

Abstract
Epigenetics are defined, in broad-terms, as alterations in gene expression without changes in DNA sequence. While histone modifications and DNA methylation are two classical means to regulate gene expression, miRNA has also recently been documented to govern gene expression in normal as well as cancer cells. In this review, we will first describe briefly histone modifications, DNA methylation and miRNAs and the functions of these epigenetic marks during different cellular processes involving DNA metabolism. We will then highlight some epigenetic changes in glioblastomas, a malignant form of brain tumor, and potential application of epigenetic means for diagnosis, prognosis, and treatment of gliomas. We expect that novel therapies will be developed to counter epigenetic changes in this deadly disease.
Keywords: PTEN, PI3K, cell cycle, P53, nuclear PTEN, glioblastoma
Introduction
In eukaryotic cells, DNA is packaged into chromatin, a complex structure of DNA and proteins. The basic repeat unit of chromatin is a nucleosome that is composed of 147 bp of DNA wrapped around a histone octamer consisting of two copies each of four core histones H2A, H2B, H3, and H4. The chromatin structures encode epigenetic information, which is defined as heritable changes of gene expression without alteration in DNA sequence.1 In the postgenomic era where DNA sequences of many organisms are known, there has been extensive interest in studying chromatin structures and their role in the regulation of gene expression.2 This interest arises from the following two facts. First, epigenetic regulation of gene expression plays a critical role in the regulation of a variety of cellular processes including gene expression, DNA replication and DNA repair, and stem cell maintenance and differentiation.3,4 Second, epigenetic alterations in gene expression play a causal role in a variety of human diseases including carcinogenesis and aging.4 While there are outstanding reviews on epigenetic changes in cancer cells, we will focus our discussion on studies related to epigenetic alteration in gliomas.5-7
Histone modifications regulate diverse cellular processes that are critical for cancer prevention
Chromatin is dynamically regulated to impact different cellular processes.8-10 One way to regulate chromatin is through post-translational modifications on histones. Histone modifications include acetylation, methylation, phosphorylation, sumoylation, poly(ADP)-ribosylation, and ubiquitination (Figure 1). Over 60 modifications on histones have been described so far. The “histone code” refers to the patterns of modifications where different combinations of histone modifications designate or regulate specific cellular processes and events.9,11-13 One modification or pattern of modifications can alter the chromatin structure and/or aid in the recruitment of other proteins involved in diverse cellular processes including gene transcription, DNA replication and DNA repair.12-14 Below we discuss histone acetylation and methylation, two well-studied modifications that have been implicated to play a role in carcinogenesis.
Figure 1.

Posttranslational modifications on histones. Four core histones H3, H4, H2A and H2B adopt a similar secondary structural fold called the histone fold domain or core domain of histones (circle). Moreover, the N-termini and C-termini of these core histones are called histone tails. Amino acid residues, represented by one letter, at the N-terminal tails of each core histones as well as posttranslational modifications including phosphorylation (blue oval) acetylation (green diamond), methylation (red circle) and ubiquitination (yellow cross) are shown.
Acetylation is the addition of an acetyl group from the co-factor acetyl coenzyme A to the amino group of lysine residues on histones catalyzed by group enzymes called histone acetyltransfereases (HAT).15 Acetylation on histones, in general, has two distinct functions. First, histone acetylation can loosen the interactions between histones and DNA because acetylation neutralizes positive charge on a lysine residue and thereby reduces electrostatic interactions between DNA and histones. Second, acetylation on a particular lysine residue can also recruit proteins to chromatin to perform specific functions.16,17 The bromodomain, the classical domain that recognizes acetyllysine and is often present on transcriptional factors and co-activators, recruits these factors to chromatin during transcription.18,19 Therefore, it is not surprising that histone acetylation is often associated with actively transcribed chromatin regions (Figure 2). In addition to its role in transcription, histone acetylation also plays an important role in regulation of DNA replication and DNA repair. For example, acetylation of a histone variant H2A.X is required for histone exchange during DNA lesions.20 Moreover, acetylation of lysine 56 of H3 in yeast has been shown to be associated with replication and repair by promoting efficient nucleosome assembly.21-25 Thus, histone acetylation can impact or regulate diverse cellular processes.
Figure 2.
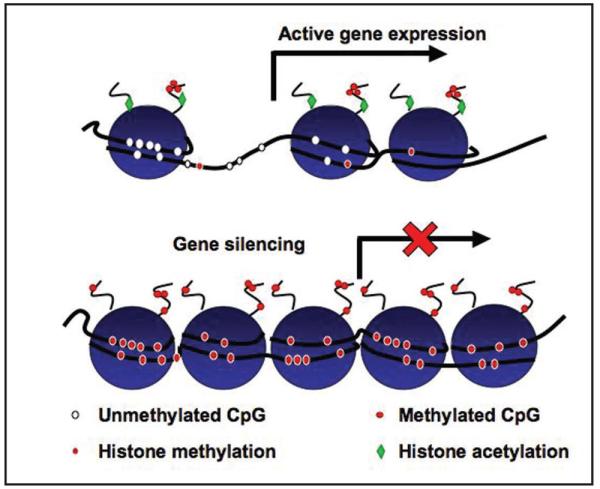
Epigenetic marks at the promoters of actively transcribed and silenced genes. When a gene is transcribed, histones are generally acetylated and methylated at lysine 4 of H3. In addition, cytosines in CpG islands are hypomethylated. On the other hand, when a gene is silenced, histones are generally hypoacetylated as well as methylated either at lysine 9 of H3 or lysine 27 of H3. Furthermore, the cytosines at the CpG islands are generally hypermethylated.
In mammalian cells, some of the primary HAT families include the GNAT (Gcn5-related N-acetyltransferase), MYST (MOZ, Ybf2/Sas3, Sas2, Tip60), and p300/CBP.15,17 Often HATs are part of larger complexes that confer specificity to the enzyme complex.17,26 Moreover, some, if not all, histone acetyltransferases also acetylate non-histone substrates.17 Thus, histone acetyltransferases have been properly renamed as lysine acetyltransferases (KAT) recently.27 Like other posttranslational modifications, histone acetylation is reversible. Removal of an acetyl group from a lysine residue or deacetylation, is carried out by histone deacetylases (HDACs).28,29 There are four different classes of HDACs, and like HATs, tend to function within larger complexes.5,6,30 While histone acetylation is linked to gene activity, histone deacetylation is generally linked to transcriptional repression.
