457, which involved a poly(C) tract. For mutations that occurred within an open reading frame, both the gene name and predicted amino acid changes (fs, frameshift) are provided. NA, not applicable. Coordinates are presented as shown in the SGD (http://www.yeastgenome.org/). The number and distribution of the sequence reads are presented for each mutation. The frameshift mutation in YLR106C/MDN1 conferred a recessive lethal phenotype (data not shown).
TABLE 3.
Mutation rates for Mut2, Mut3, and Mut4 lines grown in bottlenecks for 160 generations
| Base substitution mutations | ||||
|---|---|---|---|---|
| Strain | No. mutations | % genome ≥7× coverage | Genome Size (bp) adjusted | Mutation rate (per base per gen ×10−9) |
| Mut2 | 6 | 41 | 9,898,136 | 3.8 |
| Mut3 | 9 | 69 | 16,657,838 | 3.4 |
| Mut4 | 13 | 84 | 20,279,107 | 4.0 |
| Average | 3.7 | |||
| Single-nucleotide indel mutations in 5- to 13-nt HP tracts | ||||
| Strain |
No. mutations |
No. HP tracts >7× coverage |
Mutation rate (per HP tract/generation × 10−7) |
|
| Mut2 | 6 | 57,502 | 6.5 | |
| Mut3 | 15 | 99,714 | 9.4 | |
| Mut4 | 27 | 122,816 | 14 | |
| Average | 10 | |||
| Single-nucleotide indel mutations in 8- to 13-nt HP tracts | ||||
| Strain |
No. mutations |
No. HP tracts >7× coverage |
Mutation rate (per HP tract/generation × 10−7) |
|
| Mut2 | 4 | 2,820 | 89 | |
| Mut3 | 10 | 7,054 | 89 | |
| Mut4 | 19 | 8,696 | 140 | |
| Average | 110 | |||
The base substitution mutation rate was determined by calculating the percentage of the genome in which at least sevenfold DNA sequencing coverage to unique regions was obtained. This was done because our statistical analysis did not have sufficient power to reliably detect heterozygous mutations in regions with lower coverage. This information was used to calculate the mutation rate on the basis of the following formula: (number of mutations)/(160 generations)/(adjusted genome size), with the diploid S. cerevisiae genome size determined as 24,141,794 bp (http://www.yeastgenome.org/). To obtain indel mutation rates, we first determined the number of HP tracts of a given length in unique regions of the genome which had ≥ sevenfold sequence coverage. We then used the following equation to calculate mutation rate: (number of indels)/(160 generations)/(number of HP tracts with ≥ sevenfold coverage).
We did not detect any mutations in the wild-type generation 160 line, which was predicted on the basis of the previously calculated mutation rate of 3.3 × 10−10 mutations/base/generation (<1 expected; Lynch et al. 2008). As shown in Tables 2 and 3 and Figure 2, only heterozygous mutations, composed of 28 base substitution and 48 single-nt indel mutations, were detected in the three MMR-defective lines. All of the mutations were unique between lines except for a single-nt deletion mutation between SGD (http://www.yeastgenome.org) coordinates 92,271–92,279 on chromosome 2, which occurred independently in both Mut2 and Mut3 (Table 2). All 48 indels, composed of 46 deletions and 2 insertions, occurred in HP tracts [47 poly(A) or (T) tracts, 1 poly(G) or (C) tract] between 5 and 13 bp long (Table 2). Due to the constraints of using 36-nt Illumina GA reads, we do not have the power to detect mutations in HP tracts larger than 13 nt, but <400 such tracts are present in the yeast genome. Visual inspection of the DNA sequences surrounding the indel mutations (∼400 bp; Figure 2) suggested that they were enriched for HP runs. These are primarily poly(dA:dT) tracts that are present in the yeast genome at a 20-fold higher frequency than poly(dG:dC) tracts. Consistent with this, the AT content of the genomic regions surrounding the indel mutations was significantly higher than that for unmutated HP regions (windows up to 500 bp; data not shown). Detailed bioinformatic and genetic analyses will be required to determine if this pattern is significant; however, a previous study (Harfe and Jinks-Robertson 2000) showed that DNA polymerase slippage was not greatly influenced by sequence context, including nearby HP tracts.
Figure 2.—

The 100-bp region surrounding indel mutations in the Mut3 and Mut4 lines. The locations of the indel mutations are indicated in black boldface type. HP runs of ≥5 in this window are color coded as shown: red, An; blue, Tn; green, Cn.
