

A6-month-old male infant was sent to a quaternary referral hospital from a tertiary referral hospital with a possible diagnosis of a fatty acid oxidation disorder.
HISTORY OF PRESENTING ILLNESS
The infant was well until 4 weeks before admission at the quaternary referral hospital. He, along with the rest of his family, experienced an acute gastroenteritic illness with vomiting and watery diarrhea. Although his siblings and parents recovered, he did not, and because of concerns of a presumed secondary lactose intolerance he was changed from his usual Similac (Ross Products, Abbott Laboratories, Columbus, Oh) formula to a soy-based formula (Alsoy, Nestle Inc, Eau Claire, Wisc) about one week into his illness. He became more lethargic and hypotonic with persisting vomiting and diarrhea, and was admitted to a tertiary referral hospital.
PAST HISTORY
He was born to nonconsanguineous parents of Italian (father) and Hungarian-French (mother) descent. He had two female siblings both of whom had been well in the past, and there was no family history of notable diseases. During pregnancy his mother had a laparoscopic cholecystectomy at 20 weeks’ gestation, and he was born by Cesarean delivery at term because of fetal bradycardia. There were no other problems in the perinatal period. He had been well before this current illness, with appropriate growth parameters and development. He was fed a combination of breast milk and formula (Similac, Ross Pharmaceuticals). Cereals were introduced at 5 months along with other age-appropriate foods such as bananas and some vegetables.
TERTIARY REFERRAL HOSPITAL
Symptoms of diarrhea and poor feeding had persisted for approximately two weeks before admission to the tertiary referral hospital, where he was found to be weak, floppy, and lethargic but easily rousable. He had massive hepatomegaly, with the liver edge palpable in the right iliac fossa, which had not been previously noted on several abdominal examinations. There was no splenomegaly, ascites, or signs of chronic liver disease. He was not clinically jaundiced despite elevated conjugated bilirubin. An Escherichia coli urinary tract infection (UTI) was diagnosed, although fever was not noted. A subsequent micturating cystourethrogram was normal.
Initial hematology and biochemical results are shown in Tables I and II. Of note is the presence of hypoglycemia, hypokalemia, hypophosphatemia, low plasma carnitine levels, hepatic dysfunction and coagulopathy. Other investigations that were normal included venous blood gas, plasma lactate, amino acids, creatine kinase, galactosemia screen, and routine urinalysis (ketones negative). Gas chromatography mass spectroscopy analysis of urinary organic acids showed elevated dicarboxylic acids with hypoketosis at the time of hypoglycemia. Urinary succinylacetone was negative. Serology for hepatitis viruses A, B, and C was negative. An echocardiogram demonstrated thickening of the interventricular septum, suggestive of a possible cardiomyopathy. An abdominal ultrasonogram showed an enlarged, heterogeneous liver with increased echogenicity and normal portal flow. A liver biopsy was performed and reported marked steatosis with minimal fibrosis. No specific diagnosis was made, although a metabolic disorder was suspected.
Table I.
Initial hematologic laboratory values
| Variable | Result |
|---|---|
| Hemoglobin (100-140 g/L; 10-14 g/dL) | 108 g/L (10.8 g/dL) |
| Mean corpuscular volume (f/L) | 85.7 |
| White cell count (5-15 × 109/L) | 25 (lymph 14, neut 8) |
| Platelet count (150-400 × 109/L) | 440 |
| INR (0.9-1.1) | 1.2 |
| Partial thromboplastin time (25-35 sec) | 103 |
Table II.
Initial blood biochemical laboratory values
| Variable | Result | Normal range |
|---|---|---|
| Aspartate aminotransferase (AST) | 402 | 0-35 U/L |
| Alanine aminotransferase (ALT) | 436 | 0-35 U/L |
| Albumin | 31 (3.1) | 32-48 g/L (3.2-4.8 g/dL) |
| Gamma glutamyl transferase (GGT) | 47 | 0-45 U/L |
| Glucose | 2.2 (39.6 mg/dL) | 2.5-5 mmol/L(45-90 mg/dL) |
| Ammonium | 33 (59) | 9-33 μmol/L (16-59 μg/dL) |
| Bilirubin, conjugated/unconjugated | 31 / 1 (1.8/0.05 g/dL) | 0-17 / 0-2 μmol/L (0-1/0-0.1 mg/dL) |
| Plasma carnitine:free, total, ratio | 6, 10, 60% | 20-53 μmol/L, 25-70 μmol/L, 66-70% |
A provisional diagnosis of a fatty acid oxidation disorder (FAOD) was made. Skin fibroblasts were sent for analysis. Carnitine supplementation and feeds with Portagen (Mead Johnson Nutritionals, Indianapolis, Ind), a formula high in medium-chain triglycerides and low in long-chain triglycerides, were started. Referral to a quaternary referral hospital was arranged.
