

Abstract
Cholecystokinin (CCK), an endogenous brain-gut peptide, is released after food intake and promotes the process of satiation via activation of the vagus nerve. In vitro, CCK increases cytosolic calcium concentrations and produces membrane depolarization in a subpopulation of vagal afferent neurons. However, the specific mechanisms and ionic conductances that mediate these effects remain unclear. In this study we used calcium imaging, electrophysiological measurements, and single cell PCR analysis on cultured vagal afferent neurons to address this issue directly. A cocktail of blockers of voltage-dependent calcium channels (VDCC) failed to block CCK-induced calcium responses. In addition, SKF96365, a compound that blocks both VDCC and the C family of transient receptor potential (TRP) channels, also failed to prevent responses to CCK. Together these results suggest that CCK-induced calcium influx is not subsequent to the membrane depolarization. Ruthenium red, an inhibitor of the TRPV family and TRPA1, blocked both depolarizing responses to CCK and CCK-induced calcium increases, but had no effect on the KCl-induced calcium response. Selective block of TRPV1 and TRPA1 channels with SB366791 and HC030031, respectively, had minor effects on the CCK-induced response. Application of 2-aminoethoxydiphenyl borate, an activator of select TRPV channels but a blocker of several TRPC channels, either had no effect or enhanced the responses to CCK. Further, results from PCR experiments revealed a significant clustering of TRPV2-5 in neurons expressing CCK1 receptors. These observations demonstrate that CCK-induced increases in cytosolic calcium and membrane depolarization of vagal afferent neurons are likely mediated by TRPV channels, excluding TRPV1.
Cholecystokinin-induced activation of vagal afferent neurons is likely mediated by TRPV channels.
Cholecystokinin (CCK), released in the gastrointestinal tract after entry of protein and/or fat into the duodenum, elicits a number of biological actions, including an inhibition of gastric emptying (1), contraction of the gallbladder (2), and pancreatic secretion of enzymes (3,4,5), which altogether facilitate digestion and maximize nutrient absorption. CCK is also a robust within-meal satiety signal as established by the effects of exogenous administration of CCK (6), as well as blockade of CCK receptors (7), on food intake. Infusion of CCK into the celiac artery activates vagal afferent discharge (8) and this activation of vagal afferent fibers participates in many CCK-induced effects. For instance, the satiation response to CCK is eliminated by lesion of subdiaphragmatic vagal afferent nerves (9,10,11), and vagotomy also reduces pancreatic enzyme secretion (12) and gastric emptying (13) induced by CCK. Studies using subtype selective antagonists and agonists to CCK1 and CCK2 receptors demonstrated that CCK1 receptors (CCK1Rs) mediate CCK-induced activation of vagal afferent neurons and the specific effects of CCK on satiation (7,14).
A direct effect of CCK on vagal afferent neurons is supported by numerous observations. For example, a subpopulation of vagal afferent neurons expresses CCK1Rs (15,16), and these neurons have been shown, by both electrophysiological approaches and cytosolic calcium measurements, to respond to CCK directly when isolated and maintained in cell culture (17,18,19). The calcium increase induced by application of CCK is dependent on extracellular calcium influx rather than mobilization of intracellular stores (19). However, the precise mechanisms by which CCK depolarizes these neurons and activates a calcium influx have not been established.
A prior study has demonstrated that the depolarizing effect of CCK on these neurons involves alterations in more than one background ionic conductance because both an increase in a depolarizing conductance and an inhibition of a hyperpolarizing conductance have been observed (20). One possibility for the depolarizing conductance activated by CCK is the transient receptor potential (TRP) channels, a superfamily of nonselective cation channels (21). In support of the this hypothesis, we and others have shown that various TRP channels are indeed expressed by nodose neurons (22,23,24). The calcium influx elicited by CCK could then be either influx through voltage-dependent calcium channels (VDCC) activated by the membrane depolarization, or a direct influx through a calcium-permeable TRP channel modulated by CCK. In this study, we used patch-clamp electrophysiology as well as fluorescent single cell calcium measurements to address these questions. Through the use of various compounds that are known to interact with either VDCC or TRP channels or both, we disproved the hypothesis that CCK-induced calcium signals are due to calcium entry through VDCC. Further, we established that the depolarization and the calcium influx induced by CCK are mediated by a ruthenium red (RuR) sensitive pathway, likely to be one or more members of the TRPV family of channels, excluding TRPV1. Finally, we examined the messages of CCK1Rs and TRPV subtypes in individual nodose neurons and found that indeed a vast majority of CCK1R-expressing neurons coexpressed at least one subtype of TRPV2-5.
Materials and Methods
Animals
Adult male Sprague Dawley rats (200–240 g) were purchased from Simonsen Laboratories and used as subjects for all experiments. All animals were housed in Association for Assessment and Accreditation of Laboratory Animal Care-accredited quarters under a 12-hour light/dark cycles and had ad libitum access to pelleted chow and water. The Washington State University Institutional Animal Care and Use Committee approved all procedures performed.
Dissociation and culture of vagal afferent neurons
We obtained vagal afferent neurons from the nodose ganglia as previously described (17). Briefly, we anesthetized rats with ketamine (25 mg/100 g) and xylazine (2.5 mg/100 g) and isolated nodose ganglia under aseptic conditions. Ganglia were then placed in Hank’s balanced-salt solution (HBSS), cleaned, and desheathed. We dissociated neurons in 3 ml of digestion buffer (1 mg/ml dispase II and 1 mg/ml collagenase type Ia in HBSS) for 120 min in an incubator at 37 C then washed dispersed cells with HEPES-buffered Dulbecco’s modified Eagle’s medium (HDMEM) and plated neurons onto poly-L-lysine-coated coverslips (200 μg/ml poly-lysine for 30 min). Plated neurons were maintained in HDMEM supplemented with 10% fetal calf serum.
Intracellular calcium measurements
All experiments were performed at room temperature (22 C) within 48 h of isolation. Calcium was monitored by use of the fluorescent calcium indicator fura-2 AM (Molecular Probes, Eugene, OR). MetaFluor Software (Universal Imaging, West Chester, PA) was used to collect and analyze results. Cells were loaded with 2 μm fura-2 AM for 35 min followed by a 15 min wash, then we mounted the coverslips containing the loaded cells into an open chamber and constantly perfused the coverslips with physiological saline (in mm:140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 6 glucose, and 10 HEPES; pH adjusted to 7.4 with Tris-base). Bath containing agents were applied through a common manifold upstream of the perfusion chamber. At the end of each run a high potassium (Hi-K) solution (in mm: 90 NaCl, 55 KCl, otherwise same with physiological saline) was briefly applied to depolarize the cells and determine whether or not neurons were viable. Neurons were included if they responded to the Hi-K challenge with a rapid and reversible calcium response of at least 30 nm in amplitude. These responses are not always included in the figures presented. Image pairs (340 and 380 nm excitation, 510 emissions) were collected every 6 s or 15 s, then the ratios of the fluorescence intensity were converted to calcium concentrations using a calibration curve. Calcium responses are calculated as the maximal change in calcium concentration from the baseline level of intracellular calcium.
