

Abstract
Smokers are less likely to develop some inflammatory and allergic diseases. In Brown-Norway rats, nicotine inhibits several parameters of allergic asthma including the production of Th2 cytokines and the cysteinyl leukotriene LTC4. Cysteinyl leukotrienes are primarily produced by mast cells, and these cells play a central role in allergic asthma. Mast cells express a high affinity receptor for IgE (FcεRI). Following its cross-linking, cells degranulate and release preformed inflammatory mediators (early phase), and synthesize and secrete cytokines/chemokines and leukotrienes (late phase). The mechanism by which nicotine modulates mast cell activation is unclear. Using α-bungarotoxin binding, qPCR, and PCR product sequencing, we show that the rat mast/basophil cell line RBL-2H3 express nicotinic acetylcholine receptors (nAChRs) α7, α9, and α10, and exposure to exceedingly low concentrations of nicotine (nanomolar), but not the biologically inactive metabolite cotinine for ≥8h suppressed the late phase (leukotriene/cytokine production) but not degranulation (histamine and hexosaminidase release). These effects were unrelated to those of nicotine on intracellular free calcium concentration but causally associated with the inhibition of cPLA2 activity and PI3K/ERK/NF-κB pathway, including phosphorylation of Akt and ERK, and nuclear translocation of NF-κB. The suppressive effect of nicotine on the late-phase response was blocked by the α7/α9-nAChRs antagonist methyllycaconitine and α-bungarotoxin, and by siRNA knockdown of α7, α9, or α10 nAChRs, suggesting a functional interaction between α7, α9, and α10 nAChRs that might explain the response of RBL to nanomolar concentrations of nicotine. This “hybrid” receptor might serve as a target for novel anti-allergic/asthmatic therapies.
Introduction
The prevalence and severity of atopic diseases, including allergic asthma, rhinitis, and eczema have increased dramatically in recent years (1-5). Allergic diseases involve the allergen-induced Th2 response characterized by the production of Th2 cytokines, including IL-4, IL-5, and IL-13 critical in the development of the allergic response. Mast cells are critical tissue-based effector cells that mediate IgE-dependent allergic responses (6-8). Mast cells express IgE receptors (FcεRI) and binding of an allergen to IgE-FcεR1 induces the release of three classes of proinflammatory mediators: 1) preformed granule-associated chemical mediators; 2) newly synthesized arachidonic acid metabolites, such as leukotrienes (LTs); and 3) proinflammatory cytokines, including TNF-α and Th2 cytokines (6-8). Among these mediators, the cysteinyl LTs (cysLTs) exert a number of pathophysiological effects of allergic asthma, including proliferation and contraction of bronchial smooth muscle cells, mucus secretion, inflammatory cell migration, and increased vascular permeability (9-11). Indeed, cysLTs are important indicators of allergic asthma severity (12-14).
Several reports suggest an inverse correlation between cigarette smoking and the development of allergic diseases (15, 16). Smoking increases the risk of various diseases including infections, and development of these diseases may, in part, by the suppressive effects of nicotine on some parameters of adaptive and innate immune responses (17). Linneberg et al. (16)) reported that smoking was negatively associated with the incidence of allergic sensitization, which is consistent with another population-based study that concluded cigarette smokers were less likely to develop allergic sensitization during an 8-year follow-up period (15). Several cross-sectional studies also report a lower incidence of aeroallergen sensitization among current smokers than never smokers; even past smokers were less likely to be sensitized than never smokers (18-22).
Nicotine (NT), the major constituent of cigarette smoke, suppresses adaptive and inflammatory immune responses (23-25), and recently we demonstrated that NT pretreatment attenuated some parameters of ragweed- and house dust mite-induced allergic asthma in Brown Norway rats by primarily suppressing leukocytic infiltration and production of LTs and Th2 cytokines/chemokines without affecting the allergen-induced hexosaminidase/histamine release in the lung (26). Thus, in addition to its effects on T cells (27), and macrophages (24), NT also affects the mast cell function in the lung (26), and the presence of nicotinic acetylcholine receptors (nAChRs) on murine bone marrow derived mast cells (28) and human skin mast cells has been suggested (29). To understand the mechanism by which NT modulates mast cell function, we used the rat mast cell/basophil cell line RBL-2H3 (RBL) to show that NT in nanomolar quantities blocked the delayed phase of mast cell activation through α7/α9/α10 nAChRs and inhibited the cPLA2/MAP kinase pathway.
Materials and Methods
Reagents
RBL cells were obtained from Dr. Janet M. Oliver, University of New Mexico Health Science Center (Albuquerque, NM). The following reagents were purchased from the indicated vendors: Anti- ERK1/2, anti-phosphor-ERK1/2, anti-Akt and anti-phospho-Akt (Cell Signaling Technology, Beverly, MA); horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and goat anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA); and cell-culture reagents (Life Technologies, Grand Island, NY). The vendors for other reagents are indicated under the relevant assays.
Cell culture
RBL cells were cultured at 1-2 × 106 cells in a 75-cm2 flask at 37°C in RPMI 1640 supplemented with 10% FBS, 2 mM glutamine, and penicillin/streptomycin in a humidified 5% CO2 atmosphere. Cells were passaged every 3-4 days.
NT/cromoglycate sodium/cotinine treatment
RBL cells were treated with various concentrations of NT (MP Biomedicals, Solon, OH) or its biologically inactive metabolite cotinine (Sigma-Aldrich Co., St. Louis, MO) or 1 μM cromoglycate sodium (Sigma) for a predetermined optimal time (8 h), followed by stimulation with 10 ng of anti-FcεRI antibody for indicated times at 37°C. Cells and supernatants were collected and subjected to various assays.
Leukotriene C4 assay
Leukotriene C4 (LTC4) was determined as described previously (30). After various treatments of RBL, cell supernatants were collected and the LTC4 content determined by an enzyme immunoassay kit from Cayman Chemicals (Ann Arbor, MI) according to the manufacturer’s protocol.
Histamine and β- Hexosaminidase release assays
The amount of histamine released was measured by enzyme immunoassay according to manufacturer’s instructions (Cayman Chemical Company). The β- hexosaminidase activity was assayed as described previously (31). Briefly, RBL cells were plated (4 × 105 cells/well) in 12-well microtiter plates, and after overnight incubation, cells were washed with PIPES solution (119 mM NaCl, 5 mM KCl, 5.6 mM glucose, 1 mM CaCl2, 0.4 mM MgCl2, 25 mM PIPES and 0.1% BSA) and stimulated with anti-FcεRI for 30 min at 37°C with gentle rotation. The supernatants were collected and 0.5% Triton X-100 solution was added to the cells to quantify the unreleased β-hexosaminidase. Samples (20 μl) were transferred to a 96-well microtiter plate and 50 μl of the substrate solution (p-nitrophenyl-N-acetyl-β-D-glucosamide) was added to each well. The plates were incubated at 37°C for 60 min under gentle rotation. After the addition of 150 μl of the stop solution (0.2 M glycine, pH 10) to each well, absorbance was recorded at 405 nm. The results of cell degranulation were expressed as the percentage of degranulation in control samples.
Western blot analysis for ERK and Akt
Whole cell lysates were prepared as described (32). Briefly, cells were lysed on ice with ice-cold 1% Brij-96 containing lysis buffer (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 1 mM orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) for 15 min. After clarification at 12000 rpm, supernatants were assayed for protein concentration by the BCA method. Samples (40 μg of protein) were resolved on Criterion Gels (BioRad, Hercules, CA), and the resolved proteins were transferred onto a nitrocellulose membrane, blocked with 2% BSA (Sigma-Aldrich) and probed with the phosphor-ERK1/2 and phosphor-Akt antibodies. The membranes were washed 3 times, incubated with HRP-conjugated goat anti-rabbit secondary antibodies, developed with ECL (Amersham Biosciences, UK), and exposed to X-ray film. The same blots were stripped with Restore Western Blot Stripping Buffer (Pierce, Rockford, IL) for 15 min at room temperature, washed, and re-probed with ERK1/2, Akt and β-Actin antibodies to verify equal loading on the gel.
cPLA2 activity assay
The cPLA2 activity from homogenized cell extracts was assayed by enzyme immunoassay (Cayman Chemical Company) according to manufacturer’s instructions.
