

Abstract
Nutrient-sensitive hypothalamic neurons regulate energy balance and glucose homeostasis, but the molecular mechanisms mediating hypothalamic responses to nutritional state remain incompletely characterized. To address these mechanisms, the present studies used quantitative PCR to characterize the expression of a panel of genes the hypothalamic expression by nutritional status of which had been suggested by DNA microarray studies. Although these genes regulate a variety of function, the most prominent set regulate intermediary metabolism, and the overall pattern clearly indicated that a 48-h fast produced a metabolic reprogramming away from glucose metabolism and toward the utilization of alternative fuels, particularly lipid metabolism. This general reprogramming of intermediary metabolism by fasting was observed both in cortex and hypothalamus but most prominently in hypothalamus. The effect of fasting on the expression of these genes may be mediated by reduction in plasma glucose or glucose metabolism, rather than leptin, because they were generally recapitulated by hypoglycemia even in the presence of elevated insulin and in vitro by low glucose but were not recapitulated in ob/ob mice. These studies suggest that fasting reduces glucose metabolism and thus minimizes the production of hypothalamic malonyl-coenzyme A. However, because the reprogramming of glucose metabolism by fasting was also observed in cortex, this apparent substrate competition may mediate more general responses to nutritional deprivation, including those responsible for the protective effects of dietary restriction. The present studies also provide a large panel of novel glucose-regulated genes that can be used as markers of glucose action to address mechanisms mediating hypothalamic responses to nutritional state.
This novel panel of fasting- and glucose-regulated hypothalamic genes suggests that nutritional deprivation produces metabolic reprogramming away from glucose metabolism and toward lipid oxidation.
Although hypothalamic neurons sensitive to metabolic status have long been known to regulate energy balance, mechanisms mediating these nutritional signals are still not completely understood (1). Mechanisms mediating hypothalamic neuronal sensitivity to nutritional status are of singular significance because failure in such mechanisms are a likely cause of obesity and diabetes, which are among the greatest threats to global public health in the 21st century (2). For about 100 yr hypothalamic sensitivity to glucose was hypothesized to be important in regulating energy balance, but until recently this hypothesis had largely been abandoned (1). In part, this was because the discovery of leptin, the plasma levels of which robustly reflected adipose stores (3) and provided a much more convincing mechanism by which hypothalamic neurons sensitive to leptin monitor nutritional stores. Furthermore, restoration of leptin in fasted animals reverses many responses to fasting (4,5,6). Other studies have suggested that a fall in plasma insulin may also mediate some neuroendocrine responses to fasting (7).
Nevertheless, many responses to fasting occur in the absence of leptin or without a change in plasma leptin or insulin concentrations (8,9,10,11). Furthermore, more recent studies have supported a role for hypothalamic glucose-sensing mechanisms in regulating energy balance (12,13,14,15,16). Similarly more recent studies have suggested that malonyl-coenzyme A (CoA), which inhibits hypothalamic fatty acid oxidation, also plays a critical role in regulating energy balance (12,17,18,19,20,21,22), effects that apparently entail production of malonyl-CoA from hypothalamic glucose metabolism (23). Nevertheless, the mechanisms by which hypothalamic neurons sense nutrients remain unclear, despite some progress (24,25,26).
To address molecular mechanisms mediating hypothalamic responses to nutritional status, we carried out a series of large-scale analyses of hypothalamic gene expression during fasting and hypoglycemia and after refeeding, using DNA microarrays to provide a provisional set of candidates. To avoid problems associated with false-positive results (27), in the present study we report only those results that have been corroborated by quantitative PCR (qPCR).
Materials and Methods
Animals
All studies were approved by the appropriate institutional animal review board (Institutional Animal Care and Use Committee). All mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed five per cage with free access to food and water under 12-h light,12-h dark cycle (lights on at 0700 h).
Fasting and refeeding
C57BL/6J mice (age 12 wk) were used in the study. Mice were randomly assigned to either the fed (n = 6) or fasted (n = 18) group. Mice were fasted for 48 h, because many responses to fasting such as induction of NPY reach a maximum between 24 and 48 h. Mice were then killed except for six of the fasted animals, which were refed for 4 h using commercially available condensed sweet milk diluted 1:1 with water, and then killed. Mice were refed with diluted sweet milk rather than chow to facilitate maximum consumption of calories over the minimum period of time.
Ob/ob mice
Male ob/ob (JAX no. 000632, B6.V-Lepob/J) mice and 12-wk-old C57BL/6J wild-type controls (n = 10/group) were purchased from The Jackson Laboratory. Animals were killed in a fed state following a balanced design. In all studies, mice were killed following a balanced design during the light period (0010 h to 1400 h). Mice were killed by decapitation after a brief exposure to carbon dioxide. Hypothalamic and cortical areas, along with peripheral tissues, were quickly removed, frozen on dry ice, and stored at −70 C until extraction of RNA. Trunk blood was collected for hormone level analysis.