Patch-clamp electrophysiology
Electrical recordings were performed using a perforated patch technique with tapered borosilicate glass pipettes (World Precision Instruments, Sarasota, FL). We used a pipette solution approximating intracellular ionic concentrations (in mm: 10 NaCl, 60 KCl, 30 K2SO4, 4 MgCl2, and 10 HEPES; pH adjusted to 7.4 with Tris-base) and the standard physiological bath solution (described above) unless otherwise indicated in the results. Nystatin (50 mg/ml in DMSO, final concentration 0.5 mg/ml) was used for membrane perforation. Data were collected using pCLAMP 9 software with an Axopatch 200A amplifier and a Digidata 1322A analog-to-digital converter (all from Axon Instruments, Union City, CA). Membrane potentials were recorded in current-clamp mode (digitized at 2 kHz). Current traces were recorded at a holding potential of −60 mV using the voltage-clamp configuration (digitized at 10 kHz). In voltage-clamp experiments, leak currents (estimated from pulses to −80 mV from −60 mV) were digitally subtracted from the current traces. Agents were applied in the same manner as described for the calcium measurements.
Single-cell PCR
We performed single-cell PCR experiments as previously described (22). Briefly, we collected individual cultured nodose neurons into 11 μl of lysis buffer using sterile glass pipettes and treated single neuron lysate with DNase I (Cell-to-cDNA II kit, Ambion). After deactivating DNase, we then initiated RT according to the instruction manual. Single-cell cDNA fragments were amplified using Taq kit (Invitrogen, Carlsbad, CA) with sequence-specific oligonucleotide primers designed to reveal genes of interest. Primer sequences are listed in Table 1. PCR protocol is as follows: polymerase activation at 95 C for 5 min and 40 or 45 cycles at 95 C for 45 sec followed by incubation at 60 C for 45 sec and then 72 C for 1 min. We then ran final products on a 2% agarose gel at 110 V for 45 min. Bands were revealed under the UV light. We used PGP9.5 (Protein Gene Product 9.5) as a neuronal marker. Only single cell cDNA samples that amplified for PGP9.5 were included in the final analysis.
Table 1.
Primers for single cell RT-PCR reaction
| Gene | GenBank accession no. | Forward primer | Reverse primer |
|---|---|---|---|
| CCK1R | NM_012688 | gatcaacctcaaccttcaaaaga | cacactgagaaggaatatgatgga |
| PGP9.5 | NM_017237 | cctgctgctgctgtttcc | tgtcccttcagttcctcaattt |
| TRPV1 | NM_031982 | ggtgtgcctgcacctagc | ctcttggggtggggactc |
| TRPV2 | NM_017207 | gggatggtacccctgatga | tctcccaagcaacccaat |
| TRPV3 | NM_001025757 | cacccccaccaagaagagt | gggggagaggtcttggtaac |
| TRPV4 | NM_023970 | aagaaatgccctggagtgaa | acacaaccaccagcactgag |
| TRPV5 | NM_053787 | gaagatgatttttggagacctactg | cggtctggaagatgatatagaagg |
| TRPC1 | NM_053558 | cagcctcagacattccaggt | tcaaacgctctcagaattgga |
| TRPC3 | NM_021771 | tgctaattatggtctgggttctc | ccacagctgcacgatgtact |
| TRPM8 | NM_134371 | ctcctggtcgccatgttt | ggaacttccagacctgatcg |
| TRPA1 | NM_207608 | atttgcggcctgagttttt | gcgtacatccatcattgtcct |
Chemicals
Drugs used in this study are as follows (Name, Abbreviation): From Peptides International (Louisville, KY): Cholecystokinin-octapeptide (sulfated), CCK. From Sigma (St. Louis, MO): Capsaicin, Cap; Ruthenium Red, RuR; 2-aminoethoxydiphenyl borate, 2-APB; Diltiazem, Dil; Nifedipine, Nif; Tetrodotoxin, TTX. From Tocris (Ellisville, MO): SKF96365, SKF; SB366791, SB; HC030031, HC; ω-Agatoxin TK, Aga; ω-Conotoxin MVIIC, Cono. From Calbiochem (Gibbstown, NJ): Flunarizine, Flu. Initial stock solutions were made in distilled water except for SB, HC, Flu, and Nif (made in DMSO), and 2-APB (made in 95% ethanol).
Statistics
In general data are expressed as a percentage of control responses (average of the initial and recovery responses were calculated as the control response to minimize the influence of receptor desensitization) and presented as mean ± sem. To determine whether the effect of an agent was selective for CCK-induced responses, we used Student’s t-test to compare the action of the agent on CCK-induced responses to the action of the agent on responses elicited by Hi-K or other activators of specific pathways. In the single cell PCR study, we used Chi-square test to evaluate the coexpression of CCK1Rs with the TRP channel(s) under investigation (P < 0.05 considered statistically significant).