Another well-studied histone modification is methylation, which can occur at lysine and arginine residues.5,6,8 Up to three methyl groups can be added to a lysine residue and it appears that different states of methylation on a lysine residue have different biological outcomes.12 In contrast to histone acetylation, which is always associated with active gene expression, histone methylation can serve as a mark for active and inactive transcription. For instance, methylation of lysine 9 of histone H3 (H3K9), lysine 27 of H3 (H3K27) and lysine 20 of H4 (H4K20) is associated with transcriptional repression, whereas methylation of lysine residues of 4, 36 and 79 of H3 is associated with transcriptional activation (Table 1, Figure 2).12,13 In addition to its role in gene transcription, histone methylation is also linked to the DNA damage response. For instance, methylation of lysine 20 of H4 is involved in the recruitment of 53BP1, the DNA damage checkpoint adaptor protein to the site of DNA damage.31 Unlike acetylation, methylation usually does not modify histone and DNA interactions; instead, effector proteins or “readers” recognize a particular methylated lysine residue and thereby regulate chromatin structure and function.12,32 For example, H3K9 methylation is linked to heterochromatin formation, which is important for genome integrity and cell differentiation. H3K9 methylation is recognized by the bromodomain, like those that are present in SWI6/ heterochromatin protein 1 (HP1).33
Table 1.
The function of different histone modifications on histones H3 and H4
| Type of Modification | Residue Modified | Function |
|---|---|---|
| Acetylation | H3K9 | transcriptional activation |
| H3K27 | transcriptional activation | |
| H3K56 | transcriptional activation; nucleosome assembly | |
| H4K16 | chromatin decondensation | |
| Methylation | H3K4 | transcriptional activation |
| H3K9 | heterochromatin formation; transcriptional repression | |
| H3K27 | transcriptional repression | |
| H3K36 | transcriptional activation | |
| H3K79 | transcriptional activation; DNA damage response | |
| H4K20 | transcriptional repression; DNA damage response |
This table describes two types of modifications of Histone H3 and H4: acetylation and methylation. The function of each modification is also described.
Methylation is catalyzed by lysine or arginine methyltransferases. The majority of histone methyltransferases contain the SET domain, which is the catalytic domain of the methyltransferase. Methylation can be reversed by histone demethylases, most of which contain the catalytic domain JmjC. Most histone methyltransferases appear to target fewer lysine residues and exhibit higher specificity than histone acetyltransferases.32
DNA methylation and its role in silencing and cancer
In addition to histone modification, DNA methylation is also an epigenetic mark that is important for regulating chromatin structure and gene expression.34 In mammalian cells, DNA methylation occurs predominantly on cytosine residues, and it is estimated that about 60-80% of CpG dinucleotides are methylated.34 Most DNA methylation is found at heterochromatin regions and it is believed that DNA methylation is important for the maintenance of the heterochromatin structure and transcriptional silencing in higher eukaryotic cells (Figure 2). The enzymes that catalyze DNA methylation are DNA methyltransferases (DNMT).35 So far, three enzymes, DNMT1, DNMT3a, and DNMT3b, have DNA methyltransferase activities. DNMT1 preferentially methylates hemimethylated DNA and is responsible for maintenance of the methylation patterns during DNA replication. DNMT3a and DNMT3b methylate unmethylated DNA substrates and are responsible for de novo methylation.36,37
DNA methylation was one of the first epigenetic modifications described in cancer cells. There is a profound loss of DNA methylation in cancer cells, predominantly at repetitive DNA sequences elements.38,39 It is hypothesized that loss of DNA methylation at these DNA sequences results in genomic instability.40 In addition to a global loss of DNA methylation, DNA hypermethylation has been discovered at the promoter regions of many tumor suppressor genes.7,41,42 In mammalian cells, about half of the genes contain clusters of CpG dinucleotides called CpG islands near or within their promoter.43 Most of these CpG islands are unmethylated in normal cells. However, in cancer cells, the CpG islands of many tumor suppressor genes and growth regulator genes such as those involved in cell cycle regulation, DNA repair, cell-cell interactions, apoptosis, and angiogenesis are methylated, which leads to transcriptional silencing of these genes.
While it is not clear how cancer cells utilize the cellular silencing machinery to silence a tumor suppressor gene, several studies have led to the following general conclusions. First, it appears that DNA methylation occurs after changes in histone modifications (e.g. gain of a silencing histone mark (H3K9 methylation) and loss of histone acetylation) at the promoter regions of a tumor suppressor gene during the process of transcriptional silencing.44 Second, silencing of tumor suppressor genes such as p16 in colon cancer cells may be triggered by anti-sense transcripts that originate from the first exon of genes.45 Third, a large number of genes are required for the maintenance of genes silenced in cancer cells.46 However, it is not clear whether these genes are also involved in the maintenance of other tumor suppressor genes in other types of cancer cells.
Non-coding RNA and cancer
Non-coding RNAs have been known to play an important role in regulation of gene expression. For instance, XIST RNA is important for the initiation of heterochromatin formation during X chromosome inactivation.47 Recently, small non-coding RNAs including microRNAs (miRNAs) and small RNAs have been found to regulate gene expression.48 Intriguingly, miRNAs have been shown to play a tumor suppressing function by reducing the expression of oncogenes as well as an oncogenic role by downregulating the expression of tumor suppressors.49
In order for miRNA and small RNA to silence gene expression, these RNAs are first processed by the nuclease Dicer into 21-22 nucleotide products, which then form a complex with the RISC complex. RNA in the RISC complex then base pairs in a sequence specific manner with the 3′ untranslated region or coding regions of the target RNA and affect gene expression either by the degradation of mRNA or repression of translation (Figure 3).50,51 Not surprisingly, miRNA expression itself can be regulated via changes in chromatin structures induced by covalent modifications on histones and/or DNA methylation.50
Figure 3.
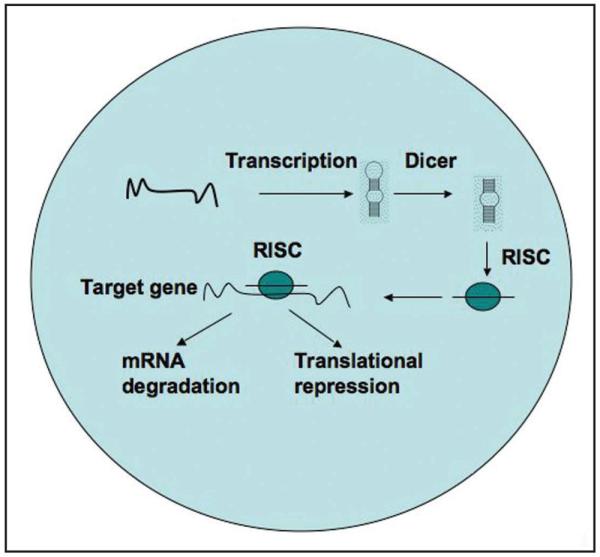
Expression of microRNA (miRNA) regulates gene expression via two mechanisms. MicroRNAs or small non-coding RNAs, when transcribed, are processed by the nuclease Dicer. The processed miRNAs bind to the RISC complex and then target the RISC complex to its target genes. The miRNAs then repress gene expression by inducing degradation of mRNA of the target genes or by inhibiting translation of the target gene.