Nuclear translocation of NF-κB
Nuclear extracts were prepared from RBL cells essentially as previously described (33). Briefly, RBL cells were lysed on ice using hypotonic cell lysis buffer [50 mM KCL, 0.5% Nonidet P-40 (NP-40), 25mM HEPES (ph 7.8), 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 20 μg/ml aprotinin, 1 mM DTT] at 4°C for 10 min. The nuclear pellet was extracted with high-salt nuclear extraction buffer (500 mM KCl, 10% glycerol, 0.5% NP-40, 25 mM HEPES, pH 7.8, 1mM PMSF, 10 μg/ml leupeptin, 20 μg/ml aprotinin, and 100 μM DTT) and the suspension was centrifuged to yield the nuclear extract. The protein content of the extracts was determined by the BCA method. For the NF-κB assay, an aliquot of the nuclear extract containing 8 μg of protein was incubated with 32P-end-labeled 22-mer double-stranded NF-κB oligonucleotide (Promega Corporation, Madison, WI) with the NF-κB consensus sequence: 5’-AGTTGAGGGGACTTTCCCAGGC-3’ and 3’-TCAACTCCCCTGAAAGGGTCCG-5’ for 30 min at 37°C. The resulting DNA-protein complex was resolved by electrophoresis on a 5% native polyacrylamide gel; after drying the gel, the radiolabeled bands were visualized by Kodak X-OMAT film (Eastman Kodak, Rochester, NY).
Assay for intracellular free calcium concentration
The assay for intracellular free calcium concentration [Ca2+]i of RBL was determined as described (34). Briefly, 5 × 106 cells were loaded with indo-1-AM (Molecular Probes, Eugene, OR) for 20 min. After washing, the cells were suspended in Ca2+-containing medium (10 mM HEPES, pH 7.4; 126 mM NaCl; 3 mM KCl; 1.25 mM NaH2PO4; 26 mM NaHCO3; 10 mM dextrose; 2 mM Ca2+ ; 1 mM Mg2+; and 5% FBS, adjusted to an osmolarity of 285). [Ca2+]i was determined by spectrofluorometry (Deltascan Model 4000; PTI). Indo-1 loaded cells were treated with the indicated reagents and [Ca2+]i calculated as previously described (35).
Treatment of RBL cells with α-bungarotoxin and methyllycaconitine
Methyllycaconitine (MLA) (Calbiochem, NJ) and α-bungarotoxin (α-BTX) (Molecular Probes, Eugene, OR) is established antagonist of α7-nAChRs; however, more recent data suggests that they also bind α9 subunits of nAChRs (36, 37). To test the effects of these antagonists on nicotine-mediated effects on mast cells, prior to nicotine and/or anti-FcεRI treatment, RBL cells were incubated with 100 nM MLA or 40 nM α-BTX for 30 min at 37°C.
Flow cytometry for α-bungarotoxin binding
To determine the presence of α7-like nAChRs on RBL cells, we used flow cytometry to determine the binding of Alexa fluor 488-conjugated α-BTX as described for T cells (27). Briefly, RBL cells (5 × 105 cells/ml) were incubated with 100 nM Alexa fluor 488-conjugated α-BTX for 1 h. After washing with cold PBS, the cells were analyzed on FACSCalibur flow cytometer (BD Biosciences, San Jose, CA).
Real-time quantitative PCR analysis
Total RNA was isolated from RBL cells as described (26). Briefly, cells were lysed in Tri-Reagent (Molecular Research Center, Cincinnati, OH) and after the addition of BCP (MRC) the lysate was centrifuged at 13,000xg for 10 min at 4°C. The aqueous layer was collected, mixed with isopropanol, and after 1 hr at -20°C, the precipitate was collected by centrifugation. The pellets were resuspended in 75% ethanol, centrifuged, and air dried. The RNA samples were dissolved in diethylpyrocarbonate (DEPC)-treated water at 55°C for 10 min and quantified spectrophotometrically. Quantitative PCR (qPCR) analysis was performed on the ABI PRISM 7900HT Real-Time PCR System using the One-Step RT-PCR Master Mix (Applied Biosystems, Foster City, CA). Primer/probes were purchased from Applied Biosystems.
Identification of nAChRs expression by PCR and sequencing
RNA from RBL was reverse transcribed using the SuperScript III First-Strand Synthesis system (Invitrogen, Carlsbad, CA) using both oligo (dT20) and random hexamers for priming. PCR was performed with cDNAs generated by both priming methods using Platinum Taq polymerase (Invitrogen) according to the manufacturer’s instructions. Magnesium, concentration was adjusted to produce the best product yields, and the primer annealing temperatures were determined using Oligo 4.0 (National Biosciences Inc, Plymouth, MN). Primers were designed using Olig 4.0 and the NCBI Primer Blast primer designing tool. Primers were synthesized by Invitrogen and PCR products were generated using both sources of cDNA with similar results.
For PCR analysis of α10-nAChR, the following three sets of primers were used: Primer 1 between bases 799-1575 (upstream: 5’-CCCTGTGTGTTCATCTCCCT-3’; downstream 5’-GGTTGTGAGGTCAGAAGCACT-3’), primer 2 between bases 799-1324 (upstream: 5’-CCCTGTGTGTTCATCTCCCT-3’; downstream: 5’-TAGCCAGACGCTTCCAATCT-3’) and primer 3 between bases 1305-1696 (upstream: 5’-AGATTGGAAGCGTCTGGCTA-3’; downstream 5’-TGCTCATCTGGCATTGAGTCTTA-3’). PCR analysis of α9 used two sets of primers: primer 1 between bases 146-654 (upstream: 5’-TCAGAGCCGTAGAGACAGCA-3’; downstream: 5’-CAGGGCATTGAATATGTCCA-3’) and primer 2 between bases 1148-1752 (upstream: 5’-TGGGTGAGAGCTGCCTTAGT-3’; downstream: 5’-CTCTCTTCCTCCAGCCCTTT-3’). To determine α7-nAChR expression, a set of primers was designed to amplify a product between 1007-1509 bases containing TM-3 and TM4 and TM4 loop (upstream: 5’-TGAACTGGTGTGCATGGTTT-3’; downstream: 5’-TTAAGCAAAGTCTTTGGACACA-3’).
The PCR products were separated on a 1.0% agarose gel, and purified using the QIAEX II gel extraction kit (Qiagen, Valencia, CA). The purified products were cloned into pCRII-TOPO vector (Invitrogen), and the clones were sequenced using a 3730 DNA Analyzer (Applied Biosystems) at the Ohio State University Plant-Microbe Genomics Facility. DNA sequences were used for a BLAST search versus the GenBank database (38). Additional sequence analysis was done using GeneWorks (Intelligenetics, Hilton head, SC).
Knockdown of α7, α9 and α10 expression by siRNA
siRNAs used in this study were purchased from Thermo Scientific (Lafayette, CO). The siRNA SMARTpool contained four pooled siRNA duplexes with “UU”overhangs and a 5′ phosphate on the antisense strand. Each siRNA was complexed with the DharmaFECT 1 transfection reagent (Thermo Fisher Scientific, Rockford, IL) for a final concentration of 100 nM per well in a 6-well microtiter plate and the cells were transfected according to the manufacturer’s instructions. Briefly, the siRNA-DharmaFECT 1 complex was added to the cells in complete medium and incubated at 37°C in 5% CO2 for 48 h. Cell viability was assessed 24 hours after transfection using a trypan blue exclusion assay. The mRNA levels of α7-, α9- and α10-nAChRs were assessed 48h post transfection using QuantiTect SYBR Green One-step RT-PCR. Oligonucleotide primers and SYBR Green One Step RT-PCR Kit were purchased from Qiagen.
Statistical Analysis
To determine statistical significance by the student’s t-test or two-way ANOVA, we used GraphPad Prism Software 3.0 (GraphPad Software, Inc. San Diego, CA). Values were considered significant at p ≤ 0.05.
Results
NT Inhibits LT and cytokine production but not histamine and β-hexosaminidase release
Inflammatory substances released by mast cells induce and maintain the allergic response. Upon stimulation of FcεRI the mast cells degranulate rapidly (in seconds) releasing the preformed chemical mediators, including histamine and β-hexosaminidase that play a critical role in the immediate-type allergic response (39). In contrast, FcεRI activation also induces de novo synthesis of LTs, such as LTC4 and cytokines such as TNF-α and IL-1β that initiate the latephase allergic response (40, 41). To determine the effects of NT on the FcεRI-mediate mast cell responses, we incubated RBL cells with indicated concentrations of NT for the predetermined optimal time (8 h) prior to stimulation with anti-ε antibodies to ligate FcεRI. Fig. 1A, a representative of 4 independent experiments, shows that the release of the LT peptide LTC4 at 30 min after FcεRI ligation (predetermined optimal time for LTC4 release) was significantly (40-60%) inhibited by an extremely low concentration of NT (10-100 nM). Under identical conditions, cromoglycate sodium (Sigma-Aldrich Chemical Co), a mast cell stabilizer and an established anti-asthma drug (42), inhibited the LTC4 response by only 10-15% (Fig. 1A). Thus, NT is relatively a strong inhibitor of mast cell LT production. The effects of NT on the mast cell LT production are specific and cotinine the biologically inactive major metabolite of NT did not affect LTC4 production/release (Fig. 1B). Indeed, cotinine was ineffective even at concentrations exceeding 1 μM (0.1-1 mM), and addition of cotinine to NT (1μM nicotine + 1 mM cotinine) did not significantly affect histamine release or the NT-induced inhibition of LTC4 (data not shown). Moreover, the kinetics of the NT response indicated that LTC4 production was not significantly affected until 8h after NT treatment and remained significantly depressed until 24 h after NT treatment (not shown). Thus, the NT-induced suppression of LT production requires long-term exposure of mast cells to NT.