Our analysis permitted the detection of up to two single-nt indels in a 36-nt read; these indels can be right next to each other to create a 2-nt indel or separated from each other. We assigned this limit because creating high-quality and unique alignments became very difficult when allowing indels larger than 2 nt. We were unable to detect indels of 2 nt in any of the lines. Such a result is not surprising due to previous studies of wild-type and MMR mutants analyzed for reversion of frameshift mutations in HP runs. In these studies the overwhelming majority of mutations involved single-nt deletions. For example Tran et al. (1997) found that 225 of 227 reversions in +1 HP tracts in wild-type, polymerase proofreading, and mismatch repair mutants were due to deletions of a single nt. For −1 HP tracts, they found that 206 of 218 reversions were due to additions of a single nt. The remaining revertants in both HP tracts involved expansions or contractions of no greater than 2 nt in size.
The predominance of single-nt deletions over single-nt insertions and base substitutions was similar to previous reports for the mutational spectra in reporter genes in MMR null mutants (Marsischky et al. 1996; Tran et al. 1997, 2001; Denver et al. 2005). The average mutation rate in the 5- to 13-bp HP tracts was 1.0 × 10−6/HP tract/generation (Table 3). The rate was an order of magnitude greater (1.1 × 10−5) if only runs between 8 and 13 bp long were considered (Table 3). These values approach the rates seen in MMR-defective yeast (mlh1, msh2) containing reporters bearing 10-bp poly(T) (2.8 × 10−4; Tran et al. 1997) and 10-bp poly(A) (7.3 × 10−5; Gragg et al. 2002) tracts. Low-sequence coverage provides one explanation for why the rate is lower than those seen previously in reporter assays. In our analysis, indels in HP tracts can be identified only if the entire tract and sequence flanking both sides are present in a 36-nt read; the longer the HP tract, the less likely it is to obtain reads that cover the entire tract. Thus higher sequence coverages are required to identify indels in HP tracts. Consistent with this, a higher indel mutation rate was seen in lines that had higher sequencing coverage (Table 3). In contrast, SNPs that occur outside of an HP tract should not be as affected by sequence coverage (aside from the relationship between coverage and probability of detecting sufficient copies of the alternate base to reliably make a call). This was seen for the analysis of base substitutions (Table 3).
The average rate of base substitution mutations in mlh1-7ts was 3.7 × 10−9 mutations/base/generation (Table 3), which is 11-fold higher than the base substitution rate observed in wild-type haploid strains (Lynch et al. 2008). Of the 28-base substitution mutations detected in the Mut2–4 lines, 16 were transitions and 12 were transversions (Table 2). Nineteen of these mutations resulted in a change from a G–C to an A–T base pair, whereas only 4 were in the opposite direction. This overall mutational bias toward A–T base pairs was seen and discussed previously (e.g., Lynch et al. 2008; Denver et al. 2009; Keightley et al. 2009). The modest increase that we observed in the base substitution rate in MMR defective strains is significantly lower than predicted (∼100-fold increase for base substitutions and frameshifts; Denver et al. 2005; Iyer et al. 2006). We suggest two reasons for these differences. First, our measurements were determined from a genome-wide measurement rather than by extrapolation from a few marker loci. Second, the mlh1-7ts allele is not a complete null mutation. It phenocopies the mlh1Δ phenotype in the CAN1 mutational assay, but has a fourfold lower mutation rate than mlh1Δ in the lys2-A14 reversion assay (Heck et al. 2006; data not shown). Because mlh1-7ts strains display residual DNA repair, it is possible that there is a bias toward the repair of specific mismatches in these strains. While we cannot rule this out, the fact that the mutation signature seen in mlh1-7ts appeared indistinguishable from mlh1 null strains argues against such a possibility (Marsischky et al. 1996; Tran et al. 1997, 2001). Finally, we cannot rule out the possibility that mutation rates in MMR-defective strains are different in haploid vs. diploid yeast, although a recent analysis of mutation rates in diploid bottleneck lines showed that wild-type diploid yeast displayed an estimated base substitution rate that was very similar to that reported previously for haploid yeast (Lynch et al. 2008; Nishant et al. 2010).