Epigenetic changes in glioblastoma multiforme
Epigenetic modifications have been of great interest as a method of diagnosing GBM as potential prognostic factors and as response factors for treatment. Gliomas are the most common type of primary brain tumor. Glioblastoma multiforme (GBM) is a form of malignant and aggressive brain tumor, with a mean survival of nine to twelve months.52 The term “glioblastoma” is synonymous with “grade IV astrocytoma” using the World Health Organization (WHO) classification and grading system.53 There are two types of GBMs: primary and secondary. Primary glioblastomas are those malignant tumors that have a short history and no prior clinical disease while secondary glioblastomas are those that develop from a lower grade gliomas.52 Secondary gliomas typically progress from grade II (astrocytoma) to grade III (anaplastic astroctyoma) and finally to grade IV (GBM) (Figure 4A).53 Seventy percent of grade II gliomas progress to grade III and IV tumors within five to ten years of diagnosis.
Figure 4.

MGMT silencing may be a useful mark for predicting the prognosis and efficacy of treatment of glioma patients treated with temozolomide. A) A diagram depicting different grades of gliomas highlighting both genetic and epigenetic changes that contribute to tumorigenesis. B) The silencing of MGMT, a protein involved in DNA repair, is associated with poor prognosis and may serve as a predictive factor for TMZ treatment.
Compared to the treatment of other types of cancer, treatment for GBM is more difficult because drugs need to be able to cross the blood brain barrier to reach the tumor and avoid protein efflux. In addition, GBM has a high level of heterogeneity, which can affect tumor response to treatment.54 Because of these complexities, the treatment of glioblastoma has been modified over the years as our understanding of this disease increases. Early treatments of gliomas included only surgery and radiation therapy, whereas chemotherapy with alkylating agents, a nitrosurea-based therapy, was introduced later. 52
Like studies in other cancer types, most studies of the epigenetic changes in glioblastomas have focused on DNA methylation as this mark is relatively easy to detect. From these studies, a large number of tumor suppressor genes silenced in glioblastoma have been identified and these genes are listed in Table 2. Below, we focus our discussion on some of these genes.
Table 2.
A list of genes that are silenced in gliomas
| Gene Name | Gene | Function | References |
|---|---|---|---|
| O6-methylguanine methyltransferase | MGMT | DNA Repair | [55-57,93] |
| mutL homolog 1 | MLH1 | DNA Repair | [94,95] |
| Target of Methylation Induced Silencing | TMS1/ASC | apoptosis regulation | [67,96] |
| Tissue factor pathway inhibitor 2 | TFPI-2 | protease inhibition | [97] |
| cyclin dependent kinase inhibitor 2A | p16/CDKN2A | cell cycle regulation | [65,93,98-101] |
| epithelial membrane protein 3 | EMP3 | proliferation | [69,70] |
| Ras association (RalGDS/AF-6) domain family member 1 | RASSF1A | tumor suppressor | |
| cell cycle regulation | [102-105] | ||
| phosphate and tensin homolog | PTEN | tumor suppressor; proliferation | [106,107] |
| tumor protein 53 | TP53 | cell cycle regulation; apoptosis | [93] |
| death associated protein kinase | DAPK | apoptosis regulation; angiogenesis inhibition | [93] |
| p14ARF | p14 | cell cycle regulation | [93,101,108-110] |
| cyclin dependent kinase inhibitor 2B | p15/CDKN2B | cell cycle regulation | [45,101,109] |
| retinablastoma 1 | RB1 | cell cycle regulation | [93,101,109, 111,112] |
| zinc finger , MYND-type containing 10 | BLU | tumor suppressor | [102,105] |
| tumor protein 73 | TP73 | cell cycle regulation; apoptosis | [93,104,109, 110,113] |
| tachykinin precursor 1 | TAC1 | neurotransmitter | [114] |
| TSPY-like 5 | TSPYL5 | nucleosome assembly | [114] |
| cystatin E/M | CST6 | protease inhibition | [114] |
| Bcl-2 interacting killer | BIK | apoptosis | [114] |
| tumor associated cancer signal transducer 2 | TACSTD2 | signal transduction | [114] |
| chloride intracellular channel 3 | CLIC3 | growth regulation | [114] |
| transcription factor DP family, member 3 | TFDP3 | transcription regulation | [114] |
| ribonuclease T2 | RNASET2 | tumorigenesis | [114] |
| cytidine deaminase | CDA | cytidine metabolism | [114] |
| cysteine deoxygenase, type 1 | CDO1 | cysteine regulation | [114] |
| TSPY-like 2 | TSPYL2 | nucleosome assembly | [114] |
| TSPY-like 3 | TSPYL3 | nucleosome assembly | [114] |
| TSPY-like 6 | TSPYL6 | nucleosome assembly | [114] |
| Glutathione S-transferase P1 | GSTP1 | detoxification | [93] |
| thrombospondin-1 | THBS1 | angiogenesis inhibition | [93,115] |
| tissue inhibitor of metalloproteinase 3 | TIMP3 | protease inhibition | [93] |
| androgen receptor | AR | transcription factor | [104] |
| cadherin type 1, E-cadherin | CDH1 | cell adhesion | [104,116] |
| POU domain 3, transcription factor 1 | OCT6 | transcription factor | [104] |
| metallothionein 1A | MT1A | antioxidant | [104] |
| wilms tumor 1 | WT1 | transcription factor; development | [104] |
| interferon regulatory factor 7 | IRF7 | apoptosis regulation | [104] |
| protocadherin gamma subfamily A, 11 | PCDH-γ-A11 | cell adhesion | [117] |
| solute carrier family 5 (iodide transporter), member 8 | SLC5A8 | ion transport; apoptosis; cell cycle regulation | [96] |
| large tumor suppressor, homolog 1 | LATS1 | cell cycle regulation | [118] |
| large tumor suppressor, homolog 2 | LATS2 | cell cycle regulation | [118] |
| testis derived transcript | TES | protein-protein interactions; tumor suppressor | [119] |
| runt related transcription factor 3 | RUNX | transcription factor; proliferation | [119] |
| ATPase, Na+/K+ transporting, beta 2 polypeptide | ATP1B2/AMOG | ion transport; cell size regulation | [120] |
| apolipoprotein D | APOD | lipid transport and metabolism | [120] |
| dmx-like 1 | DMXL1 | unknown; family members involved in cell regulatory processes |
[120] |
| family with seqeunce similiarity 107, member A | FAM107A/DRR1 | cell growth regulation | [120] |
| pleckstrin and Sec 7 domain containing 3 | PSD3 | signal transduction regulation | [120] |
| harakiri, BCL2 interacting protein | HRK | apoptosis | [121] |
| caspase 8 | caspase 8 | apoptosis | [122] |
| Estrogen Receptor 1 | ESR1 | transcription | [99,116] |
| calcitonin related polypeptide alpha | CALCA | calcium regulation, signaling, regulation | [116] |
| myogenic differentiation 1 | MYOD1 | differentiation, muscle development | [116] |
| Ras association (RalGDS/AF-6) domain family member 5 | RASSF5/NORE1A | apoptosis, signal transduction, cell cycle regulation | [105] |
| tumor suppressor candidate 3 | TUSC3/N33 | tumor suppressor | [99] |
| hypermethylated in cancer 1 | HIC1 | cell cycle and transcription regulation | [99] |
| cyclin dependent kinase inhibitor 1A | CDKN1A/p21 | cell cycle regulation | [109] |
| cyclin dependent kinase inhibitor 2B | CDKN1B/p27kip1 | cell cycle regulation | [109] |
| carboxy terminal modulator protein | CTMP/ THEM4 | Akt inhibition | [123] |
In this table, the names of genes that have been shown to be silenced in gliomas are listed. Moreover, the potential function of each gene and reference are also listed.