Figure 1. NT inhibits LT synthesis and secretion in RBL cells.
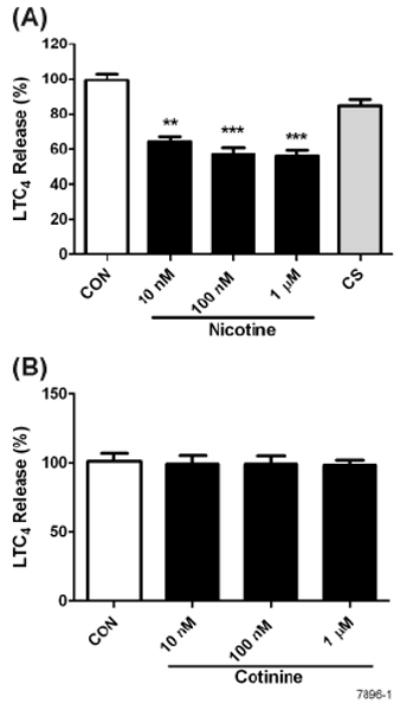
RBL cells were treated with various concentrations of NT or 1 μM cromoglycate sodium (CS) (A) or cotinine (B) for 8 h, followed by stimulation with 10 ng of anti-FcεRI antibody. LTC4 content in the supernatants was determined as described in Materials and Methods. Release of LTC4 is expressed as the percent of control RBL cells treated with anti-FcεRI alone. Data shown are the means ± SE of 4 independent experiments. * p < 0.05, ** p < 0.01 and *** p < 0.001.
After FcεRI activation, in addition to LTs, proinflammatory cytokines, such as TNF-α and IL-1β are also mostly synthesized de novo by mast cells (43). At 30 min post activation, qPCR analysis indicated that pretreatment of RBL cells with NT (1 μM) significantly inhibited the FcεRImediated mRNA expression of both TNF-α and IL-1β (Fig. 2A-B). Similarly, the concentration of released TNF-α in the culture medium at 2 h after anti-FcεRI antibody treatment was significantly reduced in NT-treated RBL cultures (Fig. 2C). These results suggest that, in addition to leukotrienes, NT also inhibits the mRNA expression and as well as the production of the proinflammatory cytokine TNF-α by mast cells i.e., both parameters of the late phase response. Under these conditions; however, the concentration of IL-1β in the culture fluids was too low to clearly evaluate the effects of NT on the release of this cytokine.
Figure 2. NT inhibits the expression of proinflammatory cytokines in RBL.
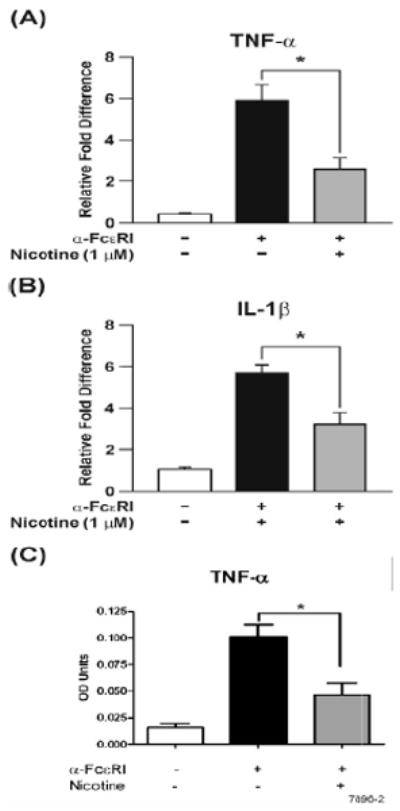
Where indicated, RBL cells were pretreated with 1 μM NT for 8 h, followed by stimulation with 10 ng of anti-FcεRI antibody. (A & B) After 30 min, total RNA was isolated and analyzed for TNF-α and IL-1β expression by qPCR, (C) 2 hrs post anti-FcεRI stimulation release of TNF-α was measured using ELISA. The results are the means ± SE of three independent experiments and expressed as the fold change in the expression and release of these cytokines. * p < 0.05.
To ascertain the effects of NT on mast cell degranulation (characterized by the release of presynthesized compounds histamine and β-hexosaminidase), RBL cells were pretreated with various NT concentrations. After activation by anti-FcεRI antibody, β-hexosaminidase enzymatic activity and histamine release were measured in cell supernatants. Results presented in Fig. 3A and 3B show that NT did not detectably affect on the release of β-hexosaminidase or histamine, suggesting thereby that NT does not significantly affect the acute phase response of the mast cells.
Figure 3. NT treatment does not affect degranulation in RBL cells.
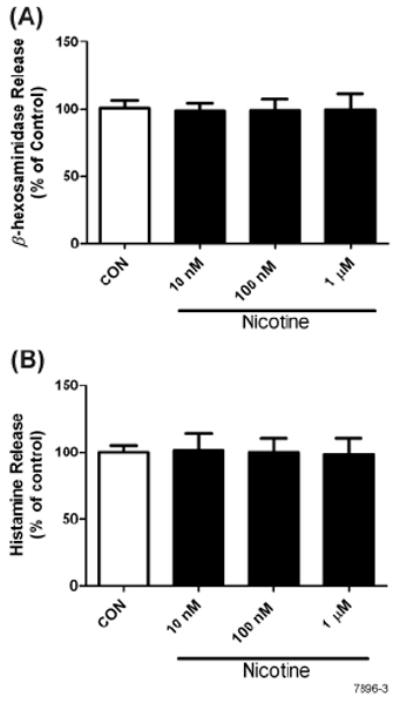
Cells were pretreated with various concentrations of NT for 8 h followed by stimulation with 10 ng of anti-FcεRI antibody for 20 min. Supernatants were analyzed for enzymatic activity of β-hexosaminidase (A) and histamine (B) as described in Materials and Methods. Data represent means ± SE for 4 independent experiments each performed in triplicate.
Nicotinic receptors on RBL cells
Leukocytes such as T cells and macrophages have functional α7-nAChRs (24, 27). There are reports that bone marrow-derived and skin mast cells express α7-nAChRs (28, 29). To determine whether RBL cells expressed α7-nAChRs, the cells were incubated with Alexa fluor 488- conjugated α-BTX and analyzed by flow cytometry. Fig. 4A shows that essentially all RBL cells bind α-BTX indicating the presence of α7-nAChR-like receptors on RBL cells. However, in addition to α7, α-BTX also binds α9 and α9/α10-nAChRs (36, 37); therefore, we examined the expression of α7-, α9-, and α10-nAChRs on RBL cells by qPCR and sequence analysis. Data presented in Fig. 4B shows that RBL cells express α7-, α9-, and α10-nAChRs.
Figure 4. RBL cells bind α-BTX and express α7-, α9-, and α10-nAChRs.
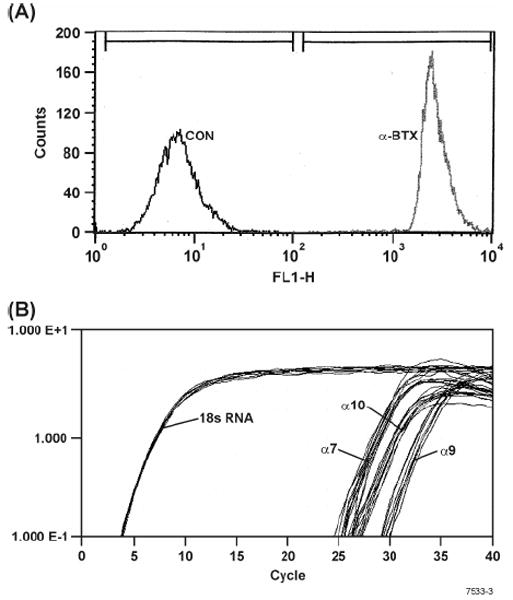
(A) RBL cells were incubated with Alexa fluor 488-conjugated α-BTX and analyzed by flow cytometry as described in Materials and Methods. Note: After α-BTX treatment there are no detectable α-BTX unbound cells as seen by lack of cells within the control (CON) region of the histogram. (B) qPCR analysis for the expression of α 7-, α9-, and α10-nAChRs on RBL. Note: On RBL cells the expression of α7-, α9-, and α10-nAChRs is approximately comparable.