In vitro responses to glucose
For the in vitro glucose/leptin responsiveness studies, brains from 1-month-old C57Bl6/J mice were used. In general, in this in vitro system, slices containing the hypothalamus are prepared as in typical electrophysiological studies (28). To prepare hypothalamic slices, mice were anesthetized by injection of urethane (1.6 g/kg, ip) followed by decapitation. Brains were promptly removed and placed in ice-cold PBS buffer. Thin coronal slices (250 μm) containing the hypothalamus were cut with a Vibratome (Leica, Inc., Deerfield, IL; series 1000). Usually eight slices were obtained per hypothalamus. Slices were then transferred into regular artificial cerebrospinal fluid (aCSF): 124 mm NaCl, 26 mM NaHPO4, mm KCl 5, 1.2 mm KH2PO4, 1.3 mm MgSO4, 2.4 mmCaCl, and either d-glucose 10 or glucose 0.1, osmolarity was adjusted by NaCl concentration. All solutions were always saturated with 95% O2 and 5% CO2 in the slice preparation and experiment at pH 7.4 and room temperature. For the in vitro responses to glucose and leptin, 24 male 1-month-old C57Bl6/J mice were fasted for 48 h to produce a uniform baseline glucose level across all mice. The animals from each strain were assigned to either the low (0.1 mm) or high (10 mm) glucose group with or without leptin (50 nm). Brain slices containing mediobasal hypothalamus for the low glucose group were incubated in oxygen-perfused aCSF containing 0.1 mm glucose with or without leptin (50 nm) for 4 h with the bath replaced after the first hour. The brain slices in the high glucose group were incubated in aCSF containing 0.1 mm glucose for 1 h and were then transferred to aCSF containing 10 mm glucose with or without leptin (50 nm) for 3 h. At the end of the incubation period, the mediobasal hypothalamus and cortical tissue was dissected from each slice, frozen on dry ice, and stored at −70 C until extraction of RNA.
Blood chemistry
Blood glucose was measured by a Bayer Contour glucose meter (Bayer, Mountain View, CA). Blood leptin and insulin levels were measured in the fasting/refeeding study using ELISA kits from Crystal Chem Inc. (Downers Grove, IL). Corticosterone levels were measured using ELISA from Assay Designs, Inc. (Ann Arbor, MI).
Extraction of hypothalamic and cortical RNA and cDNA synthesis
To obtain RNA for gene expression analysis by qPCR, hypothalamic and cortical tissue was homogenized in tubes containing RLT buffer (QIAGEN, Chatsworth, CA) supplemented with 2-mercaptoethanol, and total RNA was extracted using an RNeasy Mini Kit (QIAGEN). Although the main focus of the study was on gene expression in the hypothalamus, which contains nutrient-sensing neurons and regulates energy balance, we compared gene expression in the hypothalamus to expression in the cortex, which we have shown expresses relatively few nutrient-sensing neurons (28). The quality of total RNA was assessed using the Biophotometer (Eppendorf, Madison, WI). Due to capacity limitations of the PCR array plates, six of 10 samples from each experimental group (12 for fasting) were selected (based on superior RNA quality) and were subjected to reverse transcription. High-quality total RNA (1 μg) was used for cDNA synthesis using RT2 First Strand Kit (SABiosciences, Frederick, MD). All procedures were performed according to the manufacturers’ protocols.
Microarray analysis
RNA from fed and fasted mice, and mice 4 h after refeeding, was analyzed on MG-430 DNA microarrays (Affymetrix, Santa Clara, CA), n = 6/group. Results were exported to EXCEL spreadsheets, analyzed by ANOVA, sorted by P value, and compared with a similar study we had previously carried out to discover genes induced by hypoglycemia (27). From these lists 187 genes were chosen for quantification by qPCR based on a variety of parameters, including low P value, similar regulation by fasting and hypoglycemia, and likely relevance to metabolic regulation, similar to strategies we have previously described (27).
RT-PCR with custom RT2 Profiler PCR arrays
RT2 Profiler PCR Array (SABiosciences) technology for gene expression analysis entails a synthesis between the profiling capabilities of DNA microarray and the quantitative reliability and sensitivity of qPCR. The results are highly reproducible within the same assay run or between different assay runs. RT2 Profiler Custom PCR Arrays were used to simultaneously examine the mRNA levels of 187 genes, including seven housekeeping genes in 384-well plates according to the protocol of the manufacturer (SABiosciences). The genes were chosen based on prior DNA microarray studies, as described above. A custom PCR array with the same set of genes was used for the hypoglycemia, fasting, and ob/ob studies. For in vitro responses to glucose, a new custom PCR array was designed containing only genes that were significantly regulated in the in vivo studies. The qPCRs were carried out using an ABI Prism 7900 thermocycler. Seven genes (Rpl13a, B2m, Gusb, Hsp90ab1, Actb, and Tbp) were included in the qPCR plate to provide a composite for normalization, but because Rpl13a expression was not observed in all samples, gene expression was normalized to a composite of the other six, using the ΔΔCt method. Fasting did not influence expression of any of the normalization genes, with or without normalization using the other five genes. Data were analyzed using a web-based software program provided by the manufacturer with additional analysis using GraphPad Prism 4 (GraphPad Software, Inc., San Diego, CA) for Macintosh.