One of the most widely studied genes silenced in GBM is O6-methyl guanine methyltransferase (MGMT). MGMT is involved in DNA repair by removing alkyl groups from the O6 of guanine nucleotides. The O6-methylguanine nucleotides tend to pair with thymine and/or chemically react with other bases. Thus, the methylated guanine nucleotide will result in a base pair exchange thereby forming lethal crosslinks if not repaired. MGMT plays an important role in repairing these alterations before mutations can occur.55
DNA methylation of MGMT is a frequent event in GBM. For instance, it is estimated that 68% of glioblastoma samples exhibit MGMT promoter methylation.56,57 There are 97 CpG islands in the MGMT promoter, and these CpG islands are further divided into two hypermethylated regions.58 In addition to DNA hypermethylation, histone H3-K9 dimethylation and deacetylation, two other markers of gene silencing, have also been detected at the promoter of MGMT.59 Therefore, it is likely that multiple silencing mechanisms cooperate to silence MGMT in GBM. Because of its critical role in DNA repair, the epigenetic silencing of MGMT is associated with an increased number of mutations and poor outcome in GBM. Thus, MGMT silencing is considered a biomarker of poor prognosis.60 However, several studies have indicated that patients with silenced MGMT respond much more positively to the treatment of temozolomide than unmethylated MGMT (Figure 4B).61,62 Therefore, temozolomide has been used routinely to treat those patients exhibiting MGMT promoter methylation. Without the ability to remove methyl groups from O6-methylguanine, methylated nucleotides are introduced into DNA causing cytoxicity through the formation of lethal crosslinks. Alkylating agents such as temozolomide, carmustine (BCNU), and nimustine (ACNU) aid in such cytoxicity, thereby killing the cancer cells.63 DNA methylation of MGMT is now considered a possible predictive marker for response to temozolomide treatment despite the fact that there has been some debate as to how well MGMT methylation may predict temozolomide efficacy.54,62 The use of temozolomide based on the status of MGMT methylation highlights how important it is to understand epigenetic changes in GBM for the discovery of novel therapy and/or prognostic factors for the treatment of this deadly cancer.60-62
P16INK4A is located on chromosome 9p21 and is involved in cell cycle regulation through Cdk inactivation.64 Methylation of the p16/CDKN2 gene has been found in numerous tumors including gliomas.65 One study estimates that 73.9% of GBMs have reduced p16 protein levels, of which 76.5% of these cases exhibit hypermethylation at the promoter of p16.66 In addition, there are changes in the chromatin structure as well as DNA methylation at the promoter of p16.65
DNA promoter methylation was also observed at the promoter of the TSC1/ASC gene in association with GBM. TSC1/ASC1 is involved in the regulation of apoptosis, NF-κB activation and cytokine maturation. One study shows that CpG island hypermethylation of the TSC1 gene is observed in 43% of primary glioblastomas and suggests that silencing of TSC1 via promoter methylation contributes to GBM pathogenesis.67 Moreover, TSC1 hypermethylation seems to coincide with MGMT methylation in glioblastoma patients who show long term survival, suggesting that different epigenetic signatures may exist in different types of GBM.68
Another gene found to be epigenetically modified in gliomas is the epithelial membrane protein 3 gene (EMP3). EMP3 promoter hypermethylation is associated with reduced EMP3 mRNA expression and is considered to be an unfavorable prognostic marker in neuroblastoma patients.69 Recent studies suggest that DNA methylation of EMP3 may be an early alteration in astrocytoma, being found in 80% of anaplastic, diffuse, and secondary glioblastomas and only 17% of primary glioblastomas. Therefore, the silencing of EMP3 may be a marker that distinguishes primary from secondary glioblastomas.70
As mentioned earlier, in addition to DNA methylation and histone modifications, microRNAs have been shown to regulate gene expression.71,72 Several miRNAs including miR-128, miR-181a, miR-181b, miR-181c, and miR-221 have altered expression in GBM. MiR-221 is overexpressed while miR-128 and the miR-181 family members, normally enriched in brain, appear to be down regulated in GBM samples.73 MiR-21, another microRNA found in the brain, is thought to act as an oncogene in GBM. Elevated levels of miR-21 were found to be associated with GBM and decreased apoptosis. The inhibition of miR-21 leads to caspase activation and induction of apoptosis.74 The genes that are regulated by miR-21 are still unclear, but the proposed neural targets include SPOCK1, a proteoglycan, and two transcription regulators, ZMYND11 and RB1CC1.75 Recent evidence shows that miR-21 may be regulating genes involved in migration and invasion.76 In the future, it would be interesting to determine how alterations in these miRNA molecules as well as other epigenetic machinery such as DNA methylation and histone modification contribute to brain tumor development.
Inhibitors against DNA methyltransferase and histone deacetylases
Epigenetic silencing of tumor suppressor genes plays a causal role in carcinogenesis. Because epigenetic silencing of tumor suppressor genes is reversible, there is extensive interest in developing inhibitors against DNA methyltransferases and histone deacetylases for the re-expression of TSG silenced in cancer cells. In fact, 5-aza-2-deoxycytidine (decitabine), an inhibitor of DNA methyltransferases, has been approved to treat myelodysplastic syndrome as well as some leukemias by the FDA. However, the inhibitors of DNA methyltransferases have proved to be ineffective for solid tumors to date.77
Histone deacetylase inhibitors (HDACis) have also shown promise as the next generation of anti-cancer drugs. Five classes of HDACis including short chain fatty acids, hydroxamic acids, cyclic tetrapeptides with an amino group, cyclic peptides without an amino group and benzamides have been developed so far.78 Some of these inhibitors are now being tested in clinical trials. Most HDAC inhibitors target the active site of the HDACs and/or block interaction between the enzymes and their substrates, which in turn, results in hyperacetylation of histones and proteins.79
Several histone deacetylase inhibitors have been tested for the activation of genes silenced in glioma cells. Of these, two histone deacetylase inhibitors, FK228 and SAHA, are worth mentioning. FK228, or depsipeptide, belongs to bicyclic tetrapeptide class of histone deacetylase inhibitors and shows specificity for class I HDACs. FK228 exhibits efficacy in numerous cancer cell lines, including breast, colon, and lung, as well as growth suppression in solid tumor models including breast carcinoma and large cell lung carcinoma.80,81 In clinical trials, FK228 has shown promising results in T-cell lymphoma.82 FK228 has been hypothesized to function via the degradation of mutant p53, the promotion of Hsp90 acetylation leading to further degradation of oncogenic proteins, the activation of apoptosis, and the inhibition of angiogenesis.83 FK228 induces apoptosis as well as suppresses cell proliferation in glioblastoma cell lines and animal models through the inhibition of several cell cycle proteins, including cyclin D1 and WAF1.84 For this reason, FK228 is being studied as a potential GBM therapeutic agent.