Because the three lines showed viability that ranged from 2.5 to 15.6%, we expected to identify mutations that conferred a lethal phenotype. We examined whether any of the mutations that mapped to open reading frames in the Mut4 line (2.5% viability) were not detected in haploid progeny. This was done by sequencing DNA surrounding a particular mutation in 20 viable spore clones obtained by sporulating the Mut4 generation 160 line. Of these 14 mutations, only the frameshift mutation in MDN1 was not detected, consistent with previous work showing that mdn1Δ mutants are inviable (Giaever et al. 2002). While it is unclear how many mutations would confer lethality in the absence of other mutations, the assortment of 5 independent lethal mutations would result in 3% spore viability, similar to that seen in the Mut4 line. We hypothesize that other lethal mutations were not identified in Mut4 and other lines because:
A large number of frameshift mutations in HP tracts may not have been detected because indels can be identified only if the entire tract and sequence flanking both sides are present in a 36-nt read. Identifying indels in HP tracts is very challenging using short-read sequencing. However, increasing sequence coverage and using paired-end reads of a larger size (∼180 bp) should provide a good test of this idea.
Our sequence analysis did not cover the entire genome (84% for Mut4).
While previous CGH and PFGE analyses (∼1-kb resolution; Heck et al. 2006) did not reveal rearrangements, it is possible that mutations that involved indels larger than two nt and smaller than 1 kb occurred. However, we find this to be less likely because a previous analysis of mutation spectra in MMR mutants indicated that indels greater than two nt are extremely rare (Tran et al. 1997).
Closing thoughts:
In the S. cerevisiae S288c haploid genome there are over 77,425 HP tracts five nt or greater. Frameshift mutations in coding regions that disrupt protein function are likely to have significant effects on organism fitness. In wild-type yeast, insertion/deletion mutations appear to be relatively rare compared to base substitutions; comparative analyses of multiple domestic and wild yeast strains identified ∼14,000 indels compared to ∼235,000 SNPs (Wei et al. 2007; Liti et al. 2009). In contrast, MMR mutants display a strong bias toward frameshifts over base substitutions in the genome. Thus our data, together with previous work, illustrate the critical role that MMR plays in preventing frameshifts in HP tracts across the genome.
Acknowledgments
We thank Amit Indap and the Cornell Core Laboratory Center (CLC), especially Peter Schweitzer, James VanEe, and Tom Stelick, for preparing samples for Illumina GA sequencing and bioinformatic analyses; Julie Heck and K. T. Nishant for technical advice and providing unpublished data, and the Alani, Bustamante and Aquadro laboratories and Nadia Singh and Dan Barbash for comments on the manuscript. E.A., S.Z., and A.D. were supported by National Institutes of Health (NIH) GM53085. S.Z. was also supported by a Cornell Presidential Fellowship and an NIH training grant in Genetics and Development. A.D. was also supported by an NIH training grant in Biochemistry, Molecular and Cell Biology. X.M. and C.D.B were supported by NSF 0606461 and NSF 0701382. A.R. was supported by NIH grant RO1 HG003229. R.H. was supported by a National Science Foundation (NSF) Minority Postdoctoral Fellowship. Z.G. was supported by a startup fund from Cornell and NSF DEB-0949556. B.B. was supported by an NIH training grant to the Tri-Institutional Training Program in Computational Biology and Medicine.