QUATERNARY REFERRAL HOSPITAL
The patient had been unwell for nearly four weeks when he was admitted to our center. He continued to have frequent episodes of vomiting and diarrhea on Portagen feeds. As had been noted before, he was weak, floppy, and lethargic but responded appropriately to his environment. A firm liver edge was palpable 8 cm below the right costal margin. He was still not clinically jaundiced and the conjugated hyperbilirubinemia was improving (Tables II and III). There was no splenomegaly, ascites, or sign of chronic liver disease. Ophthalmologic examination was normal. Clinical cardiac evaluation was also normal, as was a chest radiograph and electrocardiogram.
Table III.
Abnormal laboratory values 4 weeks into the illness
| Variable | Result |
|---|---|
| Potassium | 2.5 (3.7-5.6 mmol/L), 2.5 (3.7-5.6 mEq/L) |
| Phosphate | 0.56 (1.3-2.2 mmol/L), 1.7 (4.0-6.8 mg/dL) |
| Urea | 1.3 (1.8-5.4 mmol/L), 3.6 (8.4-18.2 mg/dL) |
| Uric acid | 61 (120-360 lmol/L), 1.03 (2.02-6.05 mg/dL) |
| Carnitine: free, total, ratio |
9.9, 15.9 μmol/L, 62% |
| Urine carnitine: free, total |
2664 (8-254), 3681 (62-369) mmol/mol creat |
| AST | 559 (0-111 U/L) |
| ALT | 217 (0-60 U/L) |
| Albumin | 31 (32-48 g/L), 3.1 (3.2-4.8 g/dL) |
| Bilirubin: conjugated/ unconjugated |
14/9 (0-2/0-17 μmol/L)/ 0.8/0.5 (0-0.1/0-1 mg/dL) |
| APTT | 94.9 (25-35 seconds) |
| Factor VIII | 5.36-7.5 (0.79-1.99 IU/mL) |
| Factor XI | 0.09-0.14 (0.5-1.5 IU/mL) |
Normal values in brackets; blood unless specified.
Abnormal laboratory tests done on admission are shown in Table III. Factors V and VII levels were also low. A venous blood gas showed a compensated metabolic acidosis: pH 7.36, base excess −9, bicarbonate 15 mmol/L (15 mEq/L) and CO2 26 (40-50 mm Hg). Urinary sodium was 22 mmol/L, potassium 50 mmol/L, and uric acid:creatinine ratio was 2.16 (normal for age, 0.92-1.04).1 These findings together with a generalized aminoaciduria and relative elevation of carnitine excretion suggested a proximal renal tubulopathy. Plasma lactate, fasting free fatty acid (0.1 mmol/L), and β-hydroxybutyrate (<0.024 mmol/L) levels and blood acylcarnitine analysis were normal. Serum alkaline phosphatase and creatinine were normal. Sweat chloride was normal. Leukocyte acid lipase activity for Wolman disease was also normal. Factor IX level and fibrinogen were normal. Factor XI inhibitor was negative. Parental Factor XI levels were normal.
Abdominal ultrasonogram again showed an enlarged liver with increased echogenicity and normal portal flow. Kidneys looked normal. A skeletal survey did not show any rachitic changes, although reduced mineralization was reported “consistent with a chronic process.” In view of his diarrhea, vomiting, hepatomegaly with liver dysfunction, hypoketotic hypoglycemia, and low Factor XI levels, serum was sent for isoelectric focusing (IEF) of transferrin to exclude carbohydrate-deficient glycoprotein syndrome (or, congenital disorder of glycosylation, CDG).
