

Abstract
Over the past fifteen years, great strides have been made to understand the molecular basis of how heterotrimeric G proteins control the activity of their downstream targets. However, the mechanism by which heterotrimeric G proteins are activated by upstream G protein-coupled receptors remains obscure. Recent structural data provides more insight into this process and supports the idea that GPCRs, despite their small size, are sophisticated allosteric machines with multiple signaling outputs.
Background
Pioneering work by Rall and Sutherland in the late 1950’s revealed that hormones such as epinephrine and glucagon interact with receptors in liver cell membranes to elicit the production of cyclic-3′,5′-monophosphate (cAMP) by adenylyl cyclase 1. Over the next two decades, it was established that these integral membrane receptors, once activated, convey their signals in a GTP-dependent manner via a complex of three proteins known as the heterotrimeric G protein, or G αβγ2. The G α subunit shares homology with Ras and binds guanine nucleotides in a signal dependent manner, whereas the G βγ heterodimer has high affinity for the GDP-bound state of G α , and is released from G α · GTP after receptor activation. During the 1990’s, structural biologists began to tease apart the molecular basis for heterotrimeric G protein function, and high resolution crystal structures representing the G α subunit in its active (GTP bound), deactivated (GDP bound), and inactive (G α βγ complex) states were determined 3. In the activated state of G α , the γ-phosphate of GTP stabilizes the structure of the second helix of the Ras-like domain. This helix, known as switch II, is disordered in the GDP-bound state, and sequestered when in complex with G βγ . These observations were consistent with the idea that switch II is a critical component of the effector binding site, because it would provide an molecular explanation for how GTP hydrolysis is coupled to loss of effector binding and regulation. This hypothesis was confirmed by the structure of G α s in complex with the catalytic domains of adenylyl cyclase in 1997. Over the following ten years, structures of four other G α-effector complexes were determined, and in each case the effectors have followed suit and form extensive contacts with switch II 3
The Holy Grail
Although much progress has thus been made in understanding the molecular details of how G α subunits interact with and regulate the activity of their downstream targets, it is less clear how activated GPCRs initiate this process by catalyzing nucleotide exchange on G α βγ . The question is of great importance not only because it represents an essential, pharmacologically relevant interaction that can regulate nearly all aspects of eukaryotic cell physiology, but also because an atomic understanding of this process is key to unlocking the secrets of functional selectivity, the ability of different agonists to coerce distinct downstream effects from a single kind of GPCR 4. Functional selectivity at the level of the GPCR manifests itself in the differential ability of certain ligands to “bias” the conformation of the receptor such that it interacts better with either heterotrimeric G proteins or one of its other downstream targets 5, including G protein-coupled receptor kinases (GRKs), which phosphorylate activated GPCRs, and arrestins, which bind to these phosphorylated GPCRs and target them for clathrin-mediated endocytosis. Clearly, the conformational state of a receptor that optimally recognizes a heterotrimeric G protein is not necessarily the same as those that optimally recognize GRKs or arrestins.
The first crystal structure of a GPCR was that of bovine rhodopsin, reported in 2000 6. Whereas electron microscopy studies had successfully revealed the topology of the seven transmembrane spans of the receptor, the crystal structure provided the framework for understanding a large body of biochemical analyses and fostered many experiments aimed at understanding how specific side chains and water molecules within in the transmembrane region provide an allosteric conduit between the agonist and heterotrimeric G proteins binding sites. The rhodopsin structure, however, left the molecular mechanism of G protein coupling ambiguous, for the receptor comes with its own covalently-bound inverse agonist, 11-cis-retinal, a ligand that completely suppresses the basal activity of the receptor and locks the receptor in a rigid state. In particular, intracellular loop 3 (IL3), which connects the fifth and sixth transmembrane spans (TM5 and TM6) and is strongly implicated in heterotrimeric G protein binding, was poorly ordered. Indeed, subsequent crystal structures of rhodopsin yielded different configurations of this loop7.