The second well-studied histone deacetylase inhibitor is suberoylanilide hydroxamic acid (SAHA), a drug that has been approved by the FDA recently for the treatment of cutaneous T cell lymphoma. SAHA has anticancer activity against both hematologic and solid tumors. SAHA can induce cell cycle arrest, terminal differentiation, and cell death in a variety of transformed cells.85 Moreover, it possesses anti-glioma activity in vitro and in a rat model in vivo.86 One recent study suggests that SAHA can cross the blood-brain barrier to inhibit the growth of malignant gliomas in mouse models.87 Similarly, another independent study indicates that SAHA is effective in reducing glioma growth in vivo and increased survival when administered by continuous local intracranial delivery.88 SAHA has also been tested for the treatment of patients with recurrent GBM in a Phase II clinical trial. An early report suggests that SAHA is generally well-tolerated and has activity against GBM.89 Moreover, a Phase I trial of SAHA plus temozolomide (TMZ) in patients with malignant gliomas has reported that these drugs are well tolerated.89 Based on these studies, clinical trials soon to be started will be testing SAHA in combination with TMZ/RT in newly diagnosed GBM. SAHA will also be tested in combination with other cell cycle inhibitors, such as proteasome inhibitors in recurrent GBM (Evanthia Galanis, personal communication). However, many of the targets of many histone deacetylase inhibitors, including SAHA are unknown.
In addition to histones, histone deacetylases also deacetylate other proteins including p53. Therefore, most of histone deacetylase inhibitors not only inhibit deacetylation of histones, but other protein substrates as well. To make things more complicated, almost all of the histone deacetylase inhibitors developed so far inhibit multiple classes of histone deacetylases. While one hypothesis suggests that the chromatin structure of abnormally silenced genes is more susceptible to drugs and reactivation than those genes that have been silenced in normal cells, it is still important to use epigenomics to predict response to drugs and to access epigenetic markers for diagnostic, prognostic, and therapeutic responses.4,90 Moreover, designing and testing inhibitors against each specific class of histone deacetylases may prove useful for the treatment of cancer. Lastly, it may be worthwhile to develop inhibitors against histone methyltransferases/demethylases to regulate gene expression. Compared to histone deacetylases, histone methyltransferases/demethylases are more selective against particular lysine residues. In principle, inhibitors against these histone methyltransfeases and histone demethylases are likely to be more specific for regulation of gene expression. Recently a small molecule inhibitor was found for G9a, one of the methyltransferases that contributes to H3K9 methylation. Although G9a is not known to be involved in cancer, it is considered to represent the future of epigenetic directed therapy. 91
Conclusion
Epigenetic alterations in gene expression are becoming increasingly important for the study of cancer. Future studies should focus on documenting all the epigenetic changes in glioma, finding key molecular targets that lead to tumoriogenesis and discovering more specific inhibitors towards histone deacetylases and histone methyltransferases. A combination of these approaches as well as the anticipated advancements in epigenomics in the near future will lead to identify novel molecular marks and novel therapy for the diagnostics and prognosis and treatment of cancer.92 With the current poor prognosis of GBM, it will probably beneficial and helpful to profile epigenetic alterations in brain tumors and test whether novel epigenetic drugs as well as combinations of existing drugs have improved anti-glioma activity in the hope that these studies will find a better treatment for this deadly disease.
Acknowledgements
This work is supported by a pilot project grant from the Brain Tumor SPORE at Mayo Clinic (Z. Z) and Brain Tumor SPORE (R.J.) and from NIH grants (Z.Z). We would like to thank Dr. Evanthia Galanis for her critical comments and helpful suggestions.
References
- 1.Henikoff S, Furuyama T, Ahmad K. Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet. 2004;20:320–6. doi: 10.1016/j.tig.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Schones DE, Zhao K. Genome-wide approaches to studying chromatin modifications. Nat Rev Genet. 2008;9:179–91. doi: 10.1038/nrg2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–8. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marks PA, Richon VM, Kelly WK, Chiao JH, Miller T. Histone deacetylase inhibitors: development as cancer therapy. Novartis Found Symp. 2004;259:269–81. discussion 281-8. [PubMed] [Google Scholar]
- 6.Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137–68. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- 7.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–60. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 9.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 10.Sedivy JM, Banumathy G, Adams PD. Aging by epigenetics--a consequence of chromatin damage? Exp Cell Res. 2008;314:1909–17. doi: 10.1016/j.yexcr.2008.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 12.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 13.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 14.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 15.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 16.Carrozza MJ, Utley RT, Workman JL, Cote J. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003;19:321–9. doi: 10.1016/S0168-9525(03)00115-X. [DOI] [PubMed] [Google Scholar]
- 17.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–95. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 18.Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–7. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- 19.Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–8. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 20.Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR, 3rd, Abmayr SM, Washburn MP, Workman JL. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306:2084–7. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 21.Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–5. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- 22.Han J, Zhou H, Li Z, Xu RM, Zhang Z. The Rtt109-Vps75 histone acetyltransferase complex acetylates non-nucleosomal histone H3. J Biol Chem. 2007;282:14158–64. doi: 10.1074/jbc.M700611200. [DOI] [PubMed] [Google Scholar]