His clinical condition progressively worsened over the first week of this admission. The international normalized ratio reached a maximum of 1.25 and the activated partial thromboplastin time, 101 seconds. His serum albumin fell to 22 g/L, and he developed moderate ascites requiring albumin infusions with diuretic therapy. Despite supplementation with appropriate amounts of potassium, bicarbonate, and phosphate he continued to have a compensated metabolic acidosis with hypokalemia and hypophosphatemia. There were several episodes of hypoglycemia. He continued on the Portagen feeds with frequent vomiting, and had as many as 11 loose nonbloody stools daily.
Two weeks later, due to his deteriorating condition and persistent vomiting and diarrhea, his feeds were stopped and total parenteral nutrition (TPN) was commenced. With TPN, his vomiting and diarrhea resolved within several days, and his abnormal biochemical parameters slowly improved. After one week of TPN, feeds were reintroduced using Vivonex Pediatric (Novartis Nutrition Corp, Mississauga, Ontario, Canada) via nasogastric tube. This formula was chosen in view of the possible FAOD, plus it did not contain sucrose (unlike Portagen), which was thought to be exacerbating his diarrhea. This formula was well tolerated, and a week later his electrolytes, liver function tests, and coagulation profile had normalized, and liver size had decreased. A second UTI caused by E coli was diagnosed and treatment was started with Novo-Trimel (Trimethoprim-Sulfamethoxazole, Novopharm Ltd, Toronto, Canada) oral suspension. After one dose of Novo-Trimel the diarrhea recurred and persisted, but settled when the drug was stopped. At that time, the IEF transferrin was reported as abnormal, suggesting a possible diagnosis of CDG.
CASE DISCUSSION
This 6-month-old child presented with massive hepatomegaly, raised liver transaminases, coagulopathy, hypoglycemia (with hypoketosis), and evidence of renal tubular dysfunction. These symptoms followed an episode of gastroenteritis and were associated with the two E coli UTIs. His health and development had been previously normal and there was neither consanguinuity nor a family history of significant illness.
Subsequent review of the liver biopsy pathology slides at our institution showed the findings shown in Figure 1. There was also a sparse lymphocytic infiltrate. There was no evidence of cholestasis. Electron microscopy is shown in Figure 2. There was also slight dilatation of rough endoplasmic reticulum. It showed the presence of peroxisomes, and dark staining mitochondria without any inclusions. There were no obvious ultrastructural abnormalities of the mitochondrial cristae. Glycogen particles showed usual size and distribution.
Fig 1.
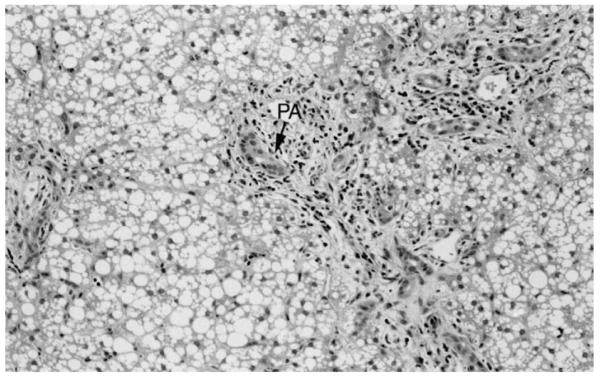
Diffuse macrovesicular fatty change with minimal portal fibrosis and ductular proliferation (arrow) in the portal area (PA) (hematoxylin and eosin staining, original magnification ×250).
Fig 2.
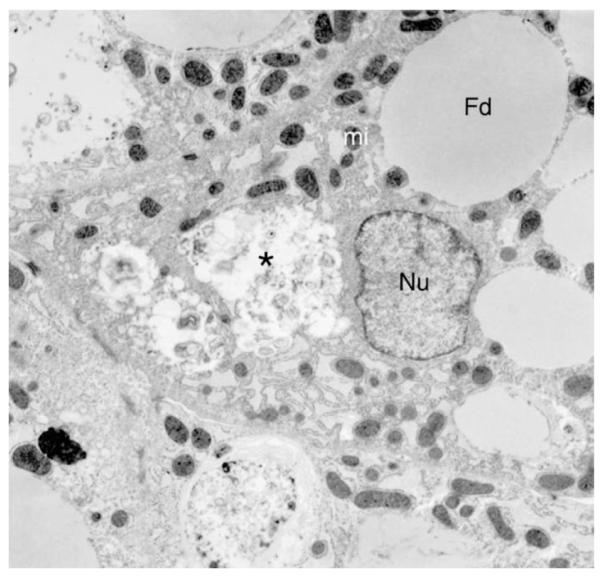
Electron micrograph of liver biopsy with microvesicular fat droplets (Fd) in hepatocyte, focal cytoplasmic necrosis (asterisk). Nucleus (Nu) and mitochondria (mi) show no specific abnormalities (TEM ×5000).