Results
CCK-induced calcium signals are independent of depolarization-driven calcium influx
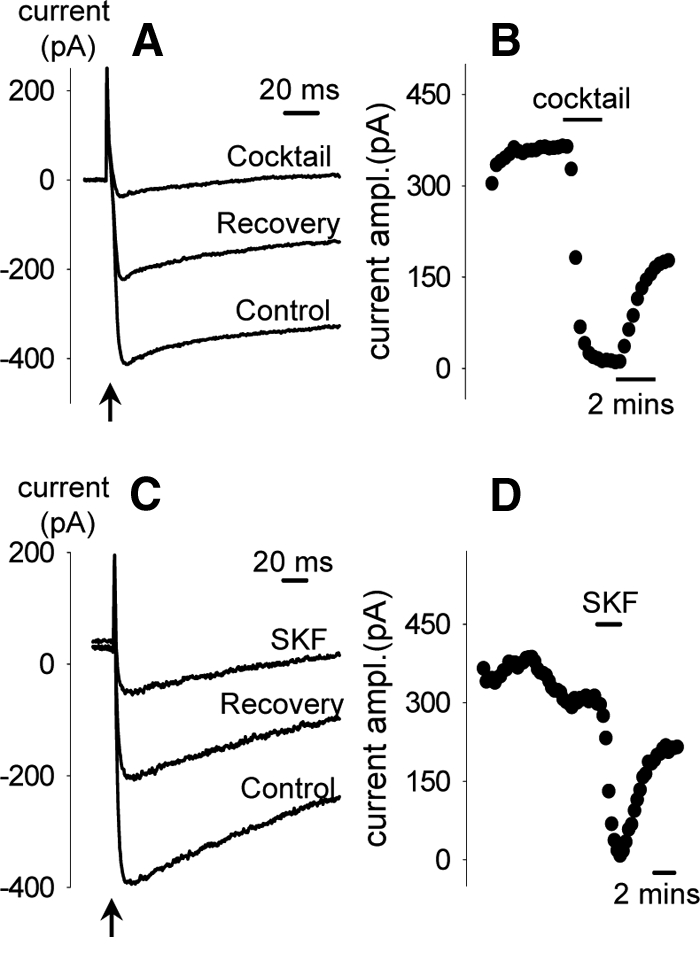
To examine whether the increase in cytosolic calcium induced by CCK is mediated via classic VDCC, we assembled a VDCC blocker cocktail (here on referred to as VDCC-cocktail) composed of 20 nm Aga (P/Q-type blocker) (25,26), 200 nm Cono (N-type blocker) (27), 20 μm Flu (T-type blocker) (28), and 100 μm Dil (L-type blocker) (29). We found that in the presence of the VDCC-cocktail, the responses to CCK (20 nm, standard dose of CCK in all experiments) were only slightly attenuated (Fig. 1A), whereas the responses to depolarization induced by Hi-K was nearly eliminated (Fig. 1, B and C). Similarly, SKF (20 μm), a compound that inhibits TRPC channels as well as voltage-gated calcium entry (30), significantly reduced the response to Hi-K solutions (Fig. 1E), with only a minimal effect on CCK-induced responses (Fig. 1, D and F).
Figure 1.

Effects of VDCC-cocktail and SKF on calcium responses to CCK and Hi-K. In this and all subsequent figures, applications of chemicals are indicated by the labeled horizontal bars above each trace. VDCC-cocktail (see Materials and Methods for composition) only slightly reduced the calcium response activated by CCK (20 nm) (A) but greatly inhibited the response to membrane depolarization induced by Hi-K (B). Similar results were observed with SKF (D and E). Quantifications of the effects of VDCC-cocktail and SKF are shown in C and F, respectively. With cocktail (C), responses to CCK were reduced to 82 ± 4% of control responses (n = 5 neurons); responses to Hi-K were reduced to 24 ± 9% of control (n = 4 neurons). With SKF (F), responses to CCK were reduced to 88 ± 5% of control (n = 20 neurons); responses to Hi-K were reduced to 14 ± 3% of control (n = 25 neurons). **, Statistically significant, P < 0.01.
To confirm that the VDCC-cocktail and SKF significantly blocked voltage-dependent calcium currents, we used patch-clamp electrophysiology to test their effects on whole-cell currents measured under ionic conditions that isolated calcium currents (Fig. 2). Calcium currents activated by depolarizing pulses were abolished by either extracellular application of the VDCC-cocktail (Fig. 2, A and B) or SKF (Fig. 2, C and D), confirming the effectiveness of these agents to block VDCC at the concentrations used.
Figure 2.
VDCC-cocktail and SKF abolished voltage-dependent calcium current. Currents were recorded every 15 s using voltage-clamp mode under ionic conditions that isolate calcium currents (in the bath: Na+ replaced on an equal molar basis with N-methyl-d-glucamine, K+ replace with Cs+, Ca2+ increased from 2 mm to 5 mm; in the pipette, Na+ and K+ replaced with 140 mm Cs+). In C and D, Ba2+ was used to replace Ca2+ as the current carrier to obtain more stable recordings. Membrane potential was held at −60 mV and at the arrow stepped to 0 mV for 200 ms. A and C, Representative traces of the depolarization-induced calcium currents with the presence and absence (including both Control and Recovery) of VDCC-cocktail and SKF, respectively. Changes in the current amplitude (average amplitude between 50 to 200 msec after onset of the depolarizing pulse) with time in two representative neurons are summarized in B and D.
Effects of RuR on CCK-induced activation
Because the actions of CCK appear to increase a Na+-dependent conductance to depolarize the neuronal membrane (20), and at the same time activate a VDCC-independent calcium-influx which is insensitive to TTX (responses to CCK in the presence of TTX averaged 56 ± 11 nm; without TTX averaged 56 ± 8 nm; n = 11, data not shown), it is highly likely that the responses to CCK are mediated by nonselective cation channels such as TRP channels. Our results with SKF (Fig. 1) demonstrate that members of the TRPC family are unlikely to be involved in the CCK-induced response. To test the involvement of other subtypes of TRP channels we used RuR, an agent widely used to block various subtypes of TRPV channels (31,32,33) and TRPA1 (34). At 1 μm, RuR abolished CCK-induced depolarization in some neurons (2 of 5; Fig. 3A), but the block was incomplete in others (3 of 5; Fig. 3B). We encountered the same scenario again in calcium imaging experiments, where 1 μm RuR blocked (representative trace not shown) or attenuated (Fig. 3C) calcium signals elicited by CCK in nearly half the neurons tested (7 of 15; on average responses reduced to 13.7 ± 8.9% of control), but was without effect in the other half (Fig. 3D). However, at a higher concentration (10 μm), RuR consistently blocked the CCK-elicited calcium signal (Fig. 4A). In electrophysiological measurements, this concentration of RuR also prevented the CCK-induced depolarization (Fig. 4C). The increase in intracellular calcium activated by CCK was suppressed to less than 20% of control (Fig. 4D). In contrast, under the same conditions, RuR did not prevent calcium signals in response to the depolarization induced by the Hi-K solutions (Fig. 4, B and D), arguing against an action of RuR to impose a global block of calcium influx. Thus, the conductance(s) that mediate CCK-induced responses are specifically inhibited by 10 μm RuR.
Figure 3.

Effects of RuR (1 μm) on CCK-induced activation. In panels A and B membrane potential recordings are shown. In panels C and D calcium responses are shown. Representative examples of a full block (n = 6 of 20) or a significant attenuation (n = 4 of 20) of CCK-induced responses by RuR are shown in A and B, respectively. In other cases (n = 10 of 20) RuR only had moderate effects (C) or was completely without effect (D). The 20 observations come from a mix of membrane potential measurements (n = 5) and calcium measurements (n = 15).