For α7-nAChRs the primer set yielded a clone covering 1007-1509, and the sequence of the amplified product matched exactly the appropriate rat α7-nAChR region. Unlike humans, the rat α7 gene does not appear to be partially duplicated, which eliminated the need to ascertain the expression of a duplicated product encoding exons 5-10. For α9-nAChR, six PCR clones, three each from the two primer sets covering sequences 146-654 and 1148-1752, were sequenced. The PCR products were identical to the corresponding regions of the rat α9-nAChR cDNA. Of the six PCR clones sequenced for α10-nAChR, one clone was derived from primer set 1, two from primer set 2, and three from primer set 3. The clones covered sequences from 799-1696, and the coding sequence ended at 1407. The sequences obtained from the PCR products were identical to the corresponding regions of the rat α10-nAChR cDNA. Together, these results suggest that RBL cells express α7, α9, and α10 nAChRs.
NT suppresses activation of NF-κB and phospho- but not total Akt and ERK1/2
Activation of mast cells through FcεRI leads to activation of PI3K-dependent activation (phosphorylation) of Akt, activation of ERK1/2 and nuclear translocation of NF-κB (44, 45); NF-κB controls the expression of proinflammatory cytokine gene products in mast cells, including RBL (46). To determine whether NT affected the components of this pathway, we tested its effects (1 μM) on the FcεRI-mediated activation of Akt, ERK1/2, and NF-κB in RBL cells. Western blot analysis indicated that NT suppressed phosphorylation but not the total content of ERK1/2 (Fig. 5A) and Akt (Fig. 5B). These effects of NT were significantly detectable at 8 h but not at 2, 4, or 6h after nicotine pretreatment (not shown). Similarly, NT inhibited the activation (nuclear translocation) of NF-κB (Fig. 5C) at 8h but not at 2h after incubation with NT. Thus, NT suppresses the PI3K/Akt/MAPK/NF-κB signaling pathway, and these effects were discernable at about 8 h after NT treatment. The inhibitory effects of NT on ERK and Akt phosphorylation were also evident at 16 and 24h after NT treatment (not shown).
Figure 5. NT treatment suppresses the activation of ERK1/2, Akt and NF-κB in RBL cells.
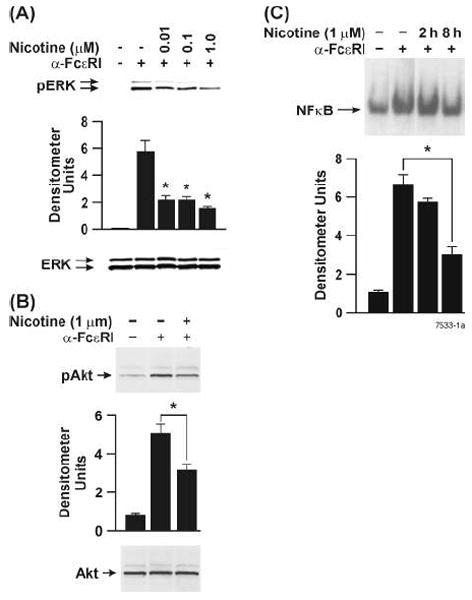
Cells were treated with indicated concentrations of NT for 8 h followed by stimulation with 10 ng of anti-FcεRI antibody for 20 min. Whole cell lysates (40 μg protein) were analyzed by Western blots as described in Materials and Methods. Blots were probed by (A) anti-phospho ERK1/2 and anti-ERK1/2 (B) anti-phospho Akt and anti-Akt. ERK1/2 and Akt were used as loading controls. Western blots shown are representative of 6 experiments. (C) Nuclear extracts (8 μg protein) from control (CON) and NT-treated RBL cells were incubated with 32P-end-labeled NF- κB oligonucleotide for 30 min at 37°C. The resulting DNA-protein complexes were analyzed by electrophoretic mobility gel shift assay as described in Material and Methods. Densitometry readings are average from 4-6 independent experiments. * p < 0.05.
Nicotine inhibits cPLA2 activity
Mast cells and RBL cells contain PLA2 of both low and high molecular weight (cPLA2) types (47); however, only cPLA2 accounts for the anti-FcεR1 stimulated arachidonic acid production and is dependent on MAP kinase activation (48). Because the inhibitory effects of NT were correlated with suppression of ERK1/2 activity, we determined whether NT affected cPLA2 activity in RBL cells. Results presented in Fig. 6, clearly shows that NT-induced inhibition of ERK1/2 is also associated with the inhibition of cPLA2 activity. Thus, it is likely that NT affects leukotriene production by regulating ERK1/2-cPLA2 activities.
Figure 6. Nicotine treatment inhibits cPLA2 activation in RBL cells.
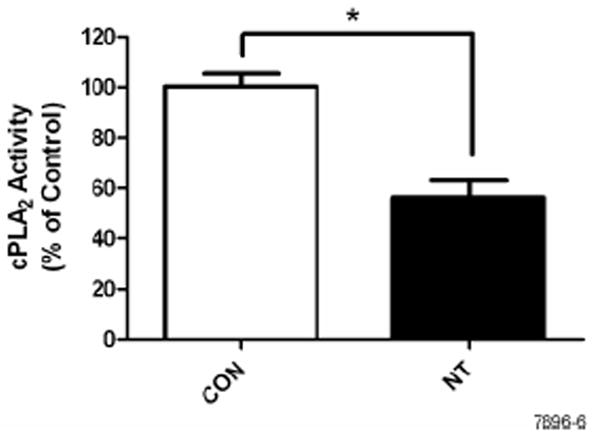
Cells were treated with NT for 8 h followed by stimulation with 10 ng of anti-FcεRI antibody for 20 min. cPLA2 activity was measured by enzyme immunoassay following manufacturer’s instructions. Data represent means ± SE of 3 independent experiments. * p < 0.05.
Antagonists of α7/α9 (α-BTX and MLA) suppress the effects of NT on FcεRI-induced ERK and Akt phosphorylation
In the absence of α9, α10-nAChR does not form a functional receptor (49). To ascertain whether the suppressive effects of NT on mast cells were mediated through the nAChRs expressed by RBL (i.e., α7, α9/α10), prior to incubation with NT, the cells were preincubated with the established antagonists of these receptors MLA or α-BTX (36). Western blots analysis suggested that both α7/α9-nAChR antagonists blocked the inhibitory effects of NT on Akt and ERK1/2 phosphorylation (Fig. 7). Thus, nAChRs α7 and/or α9/α10 are involved in mediating the effects of NT on the delayed phase of the mast cell response.
Figure 7. Antagonists of α7 and α9-nAChRs (α-BTX and MLA) attenuate the NT-induced inhibition of ERK and Akt.

Prior to NT treatment, the cells were preincubated with either 100 nM MLA or 40 nM α-BTX for 30 min. Cells were then stimulated with 10 ng of anti-FcεRI antibody for 20 min. Whole cell extracts were fractionated on SDS-PAGE, transferred onto nitrocellulose membrane and probed with anti-phospho ERK1/2 and anti-phospho Akt antibodies. Total ERK1/2 and Akt were used as loading controls. Western blots shown are representative of 3 experiments.
nAChRs α7, α9, and α10 interact to mediate the effects of NT on mast cell function
Because antagonists that can specifically inhibit α7- or α9/α10 nAChRs are not readily available, we used siRNA to selectively knockdown α7-, α9-, and α10-nAChRs on RBL cells. Depending on the siRNA used, the treatment resulted in nearly a 70% reduction in α7-, α9- (Fig. 8A), or α10-nAChRs mRNA (Fig. 9A) without significantly affecting the expression of the other mRNAs. Surprisingly, however, knocking down α7-, α9-, or α10-nAChRs resulted in blocking the response of RBL cells to NT as evidenced by the loss of the NT effect on anti-FcεRI-induced ERK1/2 phosphorylation (Fig. 8B & 9B). Thus, the lack of α7, α9, or α10 expression abrogates the effects of NT on mast cells, suggesting that a function of nAChR on these cells might involve an interaction between α7, α9, and α10 nAChRs.
Figure 8. nAChRs α7 and α9 interact to mediate the effects of NT on mast cell function.