Based on this pathology and the investigations, diseases such as galactosemia, glycogen storage diseases, tyrosinemia, urea cycle defects, organic acidemias, and Wolman disease were excluded.
One possible diagnosis was still an FAOD. Many features of this child’s illness could be explained by a defect in mobilizing fatty acids to provide energy in the face of an intercurrent illness. Hepatomegaly with hepatic dysfunction (including coagulopathy), hypoketotic hypoglycemia, possible cardiomyopathy, and low carnitine levels are consistent features of an FAOD. However, FAODs are most often associated with an elevated acylcarnitine fraction, fasting free fatty acid levels, and free fatty acid: β-hydroxybutyrate ratio which were not present in this case. In one form of FAOD, carnitine palmitoyltransferase I (CPT I) deficiency, acylcarnitine concentrations are negligible or low,2 but carnitine levels and free fatty acid: β-hydroxybutyrate ratios are usually high. Thus our case’s profile did not fit this diagnosis. In addition, blood acylcarnitine analysis was normal. The urine organic acids showed only dicarboxylic acids, which could be a feature of medium chain acyl-coenzyme A dehydrogenase (MCAD) deficiency or CPT I deficiency. However, his clinical picture was not that of MCAD, and the urinary dicarboxylic acids were thus considered to be a feature of his carnitine depletion due to renal wasting. Other aspects of this child’s illness not typical of an FAOD were persistence of his diarrhea and vomiting, and worsening of his biochemical abnormalities despite carnitine supplementation and feeding with Portagen (Mead Johnson Nutritionals). Subsequently, DNA analysis for the common MCAD and long-chain 3-hydroxyacyl-coenzyme A dehydrogenase mutations and 14C-propionate oxidation studies on fibroblasts were reported normal with no FAOD detected.
The finding of an abnormal IEF transferrin suggested the diagnosis of CDG, a group of hereditary multisystem disorders characterized by hypoglycosylation of glycoproteins.3 The defect in CDG can result in a wide range of abnormalities because of effects on enzymes, hormones, transport proteins, coagulation factors, immunoglobulins, and complement. The most common reported form, CDG type Ia, caused by phosphomannomutase deficiency, is associated with severe neurologic disease, lipodystrophy, coagulation defects, and variable gastrointestinal and hepatic abnormalities.4 The phenotype of our patient was not consistent with this form of CDG. Children with CDG type Ib have normal psychomotor development, with hepatic and gastrointestinal manifestations, because of a deficiency of phosphomannose isomerase.5 The hepatic dysfunction, hypoglycemia, and diarrhea with possible protein losing enteropathy seen in our case could be consistent with CDG Ib.6 His major abnormality was prolongation of the aPTT while the international normalized ratio was only ever slightly elevated and he had a very low Factor XI, an unusual pattern for liver disease. The low Factor XI level could be explained by CDG, because of underglycosylation of coagulation factors, which has been previously reported.7 Furthermore, abnormal glycosylation may affect lymphocytes and other immune cells, resulting in an increased incidence of infection in these patients, which may explain his two UTIs.8 Hypoglycemia has been described in CDG Ib due to varying mechanisms, including hyperinsulinism.9 More importantly, treatment with oral mannose supplementation appears to improve both the clinical and biochemical abnormalities.5,10 Treatment with mannose was fortunately not started in this child while we awaited confirmation of his diagnosis, because the potential conversion of mannose to fructose could have caused more complications. Features not consistent with CDG Ib included the improvement in his clinical features without mannose supplementation. Furthermore, pathologic findings in CDG include significant hepatic fibrosis,5,6 sometimes with ductal plate malformation suggestive of congenital hepatic fibrosis or Caroli’s disease,11 whereas only minimal portal fibrosis was seen in our patient.