References
- Arndt, P. F., T. Hwa and D. A. Petrov, 2005. Substantial regional variation in substitution rates in the human genome: importance of GC content, gene density, and telomere-specific effects. J. Mol. Evol. 60 748–763. [DOI] [PubMed] [Google Scholar]
- Datta, A., and S. Jinks-Robertson, 1995. Association of increased spontaneous mutation rates with high levels of transcription in yeast. Science 268 1616–1619. [DOI] [PubMed] [Google Scholar]
- Denver, D. R., S. Feinberg, S. Estes, W. K. Thomas and M. Lynch, 2005. Mutation rates, spectra and hotspots in mismatch repair-deficient Caenorhabditis elegans. Genetics 170 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver, D. R., P. C. Dolan, L. J. Wilhelm, W. Sung, J. I. Lucas-Lledó et al., 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc. Natl. Acad. Sci. USA 106 16310–16314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohm, J. C., C. Lottaz, T. Borodina and H. Himmelbauer, 2008. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 36 e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles et al., 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418 387–391. [DOI] [PubMed] [Google Scholar]
- Gragg, H., B. D. Harfe and S. Jinks-Robertson, 2002. Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 22 8756–8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison, R. C., K. M. Roskin, S. Yang, M. Diekhans, W. J. Kent et al, 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 13 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe, B. D., and S. Jinks-Robertson, 2000. Sequence composition and context effects on the generation and repair of frameshift intermediates in mononucleotide runs in Saccharomyces cerevisiae. Genetics 156 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawk, J. D., L. Stefanovic, J. C. Boyer, T. D. Petes and R. A. Farber, 2005. Variation in efficiency of DNA mismatch repair at diferent sites in the yeast genome. Proc. Natl. Acad. Sci. USA 102 8639–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck, J. A., D. Gresham, D. Botstein and E. Alani, 2006. Accumulation of recessive lethal mutations in Saccharomyces cerevisiae mlh1 mismatch repair mutants is not associated with gross chromosomal rearrangements. Genetics 174 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, R. R., A. Pluciennik, V. Burdett and P. L. Modrich, 2006. DNA mismatch repair: functions and mechanisms. Chem. Rev. 106 302–323. [DOI] [PubMed] [Google Scholar]
- Kadyrov, F. A., L. Dzantiev, N. Constantin and P. Modrich, 2006. Endonucleolytic function of MutLalpha in human mismatch repair. Cell 126 297–308. [DOI] [PubMed] [Google Scholar]
- Kadyrov, F. A., S. F. Holmes, M. E. Arana, O. A. Lukianova, M. O'Donnell et al., 2007. Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J. Biol. Chem. 282 37181–37190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., U. Trivedi, M. Thomson, F. Oliver, S. Kumar et al., 2009. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res. 19 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel, T. A., and D. A. Erie, 2005. DNA mismatch repair. Annu. Rev. Biochem. 74 681–710. [DOI] [PubMed] [Google Scholar]
- Liti, G., D. M. Carter, A. M Moses, J. Warringer, L. Parts, et al., 2009. Population genomics of domestic and wild yeasts. Nature 458 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. W., Sung, K. Morris, N. Coffey, C. R. Landry et al., 2008. A genome- wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. USA 105: 9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsischky, G. T., N. Filosi, M. F. Kane and R. Kolodner, 1996. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 10 407–420. [DOI] [PubMed] [Google Scholar]
- Matassi, G., P. M. Sharp and C. Gautier, 1999. Chromosomal location effects on gene sequence evolution in mammals. Curr. Biol. 9 786–791. [DOI] [PubMed] [Google Scholar]
- McCulloch, S. D., and T. A. Kunkel, 2008. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 18 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich, P., and R. S. Lahue, 1996. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Ann. Rev. Biochem. 65 101–133. [DOI] [PubMed] [Google Scholar]
- Nishant, K. T., N. D. Singh and E. Alani, 2009. Genomic mutation rates: what high-throughput methods can tell us. BioEssays 31 912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant, K. T., W. Wei, E. Mancera, J. L. Argueso, A. Schlattl et al., 2010. The baker's yeast diploid genome is remarkably stable in vegetative growth and meiosis. PLoS Genetics (in press). [DOI] [PMC free article] [PubMed]
- Stamatoyannopoulos, J. A., I. Adzhubei, R. E. Thurman, G. V. Kryukov, S. M. Mirkin et al., 2009. Human mutation rate associated with DNA replication timing. Nat. Genet. 41 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teytelman, L, M. B. Eisen and J. Rine, 2008. Silent but not static: accelerated base-pair substitution in silenced chromatin of budding yeasts. PLoS Genet. 4 e1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, H. T., J. D. Keen, M. Kricker, M. A. Resnick and D. A. Gordenin, 1997. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 17 2859–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, P. T., J. A. Simon and R. M. Liskay, 2001. Interactions of Exo1p with components of MutLa in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 98 9760–9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl, S., R. Machné and N. Goldman, 2008. Evolutionary footprints of nucleosome positions in yeast. Trends Genet. 24 583–587. [DOI] [PubMed] [Google Scholar]
- Wei, W., J. H. McCusker, R. W. Hyman, T. Jones, Y. Ning et al., 2007. Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc. Natl. Acad. Sci. USA 104 12825–12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, K. H., P. M. Sharp and W. H. Li, 1989. Mutation rates differ among regions of the mammalian genome. Nature 337 283–285. [DOI] [PubMed] [Google Scholar]