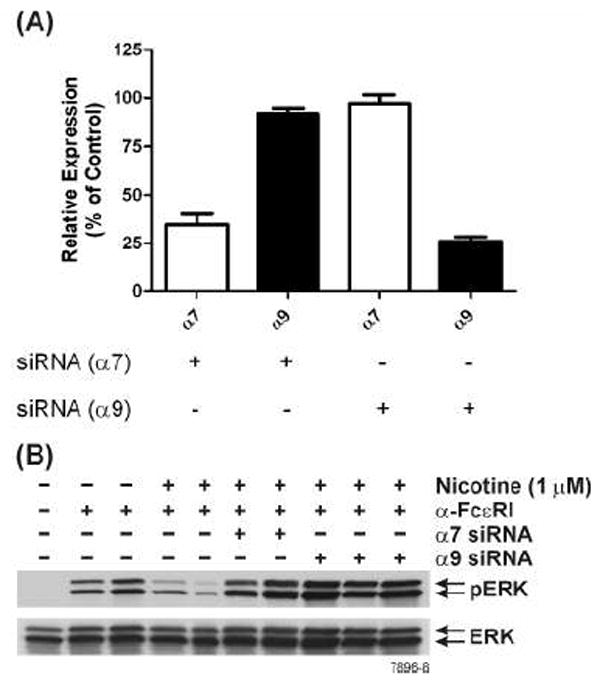
(A) Specific siRNAs selectively knockdown α7 or α9 nAChRs. RBL cells were treated with α7- or α9-specific siRNA as described in Materials and Methods. At 48 h post siRNA treatment, α7- and α9-nAChR mRNA expression was assessed by qPCR. (B) Knockdown of either α7- or α9- nAChRs attenuate NT effects on RBL cell function. At 48 h after siRNA treatment, cells were incubated with 1 μM NT and then stimulated with anti-FcεRI antibody. Whole cell extracts were run on SDS-PAGE and probed with anti-phospho ERK1/2. Total ERK1/2 was used as loading control. The results are representative of 3 independent experiments.
Figure 9. nAChRs α7, α9 and α10 interact to mediate the effects of NT in RBL cells.
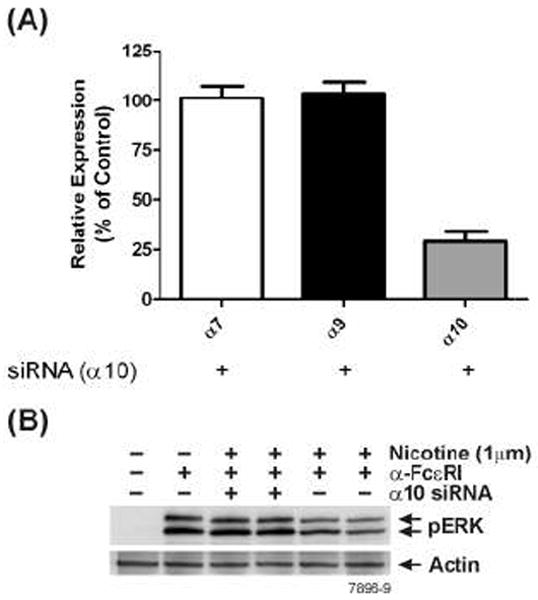
(A) Specific siRNAs selectively knockdown α10 nAChRs. RBL cells were treated with α10-specific siRNA and 48 hrs post siRNA treatment, α7-, α9- and α10-nAChRs mRNA expression was assessed by qPCR. (B) Knockdown of α10-nAChRs attenuates NT effect of RBL cells. At 48 h after siRNA treatment, cells were incubated with 1 μM NT and then stimulated with anti-FcεRI antibody. Whole cell extracts were run on SDS-PAGE and probed with anti-phospho ERK1/2. β-actin was used as loading control. Western blot shown is representative of 3 independent experiments.
Role of Ca2+ in modulating NT response
A sustained increase in [Ca2+]i is critical for both degranulation and transcriptional regulation in mast cells and, as in TCR-mediated T cell activation, crosslinking of FcεRI increases the intracellular concentration of inositol-1,4,5-trisphosphate (IP3) that releases Ca2+ from the IP3-senstive Ca2+ stores. The increased [Ca2+]i triggers Ca2+ influx that stimulates mast cell degranulation (50). We have observed that chronic exposure of animals to NT significantly reduces the TCR-mediated increase in [Ca2+]i (34); therefore it was possible that NT suppressed mast cell activation by inhibiting the FcεRI-mediated rise in [Ca2+]i. Results presented in Fig. 10 clearly show that increasing nicotine concentrations (10 nM-10 μM) progressively decreased [Ca2+]i; however, the magnitude of LTC4 inhibition by these NT concentrations was essentially identical (Fig. 1A). Moreover, none of these NT concentrations affected the release of either β- hexosaminidase or histamine (Fig. 3A & 3B), which is believed to be very sensitive to [Ca2+]i. It should be noted however, that none of these NT concentrations totally blocked the α-FcεRI-induced rise in [Ca2+]i. Thus, ligation of FcεRI increases [Ca2+]i in RBL that, even after partial inhibition by NT, is sufficient to promote degranulation and late phase response. Therefore, it is unlikely that NT-induced suppression of the late phase response is related to the reduction in the magnitude of rise in α-FcεRI-induced [Ca2+]i in NT treated mast cells.
Figure 10. Nicotine inhibits the rise in anti-FcεRI-induced [Ca2+]i.
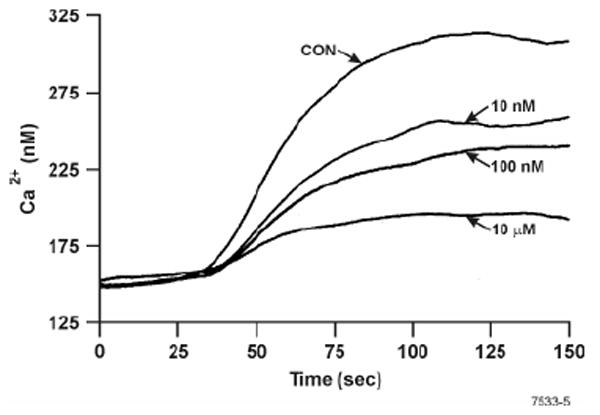
RBL cells were preincubated with indicated concentrations of NT, and after 8 h cells were loaded with Indo-1, treated with anti-FcεRI antibody, and analyzed for changes in [Ca2+]i by spectrofluorometry as described in Materials and Methods. Results shown are representative of two independent experiments.
Discussion
Mast cells play an important role in host defense, but also mediate allergic and inflammatory diseases (51). Allergic diseases are less common in smokers than in non- or ex-smokers, and we have previously reported that in animal models of allergic asthma, NT suppresses the allergeninduced production of LTC4 and inflammatory cytokines without affecting the release of β-hexosaminidase/histamine in the lung (26). The experiments presented herein with RBL cells also show that in response to crosslinking of the high-affinity FcεRI receptor, NT treatment does not affect the release of preformed compounds such as β-hexosaminidase and histamine (degranulation). On the other hand, the late-phase response, including the synthesis of LTC4 and TNF-α, as well as the mRNA expression of TNF-α and IL-1β were significantly inhibited by NT treatment. This supports the recent reports that following FcεRI activation in mast cells, the latephase response (NF-κB activation and cytokine production) is independent of degranulation (52, 53). These results suggest that NT affects the late but not the early degranulation phase of mast cell activation. Moreover, the response to NT was specific and cotinine, the major metabolic product of NT, was essentially ineffective even at concentrations that were 10 -100 fold higher than NT used to inhibit the late-phase response. Cotinine was also ineffective in modulating the effects of NT on anti-FcεRI-induced ERK1/2 activation and leukotriene production by RBL cells. Although the NT-induced inhibition of LTC4 production varied between 40-60%, nicotine was far more effective than the established anti-asthmatic drug cromoglycate sodium (42) that optimally suppressed LTC4 by only 10 to15%. Thus NT is a relatively strong suppressor of the late-phase response. A recent report (28) indicated that NT inhibited degranulation in bone marrow-derived mast cell. Although the reasons for this disparity in the NT response between RBL cells and bone marrow derived mast cells are not clear at present, in addition to the difference in the type, the concentrations of NT required to detect the inhibition of degranulation exceeded 3.0 mM (28), a concentration that would be very difficult to achieve in live animals. In present experiments, we used NT concentration from 1 nM to 1 μM that yielded the effects of NT on β-hexosaminidase and histamine release and the production of LTs akin to the in vivo effects observe after allergic sensitization of Brown Norway rats (26). Moreover, under these conditions rats exhibit the plasma NT levels around 30-50 nM that are comparable to the plasma NT levels in 1-2 pack human smokers.
The effects of NT on mast cell activation produced two surprising findings: 1. the amount of NT required for optimal inhibition was extremely low and significant inhibition was observed even at 1 nM of NT (not shown). On the other hand, the amount of NT required to elicit detectable action potential or whole cell currents in α7-nAChRs-containing interneurons is ≥5 μM (54), that is nearly 5000-fold higher than the amount of NT needed to inhibit the late-phase response of mast cells. As will be discussed later, the increased affinity of nAChRs for NT might reflect a complex interaction between three different nicotinic receptor subunits on mast cells. 2. It took 8-16 h of preincubation with NT to detect inhibition of LTC4 production in RBL cells, suggesting that the NT-induced suppression might represent the “chronic” effects of NT. Indeed in vivo, the immunosuppressive effects of NT on allergic asthma take ≥2 wk of NT exposure (26, 55).