Seven years later, two atomic structures of the β2-adrenergic receptor ( β2AR) were determined 8,9. Unlike rhodopsin, the β2 AR has significant basal activity, implying that the receptor can sample multiple conformational states in the absence of ligands, which hindered efforts to crystallize the protein. This problem was circumvented by replacing much of IL3 with the rigid structural domain of T4 lysozyme, or by using an Fab fragment that recognizes the same loop. Perhaps most importantly, both structures were determined in the presence of carazolol, a partial inverse agonist that, like 11-cis retinal for rhodopsin, helps lock the receptor in an inactive state. As a consequence, these structures could not provide much additional insight into G protein coupling. Structures soon followed for the β1-adrenergic receptor10, a squid photoreceptor that couples with G αq11, and the adenosine A2A receptor12, which were likewise determined in inactive states. Furthermore, it was not clear what conformational state any of these structures corresponded to, because the so-called “ionic lock”, a salt bridge linking residues between TM3 and TM6 that is believed to be characteristic of the inactive state of the receptor, was not formed as it is in the structure of rhodopsin.
Light at the End of the Tunnel
With five unique, but inactive, receptor structures, the preponderance of evidence appeared to suggest that only an inhibited GPCR could assume a sufficiently rigid conformation amenable for crystallization, or at least that receptors would have to be engineered in such a way (e.g. by lysozyme insertion) that it rendered it difficult to determine how heterotrimeric G proteins interact with receptors. However, a glimpse of the answer came with the crystal structure of bovine opsin13, a retinal-depleted form of rhodopsin that, like the β2AR, has constitutive activity. Surprisingly, opsin assumed a conformation consistent with properties of meta II predicted by prior biophysical measurements 14,15, including a rigid body rotation of TM6 away from the core of the receptor (thus breaking the ionic lock) to form a solvent-accessible cavity and a docking site for the C-terminus of G α on the inside of the cytoplasmic ends of TM5 and TM6. The follow-up structure of opsin in complex with a peptide derived from the C-terminus of transducin (G αt) confirmed these predictions16. The G αt peptide makes direct contact with residues known to be important for heterotrimeric G protein activation, and forms specific interactions that help to explain the molecular basis of specificity between GPCRs and heterotrimeric G proteins.
Problems Reading in Dim Light
As pointed out by others, opsin is orders of magnitude less active than the meta II state of rhodopsin 7. Therefore, what state do these structures of opsin really represent? Several observations make this difficult to assess. First of all, although opsin is far less active than meta II, the detergent-solubilized protein that was crystallized could still more closely resemble a fully active receptor than the bulk of opsin molecules in solution or in lipid bilayers. After all, GPCRs with basal activity have access to multiple conformational states. Secondly, if the G α βγ heterotrimer is docked with the opsin structure using the G αt peptide as a guide, the N-terminus of G α and the G βγ subunits overlap extensively with the expected plane of the lipid bilayer 17. This simple docking exercise could indicate either that the opsin structure is not in a fully activated, G protein-bound conformation, or that the model of G α βγ does not reflect its receptor bound conformation, or both.
New data, however, reinforce the conclusion that the opsin structures exist in a conformational state at least very close to that of an agonist-occupied receptor. Crystal structures have now been determined for both a constitutively active form of rhodopsin (G. Schertler, personal communication) and an agonist-bound β2AR (B. Kobilka, personal communication). In both cases, the receptors adopt conformations very similar to those of opsin, with the expected outward rotation of TM6. Because two distinct GPCRs with properties consistent with those of an activated receptor have now been observed in essentially the same conformation, it seems likely that we now have a reasonably accurate glimpse of what an activated GPCR looks like before it couples with a heterotrimeric G protein.
Future Directions
That is not the end of the quest by far. If one assumes that the structure of the opsin · G αt peptide complex is similar to that of a receptor bound to a heterotrimeric G protein, then G α βγ must undergo a significant conformational change in order to avoid a collision with the cell membrane. Reorganization of the heterotrimer is anticipated in any event because the guanine nucleotide needs to be ejected from the active site of G α . Several lines of experimental data support this hypothesis. Electron paramagnetic resonance studies using introduced spin-labels on G α βγ in complex with light-activated rhodopsin suggest that a rigid body rotation of the C-terminal helix of G α , along with other structural changes, leads to release of the bound nucleotide 18,19. NMR spectra of a G αt/G αi chimera in complex with detergent solubilized rhodopsin exhibited severe line broadening, consistent with a high degree of dynamic disorder when the heterotrimer is in a receptor-bound, nucleotide-free state 20 –a state that is therefore much different than has been visualized in the current repertoire of heterotrimeric G protein structures.