Figure 4.
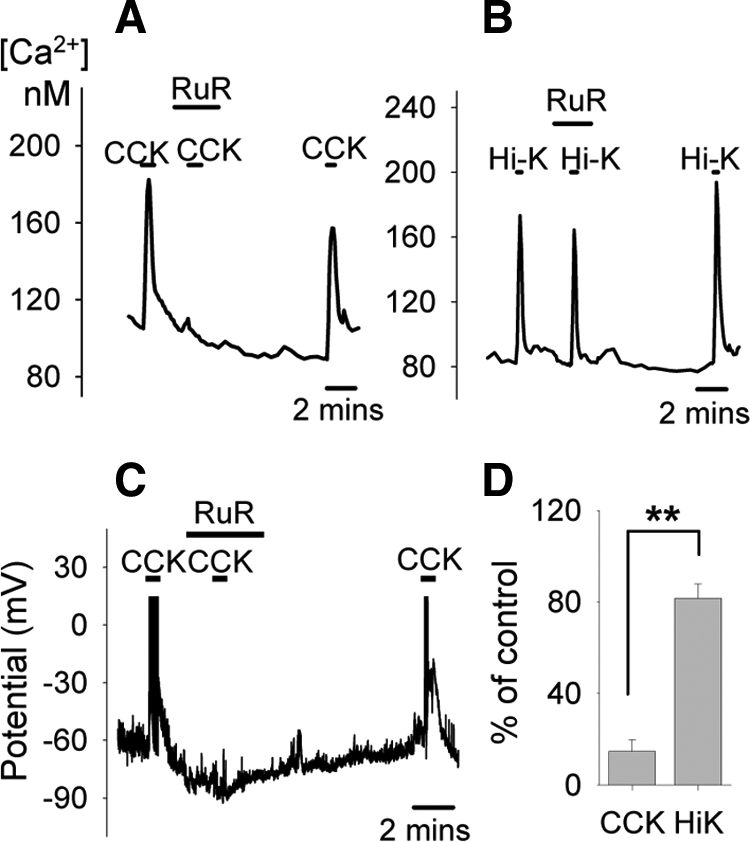
Effects of RuR (10 μm) on CCK-induced activation. At this higher concentration, RuR effectively blocked both CCK-induced calcium signal (A) and membrane depolarization (C), while the response to the Hi-K solution was minimally affected (B). Cs+ was used to replace K+ in C. Quantifications of the effects of 10 μm RuR on the calcium responses to CCK and Hi-K are shown in D. Responses to CCK were reduced to 15 ± 5% of control (n = 13 neurons); responses to Hi-K were reduced to 82 ± 6% of control (n = 20 neurons). **, Statistically significant, P < 0.01.
When we first began our series of membrane potential measurements, on occasion RuR would cause a small depolarization (two independent observations; data not shown). A possible explanation is that beyond its role of blocking TRPV and TRPA1, RuR may also inhibit the two-pore potassium channel TASK-3 (35). To eliminate this response we replaced both bath and pipette K+ with Cs+, a maneuver that would eliminate current through TASK-3 channels (36). In these conditions we no longer observed a response to RuR, yet the cells would still respond to CCK (Fig. 4C). This suggests that TASK-3 channels (or other conductance blocked by Cs+) are not important to the CCK-induced response.
Effects of selective TRP channel blockers on CCK-induced activation
To dissect the actions of RuR, we proceeded with two additional highly selective TRP channel blockers: SB, a TRPV1 antagonist (37), and HC, a TRPA1 blocker (38). SB (20 μm) failed to affect CCK-induced activation of vagal afferent neurons, while the response to Cap (50 nm), a TRPV1-specific agonist, was almost completely inhibited by SB in a reversible manner (Fig. 5, A and B). HC (20 μm) had a slight effect on the calcium signals elicited by CCK (Fig. 5, C and E), but rather minor compared with that of RuR (responses to CCK with HC: 71.6 ± 7.4% (Fig. 5E), compared with responses to CCK with RuR: 14.7 ± 5.1% (Fig. 4D); P < 0.001). However, at the same concentration, HC greatly suppressed the responses of nodose neurons to 1 μm Nif (Fig. 5, D and E), which is known to activate TRPA1 (39), confirming that HC in our preparation was able to selectively block TRPA1. The relative lack of effect seen with SB and HC indicated that TRPV1 and TRPA1 are unlikely to be the crucial targets in the CCK1 receptor signaling.
Figure 5.

Selective blockade of TRPV1 or TRPA1 in CCK-induced responses. Selective TRPV1 antagonist SB (20 μm) abolished calcium responses to Cap (50 nm) as expected but did not affect responses to CCK as shown in A. Corresponding quantifications are shown in B: With SB, responses to CCK were 104 ± 12% of control (n = 10 neurons); responses to Cap were reduced to 7 ± 5% of control (n = 7 neurons). Selective TRPA1 blocker HC (20 μm) slightly attenuated CCK-induced calcium response (C) but eliminated the response to Nif (1 μm), a previously-described TRPA1 agonist (D). Quantifications of the effects of HC are shown in E: Responses to CCK were reduced to 72 ± 7% of control (n = 12 neurons); responses to Nif were reduced to 11 ± 6% of control (n = 11 neurons). **, Statistically significant, P < 0.01.
Augmentation of CCK-induced calcium signal by 2-APB
2-APB is a membrane-permeable reagent originally described as an inhibitor of inositol 1,4,5-trisphosphate (IP3) induced calcium release (40). More recent studies, however, have shown 2-APB to be a common activator of TRPV1, TRPV2, and TRPV3 (41) and a blocker of many members of the TRPC family (21,42). In one study, in which CCK responses were monitored both in the absence and presence of 2-APB (50 μm), we never observed blockade of CCK-induced responses by 2-APB. In fact, in this group we found 5 of 9 neurons were transiently activated by 2-APB itself (Fig. 6A), while the other 4 neurons were insensitive to 2-APB (Fig. 6B). In the neurons directly activated by 2-APB, the calcium signal elicited by CCK was significantly augmented by the presence of 2-APB (Fig. 6C, also compare Fig. 6A to Fig. 6B). In a separate study, in which CCK and 2-APB were independently applied, we found that one-third of the neurons responsive to 2-APB did not respond to CCK (3 of 9, data not shown). However, the calcium response to 2-APB was always blocked by RuR (n = 9, data not shown), confirming that 2-APB induces calcium increase via TRPV channels. These results further argue against the involvement of TRPC channels in the actions of CCK but strongly support the possible participation of TRPV1-3 channels, although TRPV1 is unlikely to contribute to CCK-induced activation as implicated in the study with SB.