One further diagnosis that was entertained was that of hereditary fructose intolerance (HFI) (OMIM 22960).12 This is an autosomal recessive condition with uncertain prevalence in the population. There are reports of an estimated frequency of 1 in 23,000 live births.13 Hereditary fructose intolerance is caused by fructose-1-phosphate aldolase B (known as aldolase B) deficiency, which is expressed in the liver, small bowel, and kidney (E.C. 4.1.2.13). Under normal conditions, dietary fructose is first converted to fructose-1-phosphate, which is then catalyzed by aldolase B to form the trioses D-glyceraldehyde and dihydroxyacetone phosphate. These intermediate trioses are incorporated directly into either the glycolytic pathway (eventually forming lactate and pyruvate to act as substrate for the Krebs cycle) or the gluconeogenic pathway to form glucose or glycogen.14
In HFI, ingestion of fructose results in an exaggerated accumulation of fructose-1-phosphate and depletion of the intracellular pool of inorganic phosphate and guanosine triphosphate and hence adenosine triphosphate (ATP).14 The reduced phosphate concentration activates adenosine deaminase,15,16 resulting in degradation of purine nucleotides to uric acid,17 producing the characteristic hyperuricemia of HFI. The hypoglycemia seen in HFI results from defects in both glycogenolysis and gluconeogenesis, which are thought to be secondary to inhibition by excessive fructose-1-phosphate.18,19 Impairment of gluconeogenesis combined with fructose-1-phosphate activation of pyruvate kinase14,20 results in accumulation of Krebs cycle precursors alanine, lactate, and pyruvate, which contribute to the metabolic acidosis seen in HFI. This is exacerbated by the proximal renal tubular acidosis (Fanconi syndrome) where aminoaciduria, phosphaturia, and wasting of bicarbonate occurs, as seen in our case.21 The cause of tissue damage to the liver is not well understood but has been attributed to accumulation of osmotic substrates (such as fructose-1-phosphate) and depletion of ATP, which have been shown ultrastructurally in hepatocytes to result in membranous arrays, marked rarefaction of the hyaloplasm, formation of cytolysosomes, and damage to the endoplasmic reticulum.22-26
Our patient had many features of HFI. These included vomiting, diarrhea and lethargy, massive hepatomegaly, hypoglycemia, hepatic dysfunction, and renal tubular acidosis. All of these features resolved when fructose (in the form of sucrose in the Portagen formula) was removed from his diet. In addition, the Factor XI levels normalized. He was later inadvertently exposed to fructose when given an oral antibiotic suspension containing sorbitol (which is rapidly converted to fructose by sorbitol dehydrogenase in the liver), resulting in immediate vomiting and diarrhea and worsening of liver function. On re-review of the liver histopathology, the diagnosis of HFI would explain the diffuse steatosis, portal ductular proliferation, and mild pericholangitis, and also the distinctive electron microscopy findings of focal cytoplasmic necrosis with dilatation of the rough endoplasmic reticulum. This reinforces the need for a complete analysis of liver biopsies including electron microscopy in the diagnosis of genetic metabolic disease, as well as review by an experienced liver pathologist in unusual cases. The abnormal glycoprotein IEF has been reported in HFI,27 because fructose-1-phosphate has been shown to inhibit phosphomannose isomerase28 resulting in the synthesis of abnormal plasma glycoproteins, eg, transferrin isoforms and producing a pattern much like that seen in CDG type I. It is important to differentiate between HFI and CDG. The gastrointestinal form of CDG, CDG type Ib, is treated with mannose. Mannose can be converted to fructose by hexokinase and phosphomannoisomerase and this could have potentially worsened his symptoms, which we subsequently established were caused by HFI.
Several features in this case were not consistent with a diagnosis of HFI. The lack of lactic acidosis, a low uric acid level, and absence of hypermagnesemia are unusual in a patient with HFI and initially suggested alternative diagnoses. The low uric acid level in our patient could possibly be explained by excessive urinary losses of uric acid because the renal tubular acidosis. Another possibility is that phenotypic variability exists, although experts in this field maintain that disease severity relates less to genotype than to the individual dietary exposure.14 The source of fructose, the cause of his deterioration, was from both the soya formula (Alsoy, Nestle) and particularly the Portagen (Mead Johnson Nutritionals) that he had been given, both of which contain carbohydrate as sucrose, which is broken down to fructose and glucose in the gut. His prior diet also contained some fructose in fruit and vegetables although he was not symptomatic, which probably relates to the amount of substrate he was exposed to and possibly to his residual enzyme activity.