Data analysis
All data are presented as mean ± sem. Statistical analysis was performed using GraphPad Prism 4.0 by one-way ANOVA followed by Newman-Keuls post hoc test. P < 0.05 indicates statistical significance.
Results
Complete nomenclature of all genes discussed in the present report is given in Table 1, which also provide the relative enrichment of these genes in hypothalamus vs. cortex. Genes the hypothalamic expression of which was regulated by fasting fell into several categories that are mechanistically informative. The most prominent genes are those indicating that fasting produces a characteristic reprogramming of utilization of substrate for energy production. Thus, as shown in Figure 1, A–D, fasting produced a hypothalamic profile of gene expression (induction of Pdk4, inhibition of Pfkfb2, inhibition of Mdh1, and inhibition of GLUT-2) that would be expected to reduce glucose utilization. Conversely, fasting produced an alternative profile of gene expression (induction of Cpt1a, Gpd1, Pcx, Fatp, Pxmp2, Idh2, and Apod) that suggests increased use of fuels alternative to glucose, particularly β oxidation (Fig. 1, E–L). Finally, the induction of two transcription factors, Foxo3 and Hif3a (Fig. 1, M and N), are also consistent with reduction in glycolysis (see Discussion). Of particular interest, regulation of key genes (Pdk4, Pfkfb2, Glut2, Cpt1a, Gpd1, Pcx, and Foxo3) by fasting was observed in hypothalamus and not in cortex. Interestingly, almost all the effects of fasting were completely or largely reversed within 4 h of refeeding, the exceptions being three of the four genes implicated in glucose metabolism, Pfkfb2, Mdh1, and Glut2 (Fig. 1, B–D).
Table 1.
Gene descriptions and comparison of expression levels between hypothalamus and cortex of control (ad libitum) fed mice
| Gene | Refseq no. | Gene name | Fold difference HYPO/CTX | P value | CI |
|---|---|---|---|---|---|
| Adss | NM_007422 | Adenylosuccinate synthetase | 1.39 | 0.000 | (1.31, 1.47) |
| Agrp | NM_007427 | Agouti-related protein | 12.56 | 0.000 | (4.90, 20.22) |
| Agt | NM_007428 | Angiotensinogen | 25.01 | 0.0003 | (11.38, 38.64) |
| Angptl4 | NM_020581 | Angiopoietin-like 4 | 1.88 | 0.001 | (1.41, 2.35) |
| Apod | NM_007470 | Apolipoprotein D | 2.65 | 0.000 | (2.20, 3.10) |
| Avp | NM_009732 | Arginine vasopressin | 3008.08 | 0.000 | (409.94, 5606.22) |
| Cdkn1a(p21) | NM_007669 | Cyclin-dependent kinase inhibitor 1A | 0.46 | 0.001 | (0.32, 0.60) |
| Cpt1a | NM_013495 | Carnitine palmitoyltransferase 1a | 1.28 | 0.005 | (1.10, 1.46) |
| Cpt1c | NM_153679 | Carnitine palmitoyltransferase 1c | 1.55 | 0.007 | (1.24, 1.86) |
| Crh | NM_205769 | CRH | 0.74 | 0.182 | (0.09, 1.39) |
| Cxcl12 | NM_021704 | Chemokine (C-X-C motif) ligand 12 | 0.7 | 0.080 | (0.47, 0.93) |
| Ddc | NM_016672 | Dopa decarboxylase | 11.35 | 0.007 | (5.11, 17.59) |
| Fatp | NM_011977 | Fatty acid transporter, member 1 (Slc27a1) | 1.15 | 0.007 | (1.06, 1.24) |
| Foxo3 | NM_019740 | Forkhead box O3 | 0.48 | 0.000 | (0.39, 0.57) |
| Gabrr2 | NM_008076 | GABA C receptor, subunit ρ 2 | 4.8 | 0.000 | (2.92, 6.68) |
| Gat1 | NM_178703 | Solute carrier family 6 (GABA), member 1 (Slc6a1) | 0.97 | 0.781 | (0.80, 1.14) |
| Glut2 | NM_031197 | Facilitated glucose transporter, member 2 (Slc2a2) | 5.62 | 0.000 | (0.73, 10.51) |
| Gpd1 | NM_010271 | Glycerol-3-phosphate dehydrogenase 1 (soluble) | 1.07 | 0.525 | (0.58, 1.56) |
| Gpr12 | NM_008151 | G protein-coupled receptor 12 | 0.92 | 0.072 | (0.84, 1.00) |
| Hif3a | NM_016868 | Hypoxia-inducible factor 3, α−subunit | 4.16 | 0.003 | (1.66, 6.66) |