- 23.Han J, Zhou H, Li Z, Xu RM, Zhang Z. Acetylation of lysine 56 of histone H3 catalyzed by Rtt109 and regulated by Asf1 is required for replisome integrity. J Biol Chem. 2007;282:28587–28596. doi: 10.1074/jbc.M702496200. [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008;134:244–55. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–43. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–59. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–6. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 28.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–26. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 29.Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol. 2003;4:276–84. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]
- 30.Hildmann C, Riester D, Schwienhorst A. Histone deacetylases--an important class of cellular regulators with a variety of functions. Appl Microbiol Biotechnol. 2007;75:487–97. doi: 10.1007/s00253-007-0911-2. [DOI] [PubMed] [Google Scholar]
- 31.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–73. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–18. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 33.Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 34.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 35.Griffiths EA, Gore SD. DNA methyltransferase inhibitors: class effect or unique agents? Leuk Lymphoma. 2008;49:650–1. doi: 10.1080/10428190801947575. [DOI] [PubMed] [Google Scholar]
- 36.Hermann A, Gowher H, Jeltsch A. Biochemistry and biology of mammalian DNA methyltransferases. Cell Mol Life Sci. 2004;61:2571–87. doi: 10.1007/s00018-004-4201-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quina AS, Buschbeck M, Di Croce L. Chromatin structure and epigenetics. Biochem Pharmacol. 2006;72:1563–9. doi: 10.1016/j.bcp.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 38.Feinberg AP. The epigenetics of cancer etiology. Semin Cancer Biol. 2004;14:427–32. doi: 10.1016/j.semcancer.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 39.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 40.House MG, Herman JG, Guo MZ, Hooker CM, Schulick RD, Lillemoe KD, Cameron JL, Hruban RH, Maitra A, Yeo CJ. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg. 2003;238:423–31. doi: 10.1097/01.sla.0000086659.49569.9e. discussion 431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell. 2002;1:299–305. doi: 10.1016/s1535-6108(02)00061-2. [DOI] [PubMed] [Google Scholar]
- 42.van Noesel MM, van Bezouw S, Voute PA, Herman JG, Pieters R, Versteeg R. Clustering of hypermethylated genes in neuroblastoma. Genes Chromosomes Cancer. 2003;38:226–33. doi: 10.1002/gcc.10278. [DOI] [PubMed] [Google Scholar]
- 43.Antequera F, Bird A. CpG islands. Exs. 1993;64:169–85. doi: 10.1007/978-3-0348-9118-9_8. [DOI] [PubMed] [Google Scholar]
- 44.Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB, Kinzler KW, Vogelstein B. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell. 2003;3:89–95. doi: 10.1016/s1535-6108(02)00234-9. [DOI] [PubMed] [Google Scholar]
- 45.Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–6. doi: 10.1038/nature06468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449:1073–7. doi: 10.1038/nature06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anguera MC, Sun BK, Xu N, Lee JT. X-chromosome kiss and tell: how the Xs go their separate ways. Cold Spring Harb Symp Quant Biol. 2006;71:429–37. doi: 10.1101/sqb.2006.71.012. [DOI] [PubMed] [Google Scholar]
- 48.Costa FF. Non-coding RNAs, epigenetics and complexity. Gene. 2008;410:9–17. doi: 10.1016/j.gene.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 49.Gartel AL, Kandel ES. miRNAs: Little known mediators of oncogenesis. Semin Cancer Biol. 2008;18:103–10. doi: 10.1016/j.semcancer.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 50.Saito Y, Jones PA. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle. 2006;5:2220–2. doi: 10.4161/cc.5.19.3340. [DOI] [PubMed] [Google Scholar]
- 51.Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–26. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–33. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 53.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reardon DA, Rich JN, Friedman HS, Bigner DD. Recent advances in the treatment of malignant astrocytoma. J Clin Oncol. 2006;24:1253–65. doi: 10.1200/JCO.2005.04.5302. [DOI] [PubMed] [Google Scholar]
- 55.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59:793–7. [PubMed] [Google Scholar]
- 56.Nakamura M, Watanabe T, Yonekawa Y, Kleihues P, Ohgaki H. Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C --> A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis. 2001;22:1715–9. doi: 10.1093/carcin/22.10.1715. [DOI] [PubMed] [Google Scholar]
- 57.Blanc JL, Wager M, Guilhot J, Kusy S, Bataille B, Chantereau T, Lapierre F, Larsen CJ, Karayan-Tapon L. Correlation of clinical features and methylation status of MGMT gene promoter in glioblastomas. J Neurooncol. 2004;68:275–83. doi: 10.1023/b:neon.0000033385.37098.85. [DOI] [PubMed] [Google Scholar]
- 58.Qian XC, Brent TP. Methylation hot spots in the 5′ flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997;57:3672–7. [PubMed] [Google Scholar]
- 59.Nakagawachi T, Soejima H, Urano T, Zhao W, Higashimoto K, Satoh Y, Matsukura S, Kudo S, Kitajima Y, Harada H, Furukawa K, Matsuzaki H, Emi M, Nakabeppu Y, Miyazaki K, Sekiguchi M, Mukai T. Silencing effect of CpG island hypermethylation and histone modifications on O6-methylguanine-DNA methyltransferase (MGMT) gene expression in human cancer. Oncogene. 2003;22:8835–44. doi: 10.1038/sj.onc.1207183. [DOI] [PubMed] [Google Scholar]
- 60.Komine C, Watanabe T, Katayama Y, Yoshino A, Yokoyama T, Fukushima T. Promoter hypermethylation of the DNA repair gene O6-methylguanine-DNA methyltransferase is an independent predictor of shortened progression free survival in patients with low-grade diffuse astrocytomas. Brain Pathol. 2003;13:176–84. doi: 10.1111/j.1750-3639.2003.tb00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paz MF, Yaya-Tur R, Rojas-Marcos I, Reynes G, Pollan M, Aguirre-Cruz L, Garcia-Lopez JL, Piquer J, Safont MJ, Balana C, Sanchez-Cespedes M, Garcia-Villanueva M, Arribas L, Esteller M. CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas. Clin Cancer Res. 2004;10:4933–8. doi: 10.1158/1078-0432.CCR-04-0392. [DOI] [PubMed] [Google Scholar]
- 62.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 63.Jacinto FV, Esteller M. MGMT hypermethylation: a prognostic foe, a predictive friend. DNA Repair (Amst) 2007;6:1155–60. doi: 10.1016/j.dnarep.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 64.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 65.Costello JF, Berger MS, Huang HS, Cavenee WK. Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer Res. 1996;56:2405–10. [PubMed] [Google Scholar]