The process by which GPCRs induce this conformational change in G α βγ remains mysterious. Binding to the extended C-terminus of a G α subunit seems hardly sufficient to coerce global reconfiguration of the entire heterotrimer, particularly when this region of G α is structurally uncoupled from the rest of the Ras-like domain (the C-terminal receptor binding region of G α is almost always disordered in crystal structures of heterotrimeric G proteins). There are several mechanisms by which additional leverage could be applied. First, GPCRs may also interact with other regions of G α adjacent to the C-terminal helix, such as the α N- β1 and β2- β3 loops at the top of the Ras-like domain, which also collide with the receptor in the opsin-G α βγ docking model 21. Second, there is some evidence that regions within α N and the lipid-modified N-terminus of G α and the C-terminus of Gγ can interact with receptors 21. Because these structural elements are relatively far from the G α C-terminus (40–50 Å, i.e. greater than or equal to the width of the GPCR itself), an allosteric change would then have to be brought about via the interactions of G α βγ with receptors in an oligomeric form. Disfavoring this hypothesis are studies showing that monomeric GPCRs, including the β2AR, rhodopsin, and the μ-opioid receptor, are the minimal (albeit not necessarily optimal) unit required for heterotrimeric G protein coupling 22–25. A third option is that the lipid bilayer itself supplies the necessary leverage required for nucleotide exchange. The structural constraints imposed on G α βγ by simultaneous interactions with the lipid bilayer, via the lipid modifications at the N-terminus of G α and the C-terminus of Gγ , and with the cavity formed in the active GPCR, via the C-terminus of G α , may stabilize the nucleotide free state of the G protein. This last model is attractive because it does not require each individual GPCR, which exhibit high sequence divergence, to have evolved a unique mechanism to interact with specific G α subunits at sites other than the C-terminus of G α . GRKs and arrestins, which also selectively recognize activated GPCRs, also do so in a lipid-dependent manner, and a similar mechanism may be at work.
In the end, what will be required to truly realize the grail is the crystal structure of a GPCR in complex with a heterotrimeric G protein, as this would most clearly delineate the features of a receptor in its high-affinity, G protein-bound state. But there at least three grails. Structures of GPCRs in complex with GRKs and arrestins will also be required to stabilize the receptor in two other distinct, activated states.
Figure 1.
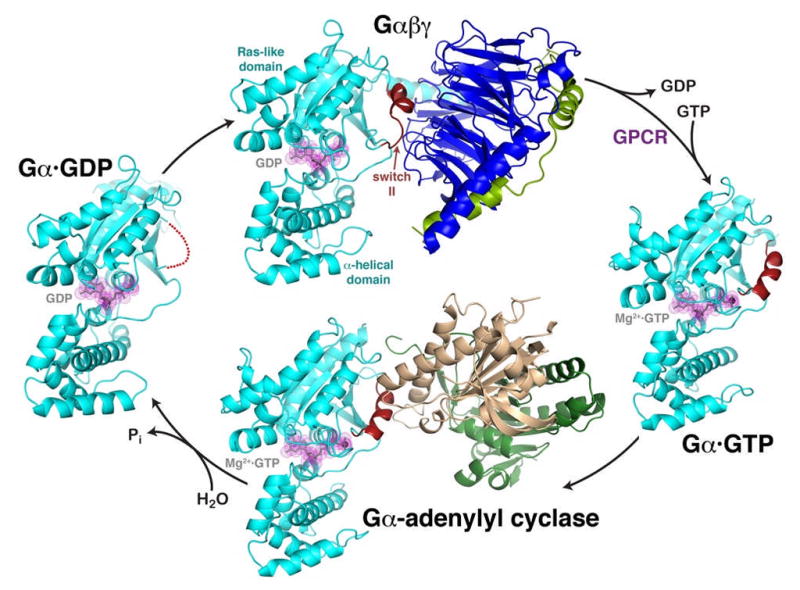
The heterotrimeric G protein cycle.