Figure 6.

Effects of 2-APB on CCK-induced calcium responses. In some neurons, 2-APB alone activated transient calcium signals and subsequently enhanced the CCK-induced responses (A). In other neurons, 2-APB was without effect (B). Quantifications are shown in C: In 2-APB–sensitive neurons (APB-S), responses to CCK were increased to 170 ± 5% of control (n = 5 neurons); in 2-APB–insensitive neurons (APB-I), responses to CCK were 103 ± 16% of control (n = 4 neurons). **, Statistically significant, P < 0.01.
Coexpression of CCK1Rs with TRPV subtypes
To further explore the possibility that TRPV channels are the critical mediators of the cellular actions of CCK, we performed single-cell PCR to examine the distribution of CCK1Rs and TRPV subtypes. Due to the limited amount of cDNA that can be produced from a single neuron, we could only examine four genes besides PGP9.5, CCK1R, and TRPV1 (three markers we tested for with every sample). Because TRPV5 and TRPV6 share many similarities in amino acid sequence and ion channel functions (21), we selected TRPV2, V3, V4, and V5 as representative members to pursue. We found that TRPV2-5 were indeed present in a subset of CCK1R positive neurons (Fig. 7A), and all showed at least a trend, which was statistically significant in the case of TRPV4, to be preferentially expressed in neurons that also expressed CCK1Rs (Fig. 7B; upper panel). On the other hand, select members from other TRP families (namely TRPC1, TRPC3, TRPM8 and TRPA1), which are unlikely to be the targets in the CCK signaling as suggested by the pharmacological study, showed no preferential expression in CCK1R positive neurons (Fig. 7B; lower panel). A majority (more than 80%) of CCK1R-expressing neurons registered positive for at least one subtype of TRPV2-5 (Fig. 7B; upper panel), significantly higher than the percentage of CCK1R-absent neurons that expressed these subtypes (∼40%), confirming a likely association between CCK signaling and TRPV2-5 channels.
Figure 7.
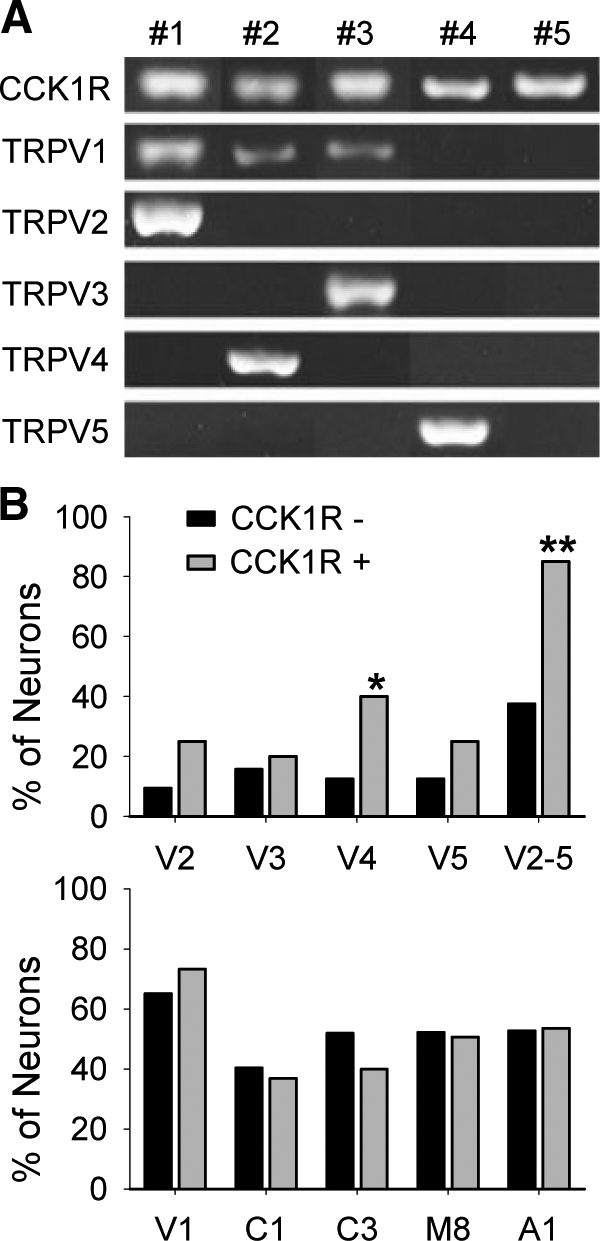
Coexpression of CCK1Rs and TRPV subtypes. Examples of CCK1R-positive neurons and expression of TRPV subtypes are presented in A. Quantifications of the distribution of each gene in relation to CCK1Rs are shown in B. Black bars represent results from neurons not expressing CCK1R (CCK1R−); Gray bars represent results from neurons expressing CCK1R (CCK1R+). Upper panel, Of 32 CCK1R- neurons, three expressed TRPV2, five expressed TRPV3, four expressed TRPV4, four expressed TRPV5, and 12 expressed at least one subtype of TRPV2-5. Of 20 CCK1R+ neurons, five expressed TRPV2, four expressed TRPV3, eight expressed TRPV4, five expressed TRPV5, and 17 expressed at least one subtype of TRPV2-5. Lower panel, In CCK1R− neurons, 56 of 86 expressed TRPV1, 23 of 57 expressed TRPC1, 27 of 52 expressed TRPC3, 23 of 44 expressed TRPM8, 39 of 74 expressed TRPA1. In CCK1R+ neurons, 85 of 116 expressed TRPV1, 21 of 57 expressed TRPC1, 20 of 50 expressed TRPC3, 40 of 79 expressed TRPM8, 60 of 112 expressed TRPA1. *, Different from CCK1R- neurons, P < 0.05; **, P < 0.01, by χ2 analysis.