The gene for human aldolase B has been mapped to chromosome 9q22.329 and fully characterized.30 More than 23 different mutations causing HFI have been identified.31 Three of these mutations, A149P, A174D, and N334K comprise about 80% of mutant alleles in the European/North American population.31,32 Our patient has been found to have the common A174D mutation on the maternal allele. The diagnosis of HFI was subsequently confirmed by the finding of a second mutation L256P inherited from the father. This mutation has previously been described in an Italian family where the proband was also a compound heterozygote for A149P/L256P.33 This T-C transition in exon 7 is in an alpha-helix, which is near one of the subunit interfaces. The L256P-mutant protein has been shown to be thermally unstable.34 It also affects aldolase B function by decreasing substrate affinity, maximal velocity, and/or enzyme stability.35 Thus, this genotype A174D/L256P is unusual and little is known about the genotype/phenotype correlation. In this case, the features of low uric acid and absence of lactic acidosis may be a reflection of this genotype. His family have declined further liver biopsy to demonstrate fructose-1-phosphate aldolase deficiency and the impact on function that these mutations may have.
SUMMARY
This case highlights a rare condition, HFI, presenting in a 6-month-old child with vomiting, diarrhea, marked hepatomegaly with steatosis, hepatic dysfunction, renal tubular dysfunction, and hypoglycemia. The last century has seen the emergence of sucrose as both a commodity and a major constituent of Western diets, resulting in an apparent increased prevalence of HFI. This case was notable for probably being precipitated by infant formulas containing sucrose that were prescribed for other perceived benefits. It also reinforces the need for close collaboration between clinician and pathologist. Clinicians should remember to consider HFI in their differential diagnosis of children presenting with steatosis and hepatic dysfunction, especially if the pathologic findings are suggestive.
Acknowledgments
We gratefully acknowledge the assistance of Dr Jennifer Ramsay (Pathology, McMaster Health Sciences Center), Drs Don Whelan and Murray Potter (Paediatrics, McMaster Health Sciences Center), Dr Gordon Forstner, Division GI Nutrition, and Dr Joe Clarke, Division of Clinical and Metabolic Genetics, (The Hospital for Sick Children) and Lorne Sargeant (Laboratory Medicine, Manitoba Health Sciences Centre).
Glossary
- ATP
Adenosine triphosphate
- HFI
Hereditary fructose intolerance
- CDG
Congenital disorder of glycosylation
- MCAD
Medium-chain acyl-coenzyme a dehydrogenase
- CPT I
Carnitine palmitoyltransferase
- TPN
Total parenteral nutrition
- FAOD
Fatty acid oxidation disorder
- UTI
Urinary tract infection
- IEF
Isoelectric focusing
REFERENCES
- 1.Kaufman JM, Greene ML, Seegmiller JE. Urine uric acid to creatinine ratio—a screening test for inherited disorders of purine metabolism. Phosphoribosyltransferase (PRT) deficiency in X-linked cerebral palsy and in a variant of gout. J Pediatr. 1968;73:583–92. doi: 10.1016/s0022-3476(68)80274-4. [DOI] [PubMed] [Google Scholar]
- 2.Olpin SE, Allen J, Bonham JR, Clark S, Clayton PT, Calvin J, et al. Features of carnitine palmitoyltransferase type I deficiency. J Inherit Metab Dis. 2001;24:35–42. doi: 10.1023/a:1005694320063. [DOI] [PubMed] [Google Scholar]
- 3.Jaeken J, Carchon H. The carbohydrate-deficient glycoprotein syndromes: an overview. J Inherit Metab Dis. 1993;16:813–20. doi: 10.1007/BF00714272. [DOI] [PubMed] [Google Scholar]