The inactive Gαβγ heterotrimer (top) is composed of two principal elements, Gα·GDP (cyan, with GDP shown in grey with magenta dot surface) and the Gβγ heterodimer (blue and green). Gβγ sequesters the switch II element (red) such that it is unable to interact with effectors. Activated GPCRs catalyze the release of GDP from Gα, allowing GTP to bind and liberate the activated Gα·GTP subunit (right). In this state, switch II forms a helix stabilized by the γ-phosphate of GTP. The activated Gαsubunit can then interact with effectors, such as the catalytic domains of adenylyl cyclase (bottom, gold and dark green). Switch II forms a major component of the interface, as it does in other characterized effector complexes. The Gαsubunit has a slow GTPase activity that converts GTP to GDP, weakening its interactions with effectors and allowing it to dissociate as a deactivated Gα·GDP subunit (left). In this state, switch II is disordered (red dotted line), and the protein has high affinity for Gβγ subunits, completing the cycle. The structures shown correspond to PDB entries 1GG2 (ref. 27) (top, cyan subunit corresponds to Gαi), 1AZT28 (right, Gαs), 1AZS4 (bottom, Gαs) and 1GDD29 (left, Gαi).
Figure 2.
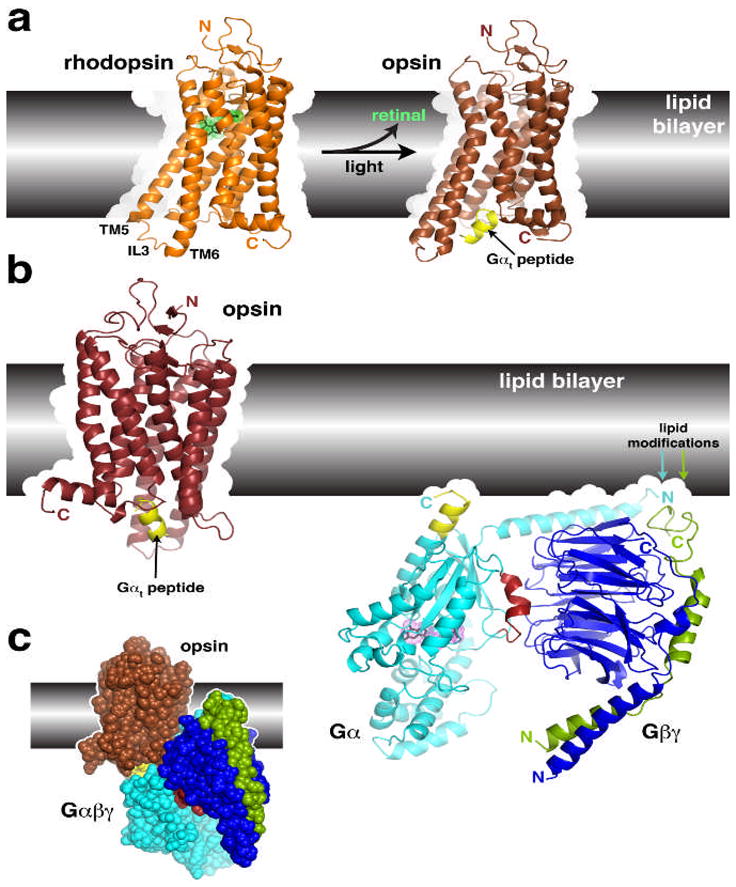
Structural transitions of GPCRs and their interactions with heterotrimeric G proteins.
(a) Comparison of the structures of rhodopsin and opsin. Opsin represents a retinal-depleted form of the GPCR with low basal activity yet shows structural features consistent with changes predicted to occur during rhodopsin activation, such as the outward twist of the third intracellular loop (IL3). Opsin can also be crystallized in complex with a peptide derived from the C-terminus of Gαt (yellow), the region of the heterotrimer most strongly linked to receptor recognition. Retinal is shown as a stick model with a green dot surface. Structures correspond to PDB entries 1GZM30 and 3DQB17. (b) Conceptual problems in docking current models of GPCRs with their heterotrimeric G protein substrates. Here we show the inactive Gαiβ1γ2 complex (PDB 1GG2 (ref. 27)), in which the C-terminus of Gαi was extended according to PDB entry 1AGR31 and the C-terminus of Gγ according to PDB entry 1OMW32. The C-terminal span of Gαi analogous to the Gαt peptide bound to opsin is colored yellow. Comparing the orientation of this helix in each model reveals an an apparent docking incompatibility, because the intact heterotrimer would have to be rotated, roughly counterclockwise, up into the plane of the lipid bilayer in order to superimpose these elements. (c) Collision of Gαβγ with the lipid bilayer when superimposed with the Gαt peptide bound to opsin. The collision suggests that either the model of opsin does not represent a GPCR in a fully activated state, or the G protein heterotrimer must undergo a significant conformational change, or both. A large conformational change is expected in Gαβγ because its interaction with receptors must induce nucleotide release.