| Hspa4l | NM_011020 | Heat shock protein 4 like | 1.2 | 0.018 | (1.04, 1.36) |
| Hsph1 | NM_013559 | Heat shock 105 kDa/110 kDa protein 1 | 1.01 | 0.822 | (0.85, 1.17) |
| Idh2 | NM_173011 | Isocitrate dehydrogenase 2 (NADP+), mitochondrial | 1.47 | 0.001 | (1.25, 1.69) |
| Mapk4 | NM_172632 | MAPK 4 | 0.62 | 0.000 | (0.52, 0.72) |
| Mdh1 | NM_008618 | Malate dehydrogenase 1, NAD (soluble) | 1.2 | 0.000 | (1.15, 1.25) |
| Mt1 | NM_013602 | Metallothionein 1 | 1.18 | 0.035 | (1.02, 1.34) |
| Mt2 | NM_008630 | Metallothionein 2 | 1.39 | 0.017 | (1.08, 1.70) |
| Npy | NM_023456 | Neuropeptide Y | 0.19 | 0.000 | (0.13, 0.25) |
| Nt5c3 | NM_026004 | 5′-Nucleotidase, cytosolic III | 0.76 | 0.000 | (0.69, 0.83) |
| Oxt | NM_011025 | Oxytocin | 1110.86 | 0.0005 | (9.42, 2212.30) |
| Pcx | NM_008797 | Pyruvate carboxylase | 0.79 | 0.011 | (0.65, 0.93) |
| Pdk4 | NM_013743 | Pyruvate dehydrogenase kinase, isoenzyme 4 | 0.97 | 0.737 | (0.73, 1.21) |
| Pfkfb2 | NM_008825 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 | 1.53 | 0.000 | (1.31, 1.75) |
| Pnpla2 | NM_025802 | Patatin-like phospholipase domain containing 2 | 1.14 | 0.131 | (0.95, 1.33) |
| Pomc | NM_008895 | Proopiomelanocortin-α | 19.28 | 0.002 | (7.37, 31.19) |
| Pxmp2 | NM_008993 | Peroxisomal membrane protein 2 | 2.66 | 0.000 | (2.29, 3.03) |
| S3-12 | NM_020568 | Perilipin 4 (Plin4) | 4.39 | 0.002 | (1.99, 6.79) |
| Sf1 | NM_139051 | Steroidogenic factor 1 (Nr5a1) | 55.59 | 0.001 | (0.78, 110.40) |
| Ucp2 | NM_011671 | Uncoupling protein 2 | 2.23 | 0.041 | (0.47, 3.99) |
CI, Confidence interval; GABA, γ-aminobutyric acid; HYPO/CTX, hypothalamus/cortex ratio; NAD, nicotinamide adenine dinucleotide.
Figure 1.

Expression of metabolism-related genes after fasting and refeeding in hypothalamus and cortex. Quantitative real-time PCR data for murine genes Pdk4 (A), Pfkfb2 (B), Mdh1 (C), Glut2 (D), Cpt1a (E), Gpd1 (F), Pcx (G), Fatp (H), Pnpla2 (I), Apod (J), S3-12 (K), Pxmp2 (L), Foxo3 (M), and Hif3a (N) in hypothalamus (left graph in each panel) and cortex (right graph in each panel). Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the FED group. Data are means ± sem (n = 6–12). *, P < 0.05 compared with FED group; #, P < 0.05 for REFED compared with FASTED group (ANOVA).
A second class of genes is of those genes mediating neuroendocrine signaling. Serving largely as positive controls, the protocol in the present study detected genes already known to be regulated by fasting and to regulate energy balance (Fig. 2, A–C), although the present studies provided novel information regarding the rapidity with which these effects of fasting were regulated by refeeding (Fig. 2, A and B). A third class of genes likely to mediate neuroendocrine responses to nutritional condition and to regulate metabolic function (for example, Angptl4, p21, and Ucp2) is shown in Figure 3. Finally, a small group of stress-associated genes was regulated similarly by fasting in both hypothalamus and cortex (Fig. 4).
Figure 2.

Expression of neuroendocrine-related genes after fasting and refeeding in hypothalamus and cortex. Quantitative real-time PCR data for murine genes Agrp (A), Npy (B), Pomc (C), Avp (D), Oxt (E), Crh (F), Agt (G), Sf1 (H), Gabrr2 (I), Gat1 (J), Ddc (K), and Gpr12 (L) in hypothalamus (left graph in each panel) and cortex (right graph in each panel). Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the FED group. Data are means ± sem (n = 6–12). *, P < 0.05 compared with FED group; #, P < 0.05 for REFED compared with FASTED group (ANOVA).