- 4.Jaeken J, Artigas J, Barone R, Fiumara A, de Koning TJ, Poll-The BT, et al. Phosphomannomutase deficiency is the main cause of carbohydrate-deficient glycoprotein syndrome with type I isoelectrofocusing pattern of serum sialotransferrins. J Inherit Metab Dis. 1997;20:447–9. doi: 10.1023/a:1005331523477. [DOI] [PubMed] [Google Scholar]
- 5.Jaeken J, Matthijs G, Saudubray JM, Dionisi-Vici C, Bertini E, de Lonlay P, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet. 1998;62:1535–9. doi: 10.1086/301873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babovic-Vuksanovic D, Patterson MC, Schwenk WF, O’Brien JF, Vockley J, Freeze HH, et al. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J Pediatr. 1999;135:775–81. doi: 10.1016/s0022-3476(99)70103-4. [DOI] [PubMed] [Google Scholar]
- 7.Young G, Driscoll MC. Coagulation abnormalities in the carbohydrate-deficient glycoprotein syndrome: case report and review of the literature. Am J Hematol. 1999;60:66–9. doi: 10.1002/(sici)1096-8652(199901)60:1<66::aid-ajh11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 8.Bergmann M, Gross HJ, Abdelatty F, Moller P, Jaeken J, Schwartz-Albiez R. Abnormal surface expression of sialoglycans on B lymphocyte cell lines from patients with carbohydrate deficient glycoprotein syndrome I A (CDGS I A) Glycobiology. 1998;8:963–72. doi: 10.1093/glycob/8.10.963. [DOI] [PubMed] [Google Scholar]
- 9.de Lonlay P, Cuer M, Vuillaumier-Barrot S, Beaune G, Castelnau P, Kretz M, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: a new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. J Pediatr. 1999;135:379–83. doi: 10.1016/s0022-3476(99)70139-3. [DOI] [PubMed] [Google Scholar]
- 10.Niehues R, Hasilik M, Alton G, Korner C, Schiebe-Sukumar M, Koch HG, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998;101:1414–20. doi: 10.1172/JCI2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Koning TJ, Dorland L, Nikkels P, van Diggelen OP, Boonman AMC, de Jong GJ, et al. Phosphomannose isomerase deficiency with cyclic vomiting and congenital hepatic fibrosis. J Inherit Metab Dis. 1998;21:96. [Google Scholar]
- 12.Gitzelmann R, Steinmann B, Van den Berghe G. Disorders of fructose metabolism. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and molecular bases of inherited disease. 7th ed McGraw-Hill, Inc; New York: 1995. pp. 905–34. [Google Scholar]
- 13.James CL, Rellos P, Ali M, Heeley AF, Cox TM. Neonatal screening for hereditary fructose intolerance: frequency of the most common mutant aldolase B allele (A149P) in the British population. J Med Genet. 1996;33:837–41. doi: 10.1136/jmg.33.10.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ali M, Rellos P, Cox TM. Hereditary fructose intolerance. J Med Genet. 1998;35:353–65. doi: 10.1136/jmg.35.5.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Berghe G, Bronfman M, Vanneste R, Hers HG. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J. 1977;162:601–9. doi: 10.1042/bj1620601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maenpaa PH, Raivio KO, Kekomaki MP. Liver adenine nucleotides: fructose-induced depletion and its effect on protein synthesis. Science. 1968;161:1253–4. doi: 10.1126/science.161.3847.1253. [DOI] [PubMed] [Google Scholar]
- 17.Bode JC, Zelder O, Rumpelt HJ, Wittkamp U. Depletion of liver adenosine phosphates and metabolic effects of intravenous infusion of fructose or sorbitol in man and in the rat. Eur J Clin Invest. 1973;3:436–41. doi: 10.1111/j.1365-2362.1973.tb02211.x. [DOI] [PubMed] [Google Scholar]
- 18.Kaufmann U, Froesch ER. Inhibition of phosphorylase-a by fructose-1-phosphate, alpha-glycerophosphate and fructose-1, 6-diphosphate: explanation for fructose-induced hypoglycaemia in hereditary fructose intolerance and fructose-1, 6-diphosphatase deficiency. Eur J Clin Invest. 1973;3:407–13. doi: 10.1111/j.1365-2362.1973.tb02208.x. [DOI] [PubMed] [Google Scholar]