- 66.Park SH, Jung KC, Ro JY, Kang GH, Khang SK. 5′ CpG island methylation of p16 is associated with absence of p16 expression in glioblastomas. J Korean Med Sci. 2000;15:555–9. doi: 10.3346/jkms.2000.15.5.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stone AR, Bobo W, Brat DJ, Devi NS, Van Meir EG, Vertino PM. Aberrant methylation and down-regulation of TMS1/ASC in human glioblastoma. Am J Pathol. 2004;165:1151–61. doi: 10.1016/S0002-9440(10)63376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martinez R, Schackert G, Esteller M. Hypermethylation of the proapoptotic gene TMS1/ASC: prognostic importance in glioblastoma multiforme. J Neurooncol. 2007;82:133–9. doi: 10.1007/s11060-006-9264-4. [DOI] [PubMed] [Google Scholar]
- 69.Alaminos M, Davalos V, Ropero S, Setien F, Paz MF, Herranz M, Fraga MF, Mora J, Cheung NK, Gerald WL, Esteller M. EMP3, a myelin-related gene located in the critical 19q13.3 region, is epigenetically silenced and exhibits features of a candidate tumor suppressor in glioma and neuroblastoma. Cancer Res. 2005;65:2565–71. doi: 10.1158/0008-5472.CAN-04-4283. [DOI] [PubMed] [Google Scholar]
- 70.Kunitz A, Wolter M, van den Boom J, Felsberg J, Tews B, Hahn M, Benner A, Sabel M, Lichter P, Reifenberger G, von Deimling A, Hartmann C. DNA hypermethylation and aberrant expression of the EMP3 gene at 19q13.3 in Human Gliomas. Brain Pathol. 2007;17:363–70. doi: 10.1111/j.1750-3639.2007.00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–22. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 73.Ciafre SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM, Farace MG. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005;334:1351–8. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 74.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 75.Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, Ambros VR, Israel MA. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67:2456–68. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- 76.Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, Krichevsky AM. MiR-21 Promotes Glioma Invasion by Targeting MMP Regulators. Mol Cell Biol. 2008;28:5369–80. doi: 10.1128/MCB.00479-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer. 2008;112:2341–51. doi: 10.1002/cncr.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mehnert JM, Kelly WK. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J. 2007;13:23–9. doi: 10.1097/PPO.0b013e31803c72ba. [DOI] [PubMed] [Google Scholar]
- 79.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–99. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 80.Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J Antibiot (Tokyo) 1994;47:301–10. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]
- 81.Ueda H, Manda T, Matsumoto S, Mukumoto S, Nishigaki F, Kawamura I, Shimomura K. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice. J Antibiot (Tokyo) 1994;47:315–23. doi: 10.7164/antibiotics.47.315. [DOI] [PubMed] [Google Scholar]
- 82.Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L, Kingma DM, Turner ML, Altemus R, Bates SE. Inhibitor of histone deacetylation, depsipeptide ( FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood. 2001;98:2865–8. doi: 10.1182/blood.v98.9.2865. [DOI] [PubMed] [Google Scholar]
- 83.Konstantinopoulos PA, Vandoros GP, Papavassiliou AG. FK228 (depsipeptide): a HDAC inhibitor with pleiotropic antitumor activities. Cancer Chemother Pharmacol. 2006;58:711–5. doi: 10.1007/s00280-005-0182-5. [DOI] [PubMed] [Google Scholar]
- 84.Sawa H, Murakami H, Kumagai M, Nakasato M, Yamauchi S, Matsuyama N, Tamura Y, Satone A, Ide W, Hashimoto I, Kamada H. Histone deacetylase inhibitor, FK228, induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo. Acta Neuropathol. 2004;107:523–31. doi: 10.1007/s00401-004-0841-3. [DOI] [PubMed] [Google Scholar]
- 85.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–9. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 86.Eyupoglu IY, Hahnen E, Buslei R, Siebzehnrubl FA, Savaskan NE, Luders M, Trankle C, Wick W, Weller M, Fahlbusch R, Blumcke I. Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J Neurochem. 2005;93:992–9. doi: 10.1111/j.1471-4159.2005.03098.x. [DOI] [PubMed] [Google Scholar]
- 87.Yin D, Ong JM, Hu J, Desmond JC, Kawamata N, Konda BM, Black KL, Koeffler HP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo. Clin Cancer Res. 2007;13:1045–52. doi: 10.1158/1078-0432.CCR-06-1261. [DOI] [PubMed] [Google Scholar]
- 88.Ugur HC, Ramakrishna N, Bello L, Menon LG, Kim SK, Black PM, Carroll RS. Continuous intracranial administration of suberoylanilide hydroxamic acid (SAHA) inhibits tumor growth in an orthotopic glioma model. J Neurooncol. 2007;83:267–75. doi: 10.1007/s11060-007-9337-z. [DOI] [PubMed] [Google Scholar]
- 89.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Giannini C, Hardwick JS, Moore DF, Zwiebel JA, Buckner JC. NCCTG phase II trial of vorinostat (suberoylanilide hydroxamic acid) in recurrent glioblastoma multiforme (GBM) J Clin Oncol. 2007;25(18S:pt 1):76s. ASCO Annual Meeting Proceedings. [Google Scholar]
- 90.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 91.Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–81. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 92.Moving AHEAD with an international human epigenome project. Nature. 2008;454:711–5. doi: 10.1038/454711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gonzalez-Gomez P, Bello MJ, Arjona D, Lomas J, Alonso ME, De Campos JM, Vaquero J, Isla A, Gutierrez M, Rey JA. Promoter hypermethylation of multiple genes in astrocytic gliomas. Int J Oncol. 2003;22:601–8. [PubMed] [Google Scholar]
- 94.Fukushima T, Katayama Y, Watanabe T, Yoshino A, Ogino A, Ohta T, Komine C. Promoter hypermethylation of mismatch repair gene hMLH1 predicts the clinical response of malignant astrocytomas to nitrosourea. Clin Cancer Res. 2005;11:1539–44. doi: 10.1158/1078-0432.CCR-04-1625. [DOI] [PubMed] [Google Scholar]
- 95.Gomori E, Pal J, Meszaros I, Doczi T, Matolcsy A. Epigenetic inactivation of the hMLH1 gene in progression of gliomas. Diagn Mol Pathol. 2007;16:104–7. doi: 10.1097/PDM.0b013e318033f140. [DOI] [PubMed] [Google Scholar]
- 96.Jiang Z, Li XG, Hu J, Lu DR, Zhou W, Jiang YQ, Li CY. The methylation and mRNA expression of SLC5A8 and TMS1/ASC genes in human glioma. Zhonghua Yi Xue Za Zhi. 2007;87:292–7. [PubMed] [Google Scholar]
- 97.Konduri SD, Srivenugopal KS, Yanamandra N, Dinh DH, Olivero WC, Gujrati M, Foster DC, Kisiel W, Ali-Osman F, Kondraganti S, Lakka SS, Rao JS. Promoter methylation and silencing of the tissue factor pathway inhibitor-2 (TFPI-2), a gene encoding an inhibitor of matrix metalloproteinases in human glioma cells. Oncogene. 2003;22:4509–16. doi: 10.1038/sj.onc.1206695. [DOI] [PubMed] [Google Scholar]