Figure 3.

Expression of genes involved in a variety of functions after fasting and refeeding in hypothalamus and cortex. Quantitative real-time PCR data for murine genes Angptl4 (A), p21 (B), Ucp2 (C), Cpt1c (D), Nt5c3 (E), Mapk4 (F), Adss (G), and Cxcl12 (H) in hypothalamus (left graph in each panel) and cortex (right graph in each panel). Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the FED group. Data are means ± sem (n = 6–12). *, P < 0.05 compared with FED group; #, P < 0.05 for REFED compared with FASTED group (ANOVA).
Figure 4.

Expression of stress-related genes after fasting and refeeding in hypothalamus and cortex. Quantitative real-time PCR data for murine genes Mt1 (A), Mt2 (B), Hsph1 (C), Hspa4l (D), and Idh2 (E) in hypothalamus (left graph in each panel) and cortex (right graph in each panel). Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the FED group. Data are means ± sem (n = 6–12). *, P < 0.05 compared with FED group; #, P < 0.05 for REFED compared with FASTED group (ANOVA).
Obviously fasting produces a complex set of signals to which the hypothalamus is sensitive, and failure to properly respond to any of these signals (e.g. leptin) could predispose to the metabolic syndrome. To clarify factors mediating effects of fasting on hypothalamic gene expression, we examined the expression of these genes in hypothalamic slices in vitro, at low and high glucose with or without leptin. As shown in Fig. 5, the response of a subset of fasting-regulated genes to low glucose was the same as that as produced by fasting, e.g. genes induced by fasting were relatively elevated at low glucose. For most of these genes the effect of leptin, although trending toward the effect of glucose, did not reach statistical significance. The addition of leptin to glucose did not produce significantly greater effect than glucose alone, although in every case leptin and glucose produced effects in the same direction. These data suggest that in general the regulation of gene expression by fasting in the present study was mediated by reduction in glucose or glucose metabolism, with leptin possibly playing a modulatory role enhancing some effects of glucose. To further examine this hypothesis, we examined the regulation of these genes in ob/ob mice (Table 2). Of the fasting-regulated genes reported here, none were differentially expressed in ob/ob mice vs. wild-type mice, other than the neuropeptide genes (e.g. Agrp, Npy, and Pomc) already known to be differentially expressed in ob/ob mice, with the interesting exception of Sf1, which was also induced in ob/ob mice, and Hsph1, the functional significance of which is unclear. However, for those genes for which there was a trend toward differential expression in ob/ob mice (0.05 < P < 0.1), the genes were all regulated in the same direction as with fasting. It should be noted that fasting, as expected, reduced plasma glucose, leptin, and insulin and increased plasma corticosterone, and these effects were completely restored to ad libitum levels within 4 h of refeeding (Fig. 6).
Figure 5.

In vitro regulation of gene expression by glucose and leptin in hypothalamic slices. Brain slices from mice previously fated for 48 h were incubated in aCSF containing either 0.1 mm glucose or 10 mm glucose with or without leptin (50 nm). Hypothalamic quantitative real-time PCR data for murine genes Gpd1 (A), S3-12 (B), Pnpla2 (C), Foxo3 (D), Hif3a (E), p21 (F), Hsph1 (G), and Idh2 (H). qPCR expression data for each gene was normalized to a panel of housekeeping transcripts and expressed as fold change compared with 0.1 mm glucose group. Data are means ± se (n = 8 for all groups). *, P < 0.05 compared with 0.1 mm glucose condition (by ANOVA).
Table 2.
Body weight and blood glucose for mice used in the gene expression studies
| Body weight (g) | Blood glucose (mg/dl) | |
|---|---|---|
| Wild-type | 28.47 ± 1.2 | 157.4 ± 11 |
| Ob/Ob | 63.57 ± 1.6 | 175.8 ± 18.9 |
| Fed | 27.95 ± 0.4 | 141.4 ± 7.6 |
| Fasted | 21.93 ± 0.3 | 69.5 ± 3.9 |
| Refed | 24.26 ± 0.4 | 145.8 ± 8.3 |
Figure 6.