- 19.Thurston JH, Jones EM, Hauhart RE. Decrease and inhibition of liver glycogen phosphorylase after fructose. An experimental model for the study of hereditary fructose intolerance. Diabetes. 1974;23:597–604. doi: 10.2337/diab.23.7.597. [DOI] [PubMed] [Google Scholar]
- 20.Eggleston LV, Woods HF. Activation of liver pyruvate kinase by fructose-1-phosphate. FEBS Lett. 1970;6:43–5. doi: 10.1016/0014-5793(70)80038-2. [DOI] [PubMed] [Google Scholar]
- 21.Morris RC., Jr An experimental renal acidification defect in patients with hereditary fructose intolerance. II. Its distinction from classic renal tubular acidosis; its resemblance to the renal acidification defect associated with the Fanconi syndrome of children with cystinosis. J Clin Invest. 1968;47:1648–63. doi: 10.1172/JCI105856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jevon GP, Dimmick JE. Histopathologic approach to metabolic liver disease: part 2. Perspect Pediatric Pathol. 1999;21:61–9. [Google Scholar]
- 23.Yu DT, Burch HB, Phillips MJ. Pathogenesis of fructose hepatotoxicity. Lab Invest. 1974;30:85–92. [PubMed] [Google Scholar]
- 24.Phillips MJ, Hetenyi G, Jr, Adachi F. Ultrastructural hepatocellular alterations induced by in vivo fructose infusion. Lab Invest. 1970;22:370–9. [PubMed] [Google Scholar]
- 25.Phillips MJ, Little JA, Ptak TW. Subcellular pathology of hereditary fructose intolerance. Am J Med. 1968;44:910–21. doi: 10.1016/0002-9343(68)90091-0. [DOI] [PubMed] [Google Scholar]
- 26.Yu DT, Phillips MJ. Hepatic ultrastructural changes in acute fructose overload. J Ultrastructure Res. 1971;36:222–36. doi: 10.1016/s0022-5320(71)80100-4. [DOI] [PubMed] [Google Scholar]
- 27.Adamowicz M, Pronicka E. Carbohydrate deficient glycoprotein syndrome—like transferrin isoelectric focusing pattern in untreated fructosaemia. Eur J Pediatr. 1996;155:347–8. doi: 10.1007/BF02002730. [DOI] [PubMed] [Google Scholar]
- 28.Jaeken J, Pirard M, Adamowicz M, Pronicka E, Van Schaftingen E. Inhibition of phosphomannose isomerase by fructose 1-phosphate: an explanation for defective N-glycosylation in hereditary fructose intolerance. Pediatr Res. 1996;40:764–6. doi: 10.1203/00006450-199611000-00017. [DOI] [PubMed] [Google Scholar]
- 29.Lebo RV, Tolan DR, Bruce BD, Cheung MC, Kan YW. Spot-blot analysis of sorted chromosomes assigns a fructose intolerance disease locus to chromosome 9. Cytometry. 1985;6:478–83. doi: 10.1002/cyto.990060513. [DOI] [PubMed] [Google Scholar]
- 30.Tolan DR, Penhoet EE. Characterization of the human aldolase B gene. Mol Biol Med. 1986;3:245–64. [PubMed] [Google Scholar]
- 31.Tolan DR. Molecular basis of hereditary fructose intolerance: mutations and polymorphisms in the human aldolase B gene. Hum Mutat. 1995;6:210–8. doi: 10.1002/humu.1380060303. [DOI] [PubMed] [Google Scholar]
- 32.Tolan DR, Brooks CC. Molecular analysis of common aldolase B alleles for hereditary fructose intolerance in North Americans. Biochem Med Metabol Biol. 1992;48:19–25. doi: 10.1016/0885-4505(92)90043-x. [DOI] [PubMed] [Google Scholar]
- 33.Ali M, Sebastio G, Cox TM. Identification of a novel mutation (Leu 256→Pro) in the human aldolase B gene associated with hereditary fructose intolerance [published erratum, Hum Mol Genet 1994;3:684] Hum Mol Genet. 1994;3:203–4. doi: 10.1093/hmg/3.1.203. [DOI] [PubMed] [Google Scholar]
- 34.Rellos P, Sygusch J, Cox TM. Expression, purification, and characterization of natural mutants of human aldolase B. J Biological Chem. 2000;275:1145–51. doi: 10.1074/jbc.275.2.1145. [DOI] [PubMed] [Google Scholar]
- 35.Esposito G, Vitagliano L, Santamaria R, Viola A, Zagari A, Salvatore F. Structural and functional analysis of aldolase B mutants related to hereditary fructose intolerance. FEBS Lett. 2002;531:152–6. doi: 10.1016/s0014-5793(02)03451-8. [DOI] [PubMed] [Google Scholar]