- 98.Fueyo J, Gomez-Manzano C, Bruner JM, Saito Y, Zhang B, Zhang W, Levin VA, Yung WK, Kyritsis AP. Hypermethylation of the CpG island of p16/CDKN2 correlates with gene inactivation in gliomas. Oncogene. 1996;13:1615–9. [PubMed] [Google Scholar]
- 99.Li Q, Jedlicka A, Ahuja N, Gibbons MC, Baylin SB, Burger PC, Issa JP. Concordant methylation of the ER and N33 genes in glioblastoma multiforme. Oncogene. 1998;16:3197–202. doi: 10.1038/sj.onc.1201831. [DOI] [PubMed] [Google Scholar]
- 100.Fan X, Munoz J, Sanko SG, Castresana JS. PTEN, DMBT1, and p16 alterations in diffusely infiltrating astrocytomas. Int J Oncol. 2002;21:667–74. [PubMed] [Google Scholar]
- 101.Yin D, Xie D, Hofmann WK, Miller CW, Black KL, Koeffler HP. Methylation, expression, and mutation analysis of the cell cycle control genes in human brain tumors. Oncogene. 2002;21:8372–8. doi: 10.1038/sj.onc.1206031. [DOI] [PubMed] [Google Scholar]
- 102.Hesson L, Bieche I, Krex D, Criniere E, Hoang-Xuan K, Maher ER, Latif F. Frequent epigenetic inactivation of RASSF1A and BLU genes located within the critical 3p21.3 region in gliomas. Oncogene. 2004;23:2408–19. doi: 10.1038/sj.onc.1207407. [DOI] [PubMed] [Google Scholar]
- 103.Horiguchi K, Tomizawa Y, Tosaka M, Ishiuchi S, Kurihara H, Mori M, Saito N. Epigenetic inactivation of RASSF1A candidate tumor suppressor gene at 3p21.3 in brain tumors. Oncogene. 2003;22:7862–5. doi: 10.1038/sj.onc.1207082. [DOI] [PubMed] [Google Scholar]
- 104.Yu J, Zhang H, Gu J, Lin S, Li J, Lu W, Wang Y, Zhu J. Methylation profiles of thirty four promoter-CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer. 2004;4:65. doi: 10.1186/1471-2407-4-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lorente A, Mueller W, Urdangarin E, Lazcoz P, Lass U, von Deimling A, Castresana JS. RASSF1A, BLU, NORE1A, PTEN and MGMT Expression and Promoter Methylation in Gliomas and Glioma Cell Lines and Evidence of Deregulated Expression of de novo DNMTs. Brain Pathol. 2008 doi: 10.1111/j.1750-3639.2008.00185.x. 10.1111/j.1750-3639.2008.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wiencke JK, Zheng S, Jelluma N, Tihan T, Vandenberg S, Tamguney T, Baumber R, Parsons R, Lamborn KR, Berger MS, Wrensch MR, Haas-Kogan DA, Stokoe D. Methylation of the PTEN promoter defines low-grade gliomas and secondary glioblastoma. Neuro Oncol. 2007;9:271–9. doi: 10.1215/15228517-2007-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Baeza N, Weller M, Yonekawa Y, Kleihues P, Ohgaki H. PTEN methylation and expression in glioblastomas. Acta Neuropathol. 2003;106:479–85. doi: 10.1007/s00401-003-0748-4. [DOI] [PubMed] [Google Scholar]
- 108.Nakamura M, Watanabe T, Klangby U, Asker C, Wiman K, Yonekawa Y, Kleihues P, Ohgaki H. p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol. 2001;11:159–68. doi: 10.1111/j.1750-3639.2001.tb00388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ohta T, Watanabe T, Katayama Y, Yoshino A, Yachi K, Ogino A, Komine C, Fukushima T. Aberrant promoter hypermethylation profile of cell cycle regulatory genes in malignant astrocytomas. Oncol Reporter. 2006;16:957–63. [PubMed] [Google Scholar]
- 110.Watanabe T, Katayama Y, Yoshino A, Yachi K, Ohta T, Ogino A, Komine C, Fukushima T. Aberrant hypermethylation of p14ARF and O6-methylguanine-DNA methyltransferase genes in astrocytoma progression. Brain Pathol. 2007;17:5–10. doi: 10.1111/j.1750-3639.2006.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H. Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest. 2001;81:77–82. doi: 10.1038/labinvest.3780213. [DOI] [PubMed] [Google Scholar]
- 112.Gonzalez-Gomez P, Bello MJ, Alonso ME, Arjona D, Lomas J, de Campos JM, Isla A, Rey JA. CpG island methylation status and mutation analysis of the RB1 gene essential promoter region and protein-binding pocket domain in nervous system tumours. Br J Cancer. 2003;88:109–14. doi: 10.1038/sj.bjc.6600737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Watanabe T, Huang H, Nakamura M, Wischhusen J, Weller M, Kleihues P, Ohgaki H. Methylation of the p73 gene in gliomas. Acta Neuropathol. 2002;104:357–62. doi: 10.1007/s00401-002-0549-1. [DOI] [PubMed] [Google Scholar]
- 114.Kim TY, Zhong S, Fields CR, Kim JH, Robertson KD. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res. 2006;66:7490–501. doi: 10.1158/0008-5472.CAN-05-4552. [DOI] [PubMed] [Google Scholar]
- 115.Li Q, Ahuja N, Burger PC, Issa JP. Methylation and silencing of the Thrombospondin-1 promoter in human cancer. Oncogene. 1999;18:3284–9. doi: 10.1038/sj.onc.1202663. [DOI] [PubMed] [Google Scholar]
- 116.Uhlmann K, Rohde K, Zeller C, Szymas J, Vogel S, Marczinek K, Thiel G, Nurnberg P, Laird PW. Distinct methylation profiles of glioma subtypes. Int J Cancer. 2003;106:52–9. doi: 10.1002/ijc.11175. [DOI] [PubMed] [Google Scholar]
- 117.Waha A, Guntner S, Huang TH, Yan PS, Arslan B, Pietsch T, Wiestler OD. Epigenetic silencing of the protocadherin family member PCDH-gamma-A11 in astrocytomas. Neoplasia. 2005;7:193–9. doi: 10.1593/neo.04490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jiang Z, Li X, Hu J, Zhou W, Jiang Y, Li G, Lu D. Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci Res. 2006;56:450–8. doi: 10.1016/j.neures.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 119.Mueller W, Nutt CL, Ehrich M, Riemenschneider MJ, von Deimling A, van den Boom D, Louis DN. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene. 2007;26:583–93. doi: 10.1038/sj.onc.1209805. [DOI] [PubMed] [Google Scholar]
- 120.van den Boom J, Wolter M, Blaschke B, Knobbe CB, Reifenberger G. Identification of novel genes associated with astrocytoma progression using suppression subtractive hybridization and real-time reverse transcription-polymerase chain reaction. Int J Cancer. 2006;119:2330–8. doi: 10.1002/ijc.22108. [DOI] [PubMed] [Google Scholar]
- 121.Nakamura M, Ishida E, Shimada K, Nakase H, Sakaki T, Konishi N. Frequent HRK inactivation associated with low apoptotic index in secondary glioblastomas. Acta Neuropathol. 2005;110:402–10. doi: 10.1007/s00401-005-1065-x. [DOI] [PubMed] [Google Scholar]
- 122.Ashley DM, Riffkin CD, Muscat AM, Knight MJ, Kaye AH, Novak U, Hawkins CJ. Caspase 8 is absent or low in many ex vivo gliomas. Cancer. 2005;104:1487–96. doi: 10.1002/cncr.21323. [DOI] [PubMed] [Google Scholar]
- 123.Knobbe CB, Reifenberger J, Blaschke B, Reifenberger G. Hypermethylation and transcriptional downregulation of the carboxyl-terminal modulator protein gene in glioblastomas. J Natl Cancer Inst. 2004;96:483–6. doi: 10.1093/jnci/djh064. [DOI] [PubMed] [Google Scholar]