Discussion
A major conclusion from our study is that the CCK-induced increase of cytosolic calcium in cultured nodose neurons is independent of depolarization, as shown by the observation that VDCC-cocktail and SKF had only minor effects on the calcium signals provoked by CCK. Further, the insensitivity of the CCK-induced calcium response to SKF also argues that TRPC channels are minimally involved in the response to CCK. The one compound that effectively eliminated both the depolarizing response and calcium influx activated by CCK was RuR, which inhibits the TRPV family of channels, as well as TRPA1. Selective blockers of TRPV1 and TRPA1, however, were ineffective, leading to the conclusion that the RuR-sensitive channels that are relevant to the CCK-induced response are in the TRPV family excluding TRPV1. We have previously shown that nodose neurons also express TRPM8 (22), but because TRPM8 is insensitive to RuR (43,44), it rules out TRPM8 as a major influx pathway for the CCK-induced response. Finally, the participation of TRPV subtypes is supported by the observations that 1) CCK-induced activation is augmented by 2-APB, which activates TRPV1-3, in a subset of CCK-responsive neurons; 2) single-cell PCR revealed a significant segregation of CCK1Rs with TRPV2, TRPV3, TRPV4, and TRPV5. However, it should be noted that we never observed a specific TRP subtype that always coexpressed with CCK1Rs. Taken together, these results suggest that multiple members of the TRPV family, excluding TRPV1, are the key pathways for calcium influx in response to CCK in vagal afferent neurons. Further, because RuR blocked the depolarization caused by CCK, it suggests that CCK-induced activation of multiple members of the TRPV family of channels significantly contributes to the depolarization that is observed in response to CCK.
TRP channels constitute a large superfamily of nonselective cation channels, organized into six families with nearly 30 members (21). They are weakly time- and voltage-dependent but responsive to a wide range of physiochemical stimuli (21,45). The TRPV family was initially described as the “Thermo TRPs,” sensing temperatures ranging from warm to hot (46), but recent investigations using knockout mice or heterologous cellular expression systems revealed the emerging features of several TRPV channels, namely TRPV1, TRPV2, and TRPV4, as being mechano- and osmo-sensitive (47). TRPM8 and TRPA1, on the other hand, are known as the “cold-sensor,” responding to innocuous cold and noxious cold temperatures, respectively (46). Most members of the TRPC family were first implicated in the store-operated calcium entry (48); however, additional studies have shown that TRPC channels are sensitive to lipids, such as diacylglycerol (49,50), and mechanical stimuli (51), independently of store depletion. Besides direct gating mechanisms, TRP channels are known to be modulated downstream of G protein–coupled receptors via a variety of second messenger pathways (52,53). Thus in neurons TRP channels can serve as both cellular sensors to a variety of physical and chemical stimuli as well as critical mediators of hormonal signaling, a role they are likely serving in vagal afferent neurons.
We have previously shown that messages of various TRP channels, including TRPC1,3,5,6, TRPV1-4, TRPA1, and TRPM8 are expressed in the nodose ganglia, and in some cases specific channels preferentially segregate within distinct neuronal populations (22). In this study, we examined the expression of CCK1Rs and TRPV subtypes in individual vagal afferent neurons. We found that the majority of neurons that expressed CCK1Rs also expressed at least one subtype of TRPV2-5, and the TRPV subtypes tended to be preferentially expressed by CCK1R positive neurons. Detection of TRP channel proteins in the vagal system has also been reported by many other investigators. Immunoreactivity for members of the TRPV family were identified in rodent vagal afferent neurons and the gut (24,54). Another group observed that TRPC channels are present in rat nodose ganglia, as well as both myelinated and unmyelinated fibers targeting the aortic arch (23). This complex expression and distribution of TRP channels in vagal afferent neurons is potentially important in the neuron-specific signaling activated by the many sensory modalities that vagal afferent neurons respond to.
Although the cellular events after CCK receptor activation in the nodose ganglia are poorly understood, intracellular signaling pathways activated by CCK receptors have been extensively studied in many other cellular models. The best known mechanism is coupling through heterotrimeric G proteins of the Gq family to activate phospholipase C leading to an increase in IP3 and diacylglycerol produced from phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis (55). Many of these pathways have been associated with activation of TRP channels; however, which pathway is more important in nodose neurons remains undefined. In our current study 2-APB either had no effect or augmented the calcium response to CCK. Because 2-APB is a known inhibitor of the IP3 receptor (40), it is not likely that IP3-sensitive intracellular stores participate in the calcium response to CCK. This conclusion is supported by a prior study in which thapsigargin, an agent that depletes intracellular stores of calcium, was also found not to affect the calcium responses to CCK (19). CCK, when applied to nodose neurons, both activates a depolarizing conductance as well as inhibits a hyperpolarizing conductance (20). However, inhibition of a hyperpolarizing conductance by itself cannot explain the calcium signal induced by CCK. Such an action would merely result in membrane depolarization, and our results with SKF and the VDCC-cocktail demonstrated that the calcium response to CCK is not secondary to depolarization. Thus while inhibition of a hyperpolarizing conductance may contribute to the membrane effect of CCK, activation of a calcium permeable pathway insensitive to the VDCC cocktail or SKF is necessary for the calcium signal induced by CCK.
An apparent paradox in our results is that both CCK and Hi-K solutions induced depolarization, yet the calcium influx pathways appear to be different, with different sensitivities to SKF and RuR. This is most likely due to both recruitment of distinct populations of calcium-permeable channels and the manner in which VDCC are recruited by CCK vs. Hi-K. When cells are exposed to the Hi-K solution, the neurons undergo a robust and prolonged membrane depolarization, which would activate a sustained calcium influx mediated by VDCC. On the other hand, CCK induces a more modest depolarization upon which spiking activity usually occurs. Because the CCK-induced calcium response is relatively insensitive to blockers of VDCC, the amount of calcium that enters the cells through VDCC during these brief spikes must be negligible. Hence another pathway, which by our observations are probably TRPV channels (excluding TRPV1), is required to mediate the calcium influx induced by CCK.
Although an isolated neuronal preparation has the advantage of allowing one to unequivocally conclude the action of a substance is mediated directly on the neurons, it suffers from the possibility that the physiologically relevant site of action may be at the neuronal terminals that are lost in the isolation procedure, and that the transduction mechanisms at the terminal are different than that observed on the cell body. For instance, Rogers and Hermann (56) examined the actions of CCK on the central vagal afferent terminals and concluded that the increase in intracellular calcium provoked by CCK is a consequence of a complex interaction between protein kinase A and phospholipase C which eventually causes a calcium influx that is sensitive to TTX and L-type calcium channel blockers (nitrendipine). Further, they concluded that calcium release from endoplasmic reticulum is also involved. These observations differ from ours in which L-type channel blocker and TTX were ineffective and our conclusion that intracellular stores are not involved. One explanation may be that the pathways that mediate the response to CCK are different at cell bodies compared with the central terminals. Indeed, at the peripheral sites, action potential initiation is likely to be the critical element in the actions of CCK, whereas at the central sites modulation of presynaptic function may be the important effect of CCK. Because these are such different processes, there would be little reason to believe that the same underlying mechanisms are invoked. Our observations on the cell bodies involve cell activation, including action potential initiation, which we believe are likely to be representative of the events that occur at peripheral terminals in the gastrointestinal tract. Our electrophysiological results, in which RuR blocked CCK-induced depolarization, support the concept that a member of the TRPV family could be involved with initiating action potential generation in peripheral terminals, but this speculation needs to be tested directly.