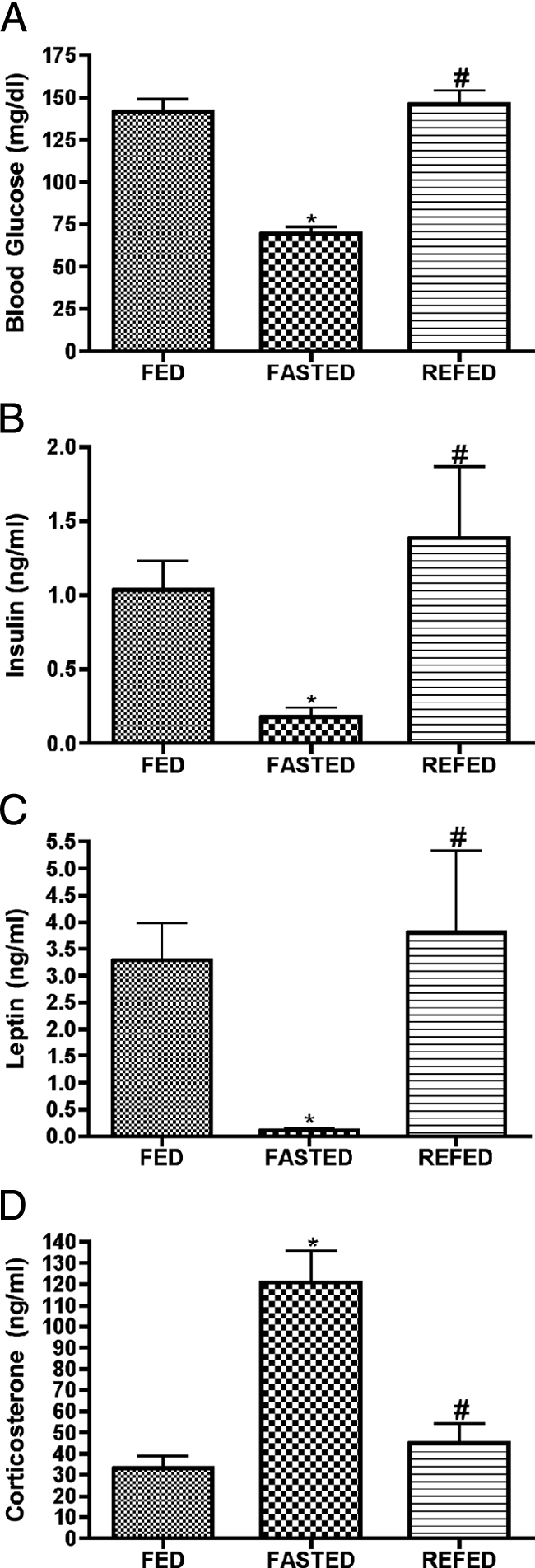
Blood glucose, insulin, leptin, and corticosterone are regulated by fasting and normalized by refeeding. Data derived from separate groups of mice killed in the fed condition, after 48 h fasting, and from fasted animals refed for 4 h. Blood glucose (A), insulin (B), and leptin (C) all decrease significantly during fasting but are completely normalized within 4 h of refeeding with a high-calorie sweet milk. Corticosterone levels (D) increase with fasting and are normalized by refeeding. Data are means ± sem (n = 6–12). *, P < 0.05 compared with FED group; #, P < 0.05 for REFED compared with FASTED group (ANOVA).
Discussion
The present studies have considerably extended the panel of genes the hypothalamic expression of which is influenced by nutritional status. Because hypothalamic gene expression plays a key role in the regulation of energy balance and glucose homeostasis, we anticipated that developing such broad panel would likely suggest novel mechanisms mediating effects of nutritional status on energy balance and glucose homeostasis. Although this is indeed the case, as described below, some changes are similarly observed in cortex, suggesting a more general role for these fasting-regulated responses.
In the present manuscript we only report results that were corroborated by qPCR, in samples independent from those on which the microarray studies were based, because microarray data alone are of limited value due to the confounding effects of even a small number of false-positive results (27). The main result in the present studies is that fasting produced a gene profile consistent with reduced glucose metabolism and increased utilization of lipid oxidation. For example, the present study demonstrates that fasting induced hypothalamic Pdk4 (Fig. 1A) consistent with in vitro studies (29). Because Pdk4 inhibits pyruvate dehydrogenase, this response would be expected to reduce glucose metabolism in the TCA cycle, although alternative fates of glucose metabolism would still be possible. Fasting also inhibited Mdh1 specifically in the hypothalamus (Fig. 1C). Mdh1 is of particular interest because it constitutes a key element in the glucose-sensing mechanism via the pyruvate shuttle (30), particularly entailing the effective transfer of NADH to NADPH, which also plausibly plays a role in hypothalamic glucose sensing (28,31). Similarly fasting reduced hypothalamic Glut2, the glucose transporter long implicated as part of the glucose-sensing apparatus of pancreatic β-cells and more recently implicated in the hypothalamic glucose-sensing mechanism regulating energy balance (15,16). Pfkb2 is inhibited by fasting and may be even further inhibited during refeeding, an unexpected result possibly related to the opposing activities of Pfkfb2, either to activate, its normal function, or inhibit glycolysis, which occurs during transient states (32). Inhibition of Pfkfb2 by chronic fasting would be expected to reduce glycolysis, but further inhibition of expression by acute refeeding may actually serve to restore glycolysis (by reducing the glycolytic inhibitory action of Pfkfb2) in this transitory state.