Because FcεRI-mediated mast cell activation is totally dependent on the rise in [Ca2+]i, it is conceivable that NT suppressed the Ca2+ response, thereby inhibiting the late-phase activation. Indeed, we observed that with increasing NT concentrations, the FcεRI-induced rise in [Ca2+]i was progressively decreased and did not affect degranulation. Moreover, there was no correlation between the concentration of NT, inhibition of [Ca2+]i, and the suppression of the late-phase response. It should be noted that because the concentrations of NT used in these experiments did not totally block the α-FcεRI-induced rise in [Ca2+]i, it is likely that the lower increases in [Ca2+]i in NT-treated cells were sufficient to activate these cells. In fact, only modest increases in [Ca2+]i are required for translocation of cPLA2 into the membrane in order to initiate mast cell activation (56) and proper intracellular compartmentalization of Ca2+ is more critical than the overall increase in [Ca2+]i (57).
The late-phase response of mast cells (i.e., production of cysLTs and proinflammatory cytokines/chemokines) requires PI3K/Akt/ERK/cPLA2/NF-κB activation (44-46, 48). We found that NT pretreatment inhibited cPLA2 activity and phosphorylation of Akt and ERK1/2, but did not affect the total content of either Akt or ERK, and in mast cells ERK activation precedes cPLA2 activation, NF-κB activation, and production of LTC4 and cytokines (48, 57, 58). Thus it is likely that NT affects the activity of kinases such as Lyn, and Fyn (59) or phosphatases such as protein tyrosine phosphatase epsilon (60) that are upstream of PI3K/ERK. Upon activation of FcεRI, Lyn is quickly phosphorylated and activates Syk to stimulate degranulation in RBL cells (61, 62). The role of Lyn in mast cell degranulation is debatable (63, 64); however there is evidence suggesting that Lyn promotes degranulation but negatively regulates ERK activation and LTC4 secretion in mast cells (65). Although Fyn is critical for cPLA2 and MAP kinase activation (65), but in Lyn-deficient cells it might initiate complementary signals for mast cell degranulation (32, 66). Thus, it is possible but not proven that nicotine suppresses Fyn but not Lyn activation to allow α-FcεRI induced mast cell degranulation while suppressing the latephase response. Interestingly, chronic NT treatment stimulates Fyn activity in rat T cells (unpublished observation) and might reflect functional differences in two cell types.
Neuronal nAChRs mediate synaptic transmission; however, nicotinic receptors are also present in peripheral cells, including the cells of the immune system (67, 68). Neuronal nAChRs are pentameric structures that function as a ligand-gated ion channels (69). Nine neuronal nAChR α subunits genes (α2-α10) and three β subunits (β2-β4) have been identified; however, α8 subunits are not found in mammals. Among the nicotinic receptors α7 is the most commonly found nAChR subunit on immune cells and α7 and α9, can form functional homomeric nAChRs (69). α10 nAChR subunit has been found to be functional only in combination with α9 subunit (49); however, α7 and α10 colocalize in rat sympathetic neurons (70). We cloned α7-nAChR from human and rat T cells and found it to interact with the T cell antigen receptor (27). Therefore, nAChRs may interact with other nAChRs or non-nAChRs to modulate cell function. Essentially all RBL cells bind α-BTX, and we identified the expression of α7, α9, and α10 nAChR subunits by qPCR and sequence analysis of the PCR products. Moreover, the effects of NT on the late phase response were inhibited by preincubation with α-BTX or MLA, and these agents have been shown to inhibit the function of both α7 and α9 nAChRs (36, 37). Thus, it was possible that either α7 or α9/α10 nAChRs was involved in NT-induced changes in mast cell function. To differentiate between these possibilities, we used α7-, α9-, or α10-specific siRNA to selectively knockdown α7, α9, or α10 nAChRs from RBL cells. Indeed the mRNA expression of siRNAtreated cells established the specificity of the knockdown method; however, to our surprise ablation of α7, α9, or α10-nAChRs by siRNA abrogated the response of RBL to NT. Therefore, it appears that on the mast cells α7, α9, and α10 subunits might interact to form a “hybrid” nAChR that strongly increases the affinity of this novel nAChR to NT by several orders of magnitude and may explain the difference of nearly 5,000 fold in the amount of NT required to activate α7 nAChRs in neurons and the suppression of α-FcεRI-induced late phase response in mast cells. This property of the nAChRs on mast cells might be accessible to novel therapeutic approaches to control allergic diseases and allergic asthma.
Acknowledgments
Authors thank Paula Bradley and Vicki Fisher for editing/preparing the manuscript and Steve Randock for his help in graphics.
Abbreviations used in this paper
- NT
nicotine
- DEPC
diethylpyrocarbonate
- qPCR
real-time PCR
- LT
leukotriene
- cysLTs
cysteinyl leukotrienes
- nAChR
nicotinic acetylcholine receptor
- RBL
RBL-2H3 cells
- LTC4
Leukotriene C4
- α-BTX
α-bungarotoxin
- MLA
methyllycaconitine
- cPLA2
cytosolic phospholipase A2
Footnotes
This work was supported in part by grants from the National Institutes of Health (RO1 DA017003, R01 DA04208-17, and RO1DA04208S).
References
- 1.Peat JK, Li J. Reversing the trend: reducing the prevalence of asthma. J Allergy Clin Iimmunol. 1999;103:1–10. doi: 10.1016/s0091-6749(99)70517-8. [DOI] [PubMed] [Google Scholar]
- 2.Beasley R, Crane J, Lai CK, Pearce N. Prevalence and etiology of asthma. J Allergy Clin Immunol. 2000;105:S466–472. doi: 10.1016/s0091-6749(00)90044-7. [DOI] [PubMed] [Google Scholar]
- 3.Wuthrich B. Epidemiology of the allergic diseases: are they really on the increase? Int Arch Allergy Appl Immunol. 1989;90(Suppl 1):3–10. doi: 10.1159/000235067. [DOI] [PubMed] [Google Scholar]
- 4.von Mutius E. The rising trends in asthma and allergic disease. Clin Exp Allergy. 1998;28(Suppl 5):45–49. doi: 10.1046/j.1365-2222.1998.028s5045.x. discussion 50-41. [DOI] [PubMed] [Google Scholar]
- 5.Burr ML, Wat D, Evans C, Dunstan FD, Doull IJ. Asthma prevalence in 1973, 1988 and 2003. Thorax. 2006;61:296–299. doi: 10.1136/thx.2005.045682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner H, Kinet JP. Signalling through the high-affinity IgE receptor Fc epsilonRI. Nature. 1999;402:B24–30. doi: 10.1038/35037021. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz LB. Mast cells: function and contents. Curr Opin Immunol. 1994;6:91–97. doi: 10.1016/0952-7915(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 8.Mekori YA, Metcalfe DD. Mast cells in innate immunity. Immunol Rev. 2000;173:131–140. doi: 10.1034/j.1600-065x.2000.917305.x. [DOI] [PubMed] [Google Scholar]
- 9.Spada CS, Nieves AL, Krauss AH, Woodward DF. Comparison of leukotriene B4 and D4 effects on human eosinophil and neutrophil motility in vitro. J Leuk Biol. 1994;55:183–191. doi: 10.1002/jlb.55.2.183. [DOI] [PubMed] [Google Scholar]
- 10.Dahlen SE, Hedqvist P, Hammarstrom S, Samuelsson B. Leukotrienes are potent constrictors of human bronchi. Nature. 1980;288:484–486. doi: 10.1038/288484a0. [DOI] [PubMed] [Google Scholar]
- 11.Henderson WR., Jr The role of leukotrienes in inflammation. Ann Int Med. 1994;121:684–697. doi: 10.7326/0003-4819-121-9-199411010-00010. [DOI] [PubMed] [Google Scholar]
- 12.Eum SY, Maghni K, Hamid Q, Campbell H, Eidelman DH, Martin JG. Involvement of the cysteinyl-leukotrienes in allergen-induced airway eosinophilia and hyperresponsiveness in the mouse. Am J Repir Cell Mol Biol. 2003;28:25–32. doi: 10.1165/rcmb.4532. [DOI] [PubMed] [Google Scholar]