In conclusion, we propose that members of the TRPV family, excluding TRPV1, are critical in mediating the effects of CCK1R activation in vagal afferent neurons. It is likely that these channels are the depolarizing conductance(s) activated by CCK in these neurons (20). We further conclude that calcium responses induced by CCK in these neurons are not dependent on depolarization, but the calcium influx pathway is likely to be a significant contributor to the depolarization. Comparison with a prior study suggests that CCK actions on central terminals and the cell bodies are not mediated by similar pathways. Finally, our results implicate that CCK1Rs are associated with multiple effectors. The significance of the diversity of coupling mechanisms is not yet clear but could be related to sensory specific information that is carried by individual neurons.
Acknowledgments
We thank Dallas Kinch for technical help with these studies and James H. Peters for critically reading and commenting on the manuscript.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant RO1 DK067146 (awarded to S.M.S.).
Disclosure Summary: The authors have nothing to declare.
First Published Online September 29, 2010
Abbreviations: CCK, Cholecystokinin; CCK1R, CCK1 receptor; Hi-K, high potassium; IP3, inositol 1,4,5-trisphosphate; RuR, ruthenium red; TRP, transient receptor potential; VDCC, voltage-dependent calcium channels.
References
- Debas HT, Farooq O, Grossman MI 1975 Inhibition of gastric emptying is a physiological action of cholecystokinin. Gastroenterology 68:1211–1217 [PubMed] [Google Scholar]
- Liddle RA, Gertz BJ, Kanayama S, Beccaria L, Coker LD, Turnbull TA, Morita ET 1989 Effects of a novel cholecystokinin (CCK) receptor antagonist, MK-329, on gallbladder contraction and gastric emptying in humans: implications for the physiology of CCK. J Clin Invest 84:1220–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosotani R, Chowdhury P, Rayford PL 1989 L-364,718, a new CCK antagonist, inhibits postprandial pancreatic secretion and PP release in dogs. Dig Dis Sci 34:462–467 [DOI] [PubMed] [Google Scholar]
- Konturek SJ, Tasler J, Cieszkowski M, Szewczyk K, Hladij M 1988 Effect of cholecystokinin receptor antagonist on pancreatic responses to exogenous gastrin and cholecystokinin and to meal stimuli. Gastroenterology 94:1014–1023 [DOI] [PubMed] [Google Scholar]
- Owyang C 1996 Physiological mechanisms of cholecystokinin action on pancreatic secretion. Am J Physiol 271:G1–G7 [DOI] [PubMed] [Google Scholar]
- Gibbs J, Young RC, Smith GP 1973 Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol 84:488–495 [DOI] [PubMed] [Google Scholar]
- Moran TH, Ameglio PJ, Schwartz GJ, McHugh PR 1992 Blockade of type A, not type B, CCK receptors attenuates satiety actions of exogenous and endogenous CCK. Am J Physiol 262:R46–R50 [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, McHugh PR, Moran TH 1991 Integration of vagal afferent responses to gastric loads and cholecystokinin in rats. Am J Physiol 261:R64–R69 [DOI] [PubMed] [Google Scholar]
- Smith GP, Jerome C, Cushin BJ, Eterno R, Simansky KJ 1981 Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science 213:1036–1037 [DOI] [PubMed] [Google Scholar]
- Smith GP, Jerome C, Norgren R 1985 Afferent axons in abdominal vagus mediate satiety effect of cholecystokinin in rats. Am J Physiol 249:R638–R641 [DOI] [PubMed] [Google Scholar]
- South EH, Ritter RC 1988 Capsaicin application to central or peripheral vagal fibers attenuates CCK satiety. Peptides 9:601–612 [DOI] [PubMed] [Google Scholar]
- Li Y, Owyang C 1993 Vagal afferent pathway mediates physiological action of cholecystokinin on pancreatic enzyme secretion. J Clin Invest 92:418–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GJ, Berkow G, McHugh PR, Moran TH 1993 Gastric branch vagotomy blocks nutrient and cholecystokinin-induced suppression of gastric emptying. Am J Physiol 264:R630–R637 [DOI] [PubMed] [Google Scholar]
- Asin KE, Bednarz L 1992 Differential effects of CCK-JMV-180 on food intake in rats and mice. Pharmacol Biochem Behav 42:291–295 [DOI] [PubMed] [Google Scholar]
- Broberger C, Holmberg K, Shi TJ, Dockray G, Hökfelt T 2001 Expression and regulation of cholecystokinin and cholecystokinin receptors in rat nodose and dorsal root ganglia. Brain Res 903:128–140 [DOI] [PubMed] [Google Scholar]
- Widdop RE, Krstew E, Mercer LD, Carlberg M, Beart PM, Jarrott B 1994 Electrophysiological and autoradiographical evidence for cholecystokinin A receptors on rat isolated nodose ganglia. J Auton Nerv Syst 46:65–73 [DOI] [PubMed] [Google Scholar]
- Simasko SM, Ritter RC 2003 Cholecystokinin activates both A- and C-type vagal afferent neurons. Am J Physiol Gastrointest Liver Physiol 285:G1204–G1213 [DOI] [PubMed] [Google Scholar]
- Lankisch TO, Tsunoda Y, Lu Y, Owyang C 2002 Characterization of CCK(A) receptor affinity states and Ca(2+) signal transduction in vagal nodose ganglia. Am J Physiol Gastrointest Liver Physiol 282:G1002–G1008 [DOI] [PubMed] [Google Scholar]
- Simasko SM, Wiens J, Karpiel A, Covasa M, Ritter RC 2002 Cholecystokinin increases cytosolic calcium in a subpopulation of cultured vagal afferent neurons. Am J Physiol Regul Integr Comp Physiol 283:R1303–R1313 [DOI] [PubMed] [Google Scholar]
- Peters JH, Ritter RC, Simasko SM 2006 Leptin and CCK modulate complementary background conductances to depolarize cultured nodose neurons. Am J Physiol Cell Physiol 290:C427–C432 [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE 2006 An introduction to TRP channels. Annu Rev Physiol 68:619–647 [DOI] [PubMed] [Google Scholar]
- Zhao H, Sprunger LK, Simasko SM 2010 Expression of transient receptor potential channels and two-pore potassium channels in subtypes of vagal afferent neurons in rat. Am J Physiol Gastrointest Liver Physiol 298:G212–G221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazebrook PA, Schilling WP, Kunze DL 2005 TRPC channels as signal transducers. Pflugers Arch 451:125–130 [DOI] [PubMed] [Google Scholar]