Conversely, whereas fasting inhibited genes that increase glucose utilization and fasting induced genes that block glucose utilization, fasting induced genes that increase the utilization of lipid oxidation. Most notably, in hypothalamus but not cortex, fasting produced an induction of Cpt1a, the rate-limiting enzyme in the utilization of free fatty acids to produce ATP through β-oxidation (Fig. 1D). Similarly, fasting (Fig. 1E) produced an induction of Gpd1 in hypothalamus but not cortex (Fig. 1F), which would facilitate the utilization of glycerol derived from the breakdown of triacylglycerides; conversely, glucose directly inhibited Gpd1 in vitro, which would be expected to reduce utilization of glycerol. Of particular interest was the induction of Pcx (Fig. 1G), which is a rate-limiting step for the anaplerotic supply of oxaloacetic acid necessary for β-oxidation (33). Furthermore, fasting induced the fatty acid transporter Fatp (Fig. 1H), which would also facilitate the utilization of free fatty acids to produce ATP. Consistent with this hypothesis, fasting also induced Pnpla2, a triglyceride lipase that would free the monoacyl glycerides and glycerol from triglycerides in preparation for β-oxidation (Fig. 1I). Fasting also induced Apod, which facilitates the release of free fatty acids from triacylglycerides (34) (Fig. 1J), S3-12 (also referred to perilipin 4) (Fig. 1K), which plays a key role in intracellular trafficking of lipids, and Pxmp2 (Fig. 1H), a channel that facilitates the exchange of substrates in the peroxisome (35), a major locus of β- oxidation.
Most of the hypothalamic responses to fasting observed in the present study may not be mediated by reduction in leptin or elevation in plasma corticosterone or free fatty acids, because, with very few exceptions including the neuropeptides, they are not observed in leptin-deficient ob/ob mice [which are also characterized by elevated corticosterone and free fatty acids (36)]. Nevertheless, dexamethasone does induce hypothalamic Pdk4 (37) as well as Angptl4 (38), so it is possible that ob/ob mice are characterized by hypothalamic resistance to glucocorticoids. Effects of fasting also may not be mediated by reduced plasma insulin, since insulin-induced hypoglycemia actually produced the same effects as fasting in a subset of these genes, rather than the opposite effects. Because hypoglycemia was produced by elevated insulin, these studies are consistent with the hypothesis that reduced blood glucose or glucose metabolism mediates many of the molecular responses to fasting observed in the present study. Indeed, this was directly demonstrated to be the case for eight genes, which in vitro responded to low glucose in the same direction as fasting. In contrast, leptin alone only regulated one gene in vitro, although this gene was not induced in ob/ob mice. It should be noted that many more genes responded to fasting than either to hypoglycemia or in vitro glucose, probably because fasting represented a chronic 48-h stimulus, whereas for technical reasons responses to hypoglycemia and in vitro leptin and/or glucose were constrained to a 4-h stimulus. By the same token, almost all of the effects of fasting observed in the present study were reversed within 4 h of refeeding, but so were plasma leptin, insulin, and corticosterone, as well as glucose, which also rapidly reversed in the same time frame. In addition to glucose, another plausible mediator of hypothalamic responses to fasting not addressed in the present study is the hormone ghrelin, which activates hypothalamic β-oxidation via elevation of uncoupling protein 2 (UCP2) (also observed to be induced by fasting in the present study), AMP-activated protein kinase, and Cpt1 (21,39,40). It is also plausible that, in addition to mediation by reduced plasma glucose, some effects of fasting are mediated by elevated plasma ketone bodies, which block molecular responses to glucose (41). In any case, to the extent that the molecular responses described here were determined by glucose and/or free fatty acids, they reflect a classic substrate competition between glucose and alternative fuels similar to that described by Randle, mainly referring to muscle metabolism, as follows: “Glucose provision promotes glucose oxidation and glucose and lipid storage, and inhibits fatty acid oxidation. Provision of free fatty acids promotes fatty acid oxidation and storage, inhibits glucose oxidation …”(42). Thus the present studies serve, in part, to extend this concept of substrate competition to the brain, which in contrast to muscle has historically not been thought to rely significantly on β-oxidation for fuel (43), although, as discussed below, may rely on β-oxidation in hypothalamic nutrient sensing.