Acknowledgments
Support was provided by NIH grants HL086865 and HL071818.
References
- 1.Rall TW, Sutherland EW. The regulatory role of adenosine-3′, 5′-phosphate. Cold Spring Harb Symp Quant Biol. 1961;26:347–54. doi: 10.1101/sqb.1961.026.01.042. [DOI] [PubMed] [Google Scholar]
- 2.Hepler JR, Gilman AG. G proteins. Trends Biochem Sci. 1992;17:383–7. doi: 10.1016/0968-0004(92)90005-t. [DOI] [PubMed] [Google Scholar]
- 3.Sprang SR, Chen Z, Du X. Structural basis of effector regulation and signal termination in heterotrimeric G proteins. Adv Protein Chem. 2007;74:1–65. doi: 10.1016/S0065-3233(07)74001-9. [DOI] [PubMed] [Google Scholar]
- 4.Kenakin T, Miller LJ. Seven Transmembrane Receptors as Shapeshifting Proteins: The Impact of Allosteric Modulation and Functional Selectivity on New Drug Discovery. Pharmacol Rev. 2010;62 doi: 10.1124/pr.108.000992. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–86. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 7.Lodowski DT, Angel TE, Palczewski K. Comparative analysis of GPCR crystal structures. Photochem Photobiol. 2009;85:425–30. doi: 10.1111/j.1751-1097.2008.00516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–65. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–7. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 10.Warne T, et al. Structure of a β1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–91. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453:363–7. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- 12.Jaakola VP, et al. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science. 2008 doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–7. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 14.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–70. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 15.Janz JM, Farrens DL. Rhodopsin activation exposes a key hydrophobic binding site for the transducin alpha-subunit C terminus. J Biol Chem. 2004;279:29767–73. doi: 10.1074/jbc.M402567200. [DOI] [PubMed] [Google Scholar]
- 16.Martin EL, Rens-Domiano S, Schatz PJ, Hamm HE. Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library. J Biol Chem. 1996;271:361–6. doi: 10.1074/jbc.271.1.361. [DOI] [PubMed] [Google Scholar]
- 17.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 18.Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat Struct Mol Biol. 2006;13:772–7. doi: 10.1038/nsmb1129. [DOI] [PubMed] [Google Scholar]
- 19.Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mapping allosteric connections from the receptor to the nucleotide-binding pocket of heterotrimeric G proteins. Proc Natl Acad Sci U S A. 2007;104:7927–32. doi: 10.1073/pnas.0702623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdulaev NG, et al. The receptor-bound “empty pocket” state of the heterotrimeric G-protein alpha-subunit is conformationally dynamic. Biochemistry. 2006;45:12986–97. doi: 10.1021/bi061088h. [DOI] [PubMed] [Google Scholar]
- 21.Oldham WM, Hamm HE. Structural basis of function in heterotrimeric G proteins. Q Rev Biophys. 2006;39:117–66. doi: 10.1017/S0033583506004306. [DOI] [PubMed] [Google Scholar]
- 22.Whorton MR, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A. 2007;104:7682–7. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J Biol Chem. 2007;282:14875–81. doi: 10.1074/jbc.M701433200. [DOI] [PubMed] [Google Scholar]
- 24.Whorton MR, et al. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem. 2008;283:4387–94. doi: 10.1074/jbc.M703346200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuszak AJ, et al. Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem. 2009;284:26732–41. doi: 10.1074/jbc.M109.026922. [DOI] [PMC free article] [PubMed] [Google Scholar]