- Ni D, Gu Q, Hu HZ, Gao N, Zhu MX, Lee LY 2006 Thermal sensitivity of isolated vagal pulmonary sensory neurons: role of transient receptor potential vanilloid receptors. Am J Physiol Regul Integr Comp Physiol 291:R541–R550 [DOI] [PubMed] [Google Scholar]
- Olivera BM, Miljanich GP, Ramachandran J, Adams ME 1994 Calcium channel diversity and neurotransmitter release: the omega-conotoxins and omega-agatoxins. Annu Rev Biochem 63:823–867 [DOI] [PubMed] [Google Scholar]
- Teramoto T, Niidome T, Miyagawa T, Nishizawa Y, Katayama K, Sawada K 1995 Two types of calcium channels sensitive to omega-agatoxin- TK in cultured rat hippocampal neurones. Neuroreport 6:1684–1688 [DOI] [PubMed] [Google Scholar]
- McDonough SI, Swartz KJ, Mintz IM, Boland LM, Bean BP 1996 Inhibition of calcium channels in rat central and peripheral neurons by omega-conotoxin MVIIC. J Neurosci 16:2612–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedding M, Paoletti R 1992 Classification of calcium channels and the sites of action of drugs modifying channel function. Pharmacol Rev 44:363–376 [PubMed] [Google Scholar]
- Kraus RL, Hering S, Grabner M, Ostler D, Striessnig J 1998 Molecular mechanism of diltiazem interaction with L-type Ca2+ channels. J Biol Chem 273:27205–27212 [DOI] [PubMed] [Google Scholar]
- Merritt JE, Armstrong WP, Benham CD, Hallam TJ, Jacob R, Jaxa-Chamiec A, Leigh BK, McCarthy SA, Moores KE, Rink TJ 1990 SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem J 271:515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D 1999 A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398:436–441 [DOI] [PubMed] [Google Scholar]
- Peier AM, Reeve AJ, Andersson DA, Moqrich A, Earley TJ, Hergarden AC, Story GM, Colley S, Hogenesch JB, McIntyre P, Bevan S, Patapoutian A 2002 A heat-sensitive TRP channel expressed in keratinocytes. Science 296:2046–2049 [DOI] [PubMed] [Google Scholar]
- Voets T, Prenen J, Vriens J, Watanabe H, Janssens A, Wissenbach U, Bödding M, Droogmans G, Nilius B 2002 Molecular determinants of permeation through the cation channel TRPV4. J Biol Chem 277:33704–33710 [DOI] [PubMed] [Google Scholar]
- Nagata K, Duggan A, Kumar G, García-Añoveros J 2005 Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci 25:4052–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musset B, Meuth SG, Liu GX, Derst C, Wegner S, Pape HC, Budde T, Preisig-Müller R, Daut J 2006 Effects of divalent cations and spermine on the K+ channel TASK-3 and on the outward current in thalamic neurons. J Physiol 572:639–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper BY, Johnson RD, Rau KK 2004 Characterization and function of TWIK-related acid sensing K+ channels in a rat nociceptive cell. Neuroscience 129:209–224 [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM, Luis Hannan S, Lappin SC, Egerton J, Smith GD, Worby A, Howett L, Owen D, Nasir S, Davies CH, Thompson M, Wyman PA, Randall AD, Davis JB 2004 Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology 46:133–149 [DOI] [PubMed] [Google Scholar]
- Eid SR, Crown ED, Moore EL, Liang HA, Choong KC, Dima S, Henze DA, Kane SA, Urban MO 2008 HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain 4:48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo O, Meseguer V, Belmonte C, Viana F 2008 TRPA1 channels: novel targets of 1,4-dihydropyridines. Channels (Austin) 2:429–438 [DOI] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K 1997 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem 122:498–505 [DOI] [PubMed] [Google Scholar]
- Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, Lee LY, Wood JD, Zhu MX 2004 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem 279:35741–35748 [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney Jr JW 2004 The mammalian TRPC cation channels. Biochim Biophys Acta 1742:21–36. [DOI] [PubMed] [Google Scholar]
- Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A 2002 A TRP channel that senses cold stimuli and menthol. Cell 108:705–715 [DOI] [PubMed] [Google Scholar]
- Wrigley PJ, Jeong HJ, Vaughan CW 2009 Primary afferents with TRPM8 and TRPA1 profiles target distinct subpopulations of rat superficial dorsal horn neurones. Br J Pharmacol 157:371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, Strübing C 2001 The TRP ion channel family. Nat Rev Neurosci 2:387–396 [DOI] [PubMed] [Google Scholar]
- Dhaka A, Viswanath V, Patapoutian A 2006 Trp ion channels and temperature sensation. Annu Rev Neurosci 29:135–161 [DOI] [PubMed] [Google Scholar]
- Liedtke W, Kim C 2005 Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci 62:2985–3001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley PF, Zeng W, Huang GN, Yuan JP, Kim JY, Lee MG, Muallem S 2007 TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 42:205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G 1999 Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397:259–263 [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bahnasi YM, Dedman AM, Al-Shawaf E 2009 TRPC channel lipid specificity and mechanisms of lipid regulation. Cell Calcium 45:583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Jian Z, Kawarabayashi Y 2009 Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther 123:371–385 [DOI] [PubMed] [Google Scholar]
- Yao X, Kwan HY, Huang Y 2005 Regulation of TRP channels by phosphorylation. Neuro-Signals 14:273–280 [DOI] [PubMed] [Google Scholar]
- Rohacs T 2009 Phosphoinositide regulation of non-canonical transient receptor potential channels. Cell Calcium 45:554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jones S, Brody K, Costa M, Brookes SJ 2004 Thermosensitive transient receptor potential channels in vagal afferent neurons of the mouse. Am J Physiol Gastrointest Liver Physiol 286:G983–G991 [DOI] [PubMed] [Google Scholar]
- Matozaki T, Williams JA 1989 Multiple sources of 1,2-diacylglycerol in isolated rat pancreatic acini stimulated by cholecystokinin. Involvement of phosphatidylinositol bisphosphate and phosphatidylcholine hydrolysis. J Biol Chem 264:14729–14734 [PubMed] [Google Scholar]
- Rogers RC, Hermann GE 2008 Mechanisms of action of CCK to activate central vagal afferent terminals. Peptides 29:1716–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]