- 13.Parameswaran K, Cox G, Radford K, Janssen LJ, Sehmi R, O’Byrne PM. Cysteinyl leukotrienes promote human airway smooth muscle migration. Am J Respir Crit Care Med. 2002;166:738–742. doi: 10.1164/rccm.200204-291OC. [DOI] [PubMed] [Google Scholar]
- 14.Parameswaran K, Radford K, Fanat A, Stephen J, Bonnans C, Levy BD, Janssen LJ, Cox PG. Modulation of human airway smooth muscle migration by lipid mediators and Th-2 cytokines. Am J Repir Cell Mol Biol. 2007;37:240–247. doi: 10.1165/rcmb.2006-0172OC. [DOI] [PubMed] [Google Scholar]
- 15.Barbee RA, Halonen M, Kaltenborn W, Lebowitz M, Burrows B. A longitudinal study of serum IgE in a community cohort: correlations with age, sex, smoking, and atopic status. J Allergy Clin Immunol. 1987;79:919–927. doi: 10.1016/0091-6749(87)90241-7. [DOI] [PubMed] [Google Scholar]
- 16.Linneberg A, Nielsen NH, Madsen F, Frolund L, Dirksen A, Jorgensen T. Factors related to allergic sensitization to aeroallergens in a cross-sectional study in adults: The Copenhagen Allergy Study. Clin Exp Allergy. 2001;31:1409–1417. doi: 10.1046/j.1365-2222.2001.01178.x. [DOI] [PubMed] [Google Scholar]
- 17.Sopori M. Effects of cigarette smoke on the immune system. Nature Rev. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 18.Jarvis D, Chinn S, Luczynska C, Burney P. The association of smoking with sensitization to common environmental allergens: results from the European Community Respiratory Health Survey. J Allergy Clin Immunol. 1999;104:934–940. doi: 10.1016/s0091-6749(99)70071-0. [DOI] [PubMed] [Google Scholar]
- 19.Wuthrich B, Schindler C, Medici TC, Zellweger JP, Leuenberger P. IgE levels, atopy markers and hay fever in relation to age, sex and smoking status in a normal adult Swiss population. SAPALDIA (Swiss Study on Air Pollution and Lung Diseases in Adults) Team. Int Arch Allergy Immunol. 1996;111:396–402. doi: 10.1159/000237398. [DOI] [PubMed] [Google Scholar]
- 20.Mensinga TT, Schouten JP, Rijcken B, Weiss ST, Speizer FE, van der Lende R. The relationship of eosinophilia and positive skin test reactivity to respiratory symptom prevalence in a community-based population study. J Allergy Clin Immunol. 1990;86:99–107. doi: 10.1016/s0091-6749(05)80129-0. [DOI] [PubMed] [Google Scholar]
- 21.Baldacci S, Modena P, Carrozzi L, Pedreschi M, Vellutini M, Biavati P, Simoni M, Sapigni T, Viegi G, Paoletti P, Giuntini C. Skin prick test reactivity to common aeroallergens in relation to total IgE, respiratory symptoms, and smoking in a general population sample of northern Italy. Allergy. 1996;51:149–156. doi: 10.1111/j.1398-9995.1996.tb04579.x. [DOI] [PubMed] [Google Scholar]
- 22.Omenaas E, Bakke P, Elsayed S, Hanoa R, Gulsvik A. Total and specific serum IgE levels in adults: relationship to sex, age and environmental factors. Clin Exp Allergy. 1994;24:530–539. doi: 10.1111/j.1365-2222.1994.tb00950.x. [DOI] [PubMed] [Google Scholar]
- 23.Sopori ML, Kozak W, Savage SM, Geng Y, Soszynski D, Kluger MJ, Perryman EK, Snow GE. Effect of nicotine on the immune system: possible regulation of immune responses by central and peripheral mechanisms. Psychoneuroendocrinology. 1998;23:189–204. doi: 10.1016/s0306-4530(97)00076-0. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 25.Razani-Boroujerdi S, Singh SP, Knall C, Hahn FF, Pena-Philippides JC, Kalra R, Langley RJ, Sopori ML. Chronic nicotine inhibits inflammation and promotes influenza infection. Cell Immunol. 2004;230:1–9. doi: 10.1016/j.cellimm.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Mishra NC, Rir-Sima-Ah J, Langley RJ, Singh SP, Pena-Philippides JC, Koga T, Razani-Boroujerdi S, Hutt J, Campen M, Kim KC, Tesfaigzi Y, Sopori ML. Nicotine primarily suppresses lung Th2 but not goblet cell and muscle cell responses to allergens. J Immunol. 2008;180:7655–7663. doi: 10.4049/jimmunol.180.11.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Razani-Boroujerdi S, Boyd RT, Davila-Garcia MI, Nandi JS, Mishra NC, Singh SP, Pena-Philippides JC, Langley R, Sopori ML. T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol. 2007;179:2889–2898. doi: 10.4049/jimmunol.179.5.2889. [DOI] [PubMed] [Google Scholar]
- 28.Kageyama-Yahara N, Suehiro Y, Yamamoto T, Kadowaki M. IgE-induced degranulation of mucosal mast cells is negatively regulated via nicotinic acetylcholine receptors. Biochem Biophys Res Commun. 2008;377:321–325. doi: 10.1016/j.bbrc.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Kindt F, Wiegand S, Niemeier V, Kupfer J, Loser C, Nilles M, Kurzen H, Kummer W, Gieler U, Haberberger RV. Reduced expression of nicotinic alpha subunits 3, 7, 9 and 10 in lesional and nonlesional atopic dermatitis skin but enhanced expression of alpha subunits 3 and 5 in mast cells. Br J Dermatol. 2008;159:847–857. doi: 10.1111/j.1365-2133.2008.08774.x. [DOI] [PubMed] [Google Scholar]
- 30.Yoshimaru T, Suzuki Y, Matsui T, Yamashita K, Ochiai T, Yamaki M, Shimizu K. Blockade of superoxide generation prevents high-affinity immunoglobulin E receptor-mediated release of allergic mediators by rat mast cell line and human basophils. Clin Exp Allergy. 2002;32:612–618. doi: 10.1046/j.0954-7894.2002.01263.x. [DOI] [PubMed] [Google Scholar]
- 31.Ortega E, Hazan B, Zor U, Pecht I. Mast cell stimulation by monoclonal antibodies specific for the Fc epsilon receptor yields distinct responses of arachidonic acid and leukotriene C4 secretion. Eu J Immunol. 1989;19:2251–2256. doi: 10.1002/eji.1830191211. [DOI] [PubMed] [Google Scholar]
- 32.Hernandez-Hansen V, Smith AJ, Surviladze Z, Chigaev A, Mazel T, Kalesnikoff J, Lowell CA, Krystal G, Sklar LA, Wilson BS, Oliver JM. Dysregulated FcepsilonRI signaling and altered Fyn and SHIP activities in Lyn-deficient mast cells. J Immunol. 2004;173:100–112. doi: 10.4049/jimmunol.173.1.100. [DOI] [PubMed] [Google Scholar]
- 33.Chaturvedi MM, Mukhopadhyay A, Aggarwal BB. Assay for redoxsensitive transcription factor. Meth Enzymol. 2000;319:585–602. doi: 10.1016/s0076-6879(00)19055-x. [DOI] [PubMed] [Google Scholar]
- 34.Geng Y, Savage SM, Razani-Boroujerdi S, Sopori ML. Effects of nicotine on the immune response. II. Chronic nicotine treatment induces T cell anergy. J Immunol. 1996;156:2384–2390. [PubMed] [Google Scholar]
- 35.Razani-Boroujerdi S, Partridge LD, Sopori ML. Intracellular calcium signaling induced by thapsigargin in excitable and inexcitable cells. Cell Calcium. 1994;16:467–474. doi: 10.1016/0143-4160(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 36.Bray C, Son JH, Meizel S. Acetylcholine causes an increase of intracellular calcium in human sperm. Mol Hum Repro. 2005;11:881–889. doi: 10.1093/molehr/gah245. [DOI] [PubMed] [Google Scholar]
- 37.Osman AA, Schrader AD, Hawkes AJ, Akil O, Bergeron A, Lustig LR, Simmons DD. Muscle-like nicotinic receptor accessory molecules in sensory hair cells of the inner ear. Mol Cell Neurosci. 2008;38:153–169. doi: 10.1016/j.mcn.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz LB, Lewis RA, Seldin D, Austen KF. Acid hydrolases and tryptase from secretory granules of dispersed human lung mast cells. J Immunol. 1981;126:1290–1294. [PubMed] [Google Scholar]
- 40.Wershil BK, Mekori YA, Murakami T, Galli SJ. 125I-fibrin deposition in IgE-dependent immediate hypersensitivity reactions in mouse skin. Demonstration of the role of mast cells using genetically mast cell-deficient mice locally reconstituted with cultured mast cells. J Immunol. 1987;139:2605–2614. [PubMed] [Google Scholar]
- 41.Galli SJ. New concepts about the mast cell. New Eng J Med. 1993;328:257–265. doi: 10.1056/NEJM199301283280408. [DOI] [PubMed] [Google Scholar]
- 42.Mancel E, Drouet M, Sabbah A. Membrane stabilizers (chromones and ketotifen) Allergie Immunol. 1999;31:103–105. [PubMed] [Google Scholar]
