

Abstract
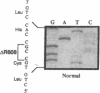
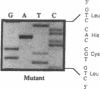
Types A and B Niemann-Pick disease both result from the deficient activity of the lysosomal hydrolase, acid sphingomyelinase (E.C. 3.1.4.12). Type A Niemann-Pick disease is a severe neurodegenerative disorder of infancy which leads to death by three years of age, whereas Type B disease has a later age at onset, little or no neurologic involvement, and most patients survive into adulthood. To investigate the molecular basis for the remarkable phenotypic heterogeneity, the nature of the mutations causing Type B Niemann-Pick disease in Ashkenazi Jewish patients was determined. The entire acid sphingomyelinase coding region from an Ashkenazi Jewish Type B patient was polymerase chain reaction-amplified, subcloned, and completely sequenced. A three-base deletion was identified of nucleotides 1821-1823 in the cDNA which predicted the removal of an arginine residue from position 608 of the acid sphingomyelinase polypeptide (delta R608). The other cDNA clones from this patient had the R496L mutation previously identified in Type A Niemann-Pick disease patients. Both Ashkenazi Jewish Type B patients were heteroallelic for the delta R608 mutation, whereas this allele was not present in 15 unrelated non-Jewish Type B patients, with the notable exception of one mildly affected patient of Arabic descent who was homoallelic for the delta R608 mutation. These results indicate that the delta R608 mutation predicts the Type B Niemann-Pick disease phenotype, even in the presence of the R496L Type A allele, thereby providing the first genotype/phenotype correlation for this lysosomal storage disease. Although only two patients have been studied, it appears that the delta R608 mutation occurs frequently in Type B Niemann-Pick disease patients of Ashkenazi Jewish descent.
Full text
PDF





Images in this article




Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bernstein H. S., Bishop D. F., Astrin K. H., Kornreich R., Eng C. M., Sakuraba H., Desnick R. J. Fabry disease: six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene. J Clin Invest. 1989 Apr;83(4):1390–1399. doi: 10.1172/JCI114027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besley G. T., Elleder M. Enzyme activities and phospholipid storage patterns in brain and spleen samples from Niemann-Pick disease variants: a comparison of neuropathic and non-neuropathic forms. J Inherit Metab Dis. 1986;9(1):59–71. doi: 10.1007/BF01813904. [DOI] [PubMed] [Google Scholar]
- Besley G. T., Hoogeboom A. J., Hoogeveen A., Kleijer W. J., Galjaard H. Somatic cell hybridisation studies showing different gene mutations in Niemann-Pick variants. Hum Genet. 1980;54(3):409–412. doi: 10.1007/BF00291589. [DOI] [PubMed] [Google Scholar]
- Bishop D. F., Desnick R. J. Affinity purification of alpha-galactosidase A from human spleen, placenta, and plasma with elimination of pyrogen contamination. Properties of the purified splenic enzyme compared to other forms. J Biol Chem. 1981 Feb 10;256(3):1307–1316. [PubMed] [Google Scholar]
- Brady R. O., Kanfer J. N., Mock M. B., Fredrickson D. S. The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick diseae. Proc Natl Acad Sci U S A. 1966 Feb;55(2):366–369. doi: 10.1073/pnas.55.2.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROCKER A. C. The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J Neurochem. 1961 Apr;7:69–80. doi: 10.1111/j.1471-4159.1961.tb13499.x. [DOI] [PubMed] [Google Scholar]
- Chatterjee S., Ghosh N. Neutral sphingomyelinase from human urine. Purification and preparation of monospecific antibodies. J Biol Chem. 1989 Jul 25;264(21):12554–12561. [PubMed] [Google Scholar]
- Gal A. E., Fash F. J. Synthesis of 2-n-(hexadecanoyl)-amino-4-nitrophenyl phosphorylcholine-hydroxide, a chromogenic substrate for assaying sphingomyelinase activity. Chem Phys Lipids. 1976 Feb;16(1):71–79. doi: 10.1016/0009-3084(76)90015-3. [DOI] [PubMed] [Google Scholar]
- Itakura K., Rossi J. J., Wallace R. B. Synthesis and use of synthetic oligonucleotides. Annu Rev Biochem. 1984;53:323–356. doi: 10.1146/annurev.bi.53.070184.001543. [DOI] [PubMed] [Google Scholar]
- Klar R., Levade T., Gatt S. Synthesis of pyrenesulfonylamido-sphingomyelin and its use as substrate for determining sphingomyelinase activity and diagnosing Niemann-Pick disease. Clin Chim Acta. 1988 Sep 15;176(3):259–267. doi: 10.1016/0009-8981(88)90185-4. [DOI] [PubMed] [Google Scholar]
- Levade T., Salvayre R., Douste-Blazy L. Sphingomyelinases and Niemann-Pick disease. J Clin Chem Clin Biochem. 1986 Apr;24(4):205–220. [PubMed] [Google Scholar]
- Levran O., Desnick R. J., Schuchman E. H. Niemann-Pick disease: a frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc Natl Acad Sci U S A. 1991 May 1;88(9):3748–3752. doi: 10.1073/pnas.88.9.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navon R., Proia R. L. The mutations in Ashkenazi Jews with adult GM2 gangliosidosis, the adult form of Tay-Sachs disease. Science. 1989 Mar 17;243(4897):1471–1474. doi: 10.1126/science.2522679. [DOI] [PubMed] [Google Scholar]
- Neufeld E. F. Natural history and inherited disorders of a lysosomal enzyme, beta-hexosaminidase. J Biol Chem. 1989 Jul 5;264(19):10927–10930. [PubMed] [Google Scholar]
- Poulos A., Ranieri E., Shankaran P., Callahan J. W. Studies on the activation of sphingomyelinase activity in Niemann-Pick type A, B, and C fibroblasts: enzymological differentiation of types A and B. Pediatr Res. 1984 Nov;18(11):1088–1093. doi: 10.1203/00006450-198411000-00006. [DOI] [PubMed] [Google Scholar]
- Quintern L. E., Schuchman E. H., Levran O., Suchi M., Ferlinz K., Reinke H., Sandhoff K., Desnick R. J. Isolation of cDNA clones encoding human acid sphingomyelinase: occurrence of alternatively processed transcripts. EMBO J. 1989 Sep;8(9):2469–2473. doi: 10.1002/j.1460-2075.1989.tb08382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki R. K., Gelfand D. H., Stoffel S., Scharf S. J., Higuchi R., Horn G. T., Mullis K. B., Erlich H. A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988 Jan 29;239(4839):487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977 Dec;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider P. B., Kennedy E. P. Sphingomyelinase in normal human spleens and in spleens from subjects with Niemann-Pick disease. J Lipid Res. 1967 May;8(3):202–209. [PubMed] [Google Scholar]
- Schuchman E. H., Suchi M., Takahashi T., Sandhoff K., Desnick R. J. Human acid sphingomyelinase. Isolation, nucleotide sequence and expression of the full-length and alternatively spliced cDNAs. J Biol Chem. 1991 May 5;266(13):8531–8539. [PubMed] [Google Scholar]
- da Veiga Pereira L., Desnick R. J., Adler D. A., Disteche C. M., Schuchman E. H. Regional assignment of the human acid sphingomyelinase gene (SMPD1) by PCR analysis of somatic cell hybrids and in situ hybridization to 11p15.1----p15.4. Genomics. 1991 Feb;9(2):229–234. doi: 10.1016/0888-7543(91)90246-b. [DOI] [PubMed] [Google Scholar]