- 43.Shakoory B, Fitzgerald SM, Lee SA, Chi DS, Krishnaswamy G. The role of human mast cell-derived cytokines in eosinophil biology. J Interferon Cytokine Res. 2004;24:271–281. doi: 10.1089/107999004323065057. [DOI] [PubMed] [Google Scholar]
- 44.Lorentz A, Klopp I, Gebhardt T, Manns MP, Bischoff SC. Role of activator protein 1, nuclear factor-kappaB, and nuclear factor of activated T cells in IgE receptor-mediated cytokine expression in mature human mast cells. J Allergy Clin Immunol. 2003;111:1062–1068. doi: 10.1067/mai.2003.1342. [DOI] [PubMed] [Google Scholar]
- 45.Ali K, Bilancio A, Thomas M, Pearce W, Gilfillan AM, Tkaczyk C, Kuehn N, Gray A, Giddings J, Peskett E, Fox R, Bruce I, Walker C, Sawyer C, Okkenhaug K, Finan P, Vanhaesebroeck B. Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature. 2004;431:1007–1011. doi: 10.1038/nature02991. [DOI] [PubMed] [Google Scholar]
- 46.Jeong HJ, Koo HN, Na HJ, Kim MS, Hong SH, Eom JW, Kim KS, Shin TY, Kim HM. Inhibition of TNF-alpha and IL-6 production by Aucubin through blockade of NF-kappaB activation RBL-2H3 mast cells. Cytokine. 2002;18:252–259. doi: 10.1006/cyto.2002.0894. [DOI] [PubMed] [Google Scholar]
- 47.Murakami M, Kudo I, Umeda M, Matsuzawa A, Takeda M, Komada M, Fujimori K, Takahashi K, Inoue K. Detection of three distinct phospholipases A2 in cultured mast cells. Journal of biochemistry. 1992;111:175–181. doi: 10.1093/oxfordjournals.jbchem.a123733. [DOI] [PubMed] [Google Scholar]
- 48.Hirasawa N, Santini F, Beaven MA. Activation of the mitogen-activated protein kinase/cytosolic phospholipase A2 pathway in a rat mast cell line. Indications of different pathways for release of arachidonic acid and secretory granules. J Immunol. 1995;154:5391–5402. [PubMed] [Google Scholar]
- 49.Sgard F, Charpantier E, Bertrand S, Walker N, Caput D, Graham D, Bertrand D, Besnard F. A novel human nicotinic receptor subunit, alpha10, that confers functionality to the alpha9-subunit. Mol Pharmacol. 2002;61:150–159. doi: 10.1124/mol.61.1.150. [DOI] [PubMed] [Google Scholar]
- 50.Wilson BS, Oliver JM. Effector roles of IgE antibodies: targeting allergen to the high-affinity IgE receptor for Fc epsilon RI-dependent signaling and antigen presentation. Clin Allergy Immunol. 2002;16:197–232. [PubMed] [Google Scholar]
- 51.Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Ann Rev Immunol. 2005;23:749–78. doi: 10.1146/annurev.immunol.21.120601.141025. [DOI] [PubMed] [Google Scholar]
- 52.Jackson JA, Riggs MW, Spiekerman AM. Testosterone deficiency as a risk factor for hip fractures in men: a case-control study. Am J Med Sci. 1992;304:4–8. doi: 10.1097/00000441-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 53.Chen Y, Pappu BP, Zeng H, Xue L, Morris SW, Lin X, Wen R, Wang D. B cell lymphoma 10 is essential for FcepsilonR-mediated degranulation and IL-6 production in mast cells. J Immunol. 2007;178:49–57. doi: 10.4049/jimmunol.178.1.49. [DOI] [PubMed] [Google Scholar]
- 54.Alkondon M, Pereira EF, Almeida LE, Randall WR, Albuquerque EX. Nicotine at concentrations found in cigarette smokers activates and desensitizes nicotinic acetylcholine receptors in CA1 interneurons of rat hippocampus. Neuropharmacology. 2000;39:2726–2739. doi: 10.1016/s0028-3908(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 55.Geng Y, Savage SM, Johnson LJ, Seagrave J, Sopori ML. Effects of nicotine on the immune response. I. Chronic exposure to nicotine impairs antigen receptor-mediated signal transduction in lymphocytes. Toxicol Appl Pharmacol. 1995;135:268–278. doi: 10.1006/taap.1995.1233. [DOI] [PubMed] [Google Scholar]
- 56.Glover S, de Carvalho MS, Bayburt T, Jonas M, Chi E, Leslie CC, Gelb MH. Translocation of the 85-kDa phospholipase A2 from cytosol to the nuclear envelope in rat basophilic leukemia cells stimulated with calcium ionophore or IgE/antigen. J Biol Chem. 1995;270:15359–15367. doi: 10.1074/jbc.270.25.15359. [DOI] [PubMed] [Google Scholar]
- 57.Chang WC, Di Capite J, Singaravelu K, Nelson C, Halse V, Parekh AB. Local Ca2+ influx through Ca2+ release-activated Ca2+ (CRAC) channels stimulates production of an intracellular messenger and an intercellular pro-inflammatory signal. J Biol Chem. 2008;283:4622–4631. doi: 10.1074/jbc.M705002200. [DOI] [PubMed] [Google Scholar]
- 58.Zaidi AK, Thangam ER, Ali H. Distinct roles of Ca2+ mobilization and G protein usage on regulation of Toll-like receptor function in human and murine mast cells. Immunology. 2006;119:412–420. doi: 10.1111/j.1365-2567.2006.02450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rivera J, Olivera A. A current understanding of Fc epsilon RI-dependent mast cell activation. Current Allergy Asthma Rep. 2008;8:14–20. doi: 10.1007/s11882-008-0004-z. [DOI] [PubMed] [Google Scholar]
- 60.Akimoto M, Mishra K, Lim KT, Tani N, Hisanaga SI, Katagiri T, Elson A, Mizuno K, Yakura H. Protein tyrosine phosphatase epsilon is a negative regulator of FcepsilonRI-mediated mast cell responses. Scan J Immunol. 2009;69:401–411. doi: 10.1111/j.1365-3083.2009.02235.x. [DOI] [PubMed] [Google Scholar]
- 61.Oliver JM, Burg DL, Wilson BS, McLaughlin JL, Geahlen RL. Inhibition of mast cell Fc epsilon R1-mediated signaling and effector function by the Syk-selective inhibitor, piceatannol. J Biol Chem. 1994;269:29697–29703. [PubMed] [Google Scholar]
- 62.Zhang J, Berenstein EH, Evans RL, Siraganian RP. Transfection of Syk protein tyrosine kinase reconstitutes high affinity IgE receptor-mediated degranulation in a Syk-negative variant of rat basophilic leukemia RBL-2H3 cells. J Exp Med. 1996;184:71–79. doi: 10.1084/jem.184.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kawakami Y, Kitaura J, Satterthwaite AB, Kato RM, Asai K, Hartman SE, Maeda-Yamamoto M, Lowell CA, Rawlings DJ, Witte ON, Kawakami T. Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cell activation. J Immunol. 2000;165:1210–1219. doi: 10.4049/jimmunol.165.3.1210. [DOI] [PubMed] [Google Scholar]
- 64.Nishizumi H, Yamamoto T. Impaired tyrosine phosphorylation and Ca2+ mobilization, but not degranulation, in lyn-deficient bone marrow-derived mast cells. J Immunol. 1997;158:2350–2355. [PubMed] [Google Scholar]
- 65.Vonakis BM, Gibbons SP, Jr, Rotte MJ, Brothers EA, Kim SC, Chichester K, MacDonald SM. Regulation of rat basophilic leukemia-2H3 mast cell secretion by a constitutive Lyn kinase interaction with the high affinity IgE receptor (Fc epsilon RI) J Immunol. 2005;175:4543–4554. doi: 10.4049/jimmunol.175.7.4543. [DOI] [PubMed] [Google Scholar]
- 66.Parravicini V, Gadina M, Kovarova M, Odom S, Gonzalez-Espinosa C, Furumoto Y, Saitoh S, Samelson LE, O’Shea JJ, Rivera J. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nature Immunol. 2002;3:741–748. doi: 10.1038/ni817. [DOI] [PubMed] [Google Scholar]
- 67.Kawashima K, Fujii T. Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front Biosci. 2004;9:2063–2085. doi: 10.2741/1390. [DOI] [PubMed] [Google Scholar]
- 68.Kummer W, Lips KS, Pfeil U. The epithelial cholinergic system of the airways. Histochem Cell Biol. 2008;130:219–234. doi: 10.1007/s00418-008-0455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boyd RT. The molecular biology of neuronal nicotinic acetylcholine receptors. Cr Rev Toxicol. 1997;27:299–318. doi: 10.3109/10408449709089897. [DOI] [PubMed] [Google Scholar]
- 70.Lips KS, Konig P, Schatzle K, Pfeil U, Krasteva G, Spies M, Haberberger RV, Grando SA, Kummer W. Coexpression and spatial association of nicotinic acetylcholine receptor subunits alpha7 and alpha10 in rat sympathetic neurons. J Mol Neurosci. 2006;30:15–16. doi: 10.1385/JMN:30:1:15. [DOI] [PubMed] [Google Scholar]