The substrate competition implied by these studies is probably most relevant to mechanisms by which hypothalamic malonyl-CoA exerts its profound effects on energy balance (17,19,21,23,40). The role of hypothalamic malonyl-CoA to regulate energy balance was discovered serendipitously when the inhibitor of fatty acid synthase C75 was discovered to profoundly reduce food intake and body weight (17). Further analysis demonstrated that this effect required an increase in hypothalamic malonyl-CoA (17). Subsequent studies suggested that the anorectic effect of C75 also required hypothalamic glucose metabolism (23). In peripheral tissues, malonyl-CoA effectively serves as a cellular indicator of glucose metabolism (produced as an anaplerotic result of glucose metabolism in the TCA cycle) that inhibits Cpt1, thus β-oxidation, to promote continued glucose metabolism at the expense of β-oxidation (44). However, classically the brain has been thought not to support a significant amount of β-oxidation (43), subsisting instead on glucose utilization in the fed state and ketone metabolism in the fasted state, so the relevance of the malonyl-CoA regulation of Cpt1 in the brain has been unclear. Although the role of malonyl-CoA in regulating energy balance was discovered based on the pharmacological effects of C75, subsequent studies have strongly supported the physiological relevance of this mechanism, because hypothalamic overexpression of malonyl-CoA decarboxylase, which degrades malonyl-CoA, produces obesity (19), whereas direct inhibition of hypothalamic Cpt1a ameliorates obesity (45). Thus the hypothalamic metabolic profile produced by fasting in the present study would effectively reduce hypothalamic malonyl-CoA by reducing glucose metabolism and would therefore be expected to minimize the anorectic and thermogenic effects of hypothalamic malonyl-CoA. Conversely, of course, the proposed studies suggest that reversal of this profile, as occurs rapidly after refeeding, could serve as a physiological indicator of nutritional, particularly glucose, sufficiency, failure in the sensing of which could plausibly cause obesity (19).
Other hypothalamic responses to fasting observed in the present study also suggest novel aspects of energy balance regulation. Thus genes induced by fasting could plausibly promote obese phenotypes if overexpressed, so, if inhibited, might reduce obese phenotypes; conversely, genes inhibited by fasting could, if inhibited, promote obesity, and, if overexpressed, could ameliorate obesity. This hypothesis is supported by the neuropeptide gene expression shown in Fig. 2, A–C, although, of course, the hypothesis has not been corroborated for the majority of the genes reported here. Interestingly, however, genetic ablation of Gpr12 (Fig. 2L) does produce obesity (46), as predicted. Furthermore, ablation of Cpt1c does enhance sensitivity to diet-induced obesity (22), consistent with expression profiles in the hypothalamus, but not cortex (Fig. 3D), and consistent with the opposite hypothalamic responses of Cpt1c (Fig. 3D) vs. Cpt1a (Fig. 1E). Angpl4 (Fig. 3B) is robustly induced by fasting in other tissues (38,47) and participates in glucocorticoid-regulated lipid metabolism (48), consistent with a role in metabolic regulation in the hypothalamus. Ucp2 (Fig. 3C) mediates effects of T3 on hypothalamic regulation of feeding behavior (49) and the effects of the fasting-induced hormone ghrelin on hypothalamic NPY/Agrp neurons through a mechanism involving Cpt1a (39). Ucp2 also negatively regulates glucose sensitivity in proopiomelanocortin (POMC) neurons and is induced in leptin-deficient mice (50), which could also contribute to the reduced hypothalamic sensitivity produced by fasting as described above.
In conclusion, the present studies suggest that fasting reprograms metabolic substrate utilization away from glycolysis and toward lipid oxidation, with implications for energy balance via reduced production of malonyl-CoA. In addition, these studies provide a large panel of glucose-sensitive genes that will be useful to assess this and related mechanisms regulating energy balance, glucose homeostasis, and aging.
Footnotes
This work was supported by a grant from the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (1R56DK072033-01A1).
Disclosure Summary: The authors have nothing to disclose.
First Published Online September 29, 2010
Abbreviations: aCSF, Artificial cerebrospinal fluid; CoA, coenzyme A; NPY, neuropeptide Y; POMC, proopiomelanocortin; qPCR, quantitative PCR; UCP2, uncoupling protein 2.
References
- Mobbs CV, Mizuno T, Isoda F, Mastaitis J, Yang XJ 2005 Impaired glucose signaling as a cause of obesity and the metabolic syndrome: the glucoadipostatic hypothesis. Physiol Behav 85:3–23 [DOI] [PubMed] [Google Scholar]
- James WP 2008 WHO recognition of the global obesity epidemic. Int J Obes (Lond) 32(Suppl 7):S120–S126 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM 1994 Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432 [DOI] [PubMed] [Google Scholar]
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS 1996 Role of leptin in the neuroendocrine response to fasting. Nature 382:250–252 [DOI] [PubMed] [Google Scholar]
- Ahima RS, Kelly J, Elmquist JK, Flier JS 1999 Distinct physiologic and neuronal responses to decreased leptin and mild hyperleptinemia. Endocrinology 140:4923–4931 [DOI] [PubMed] [Google Scholar]
- Ahima RS, Flier JS 2000 Leptin. Annu Rev Physiol 62:413–437 [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Marks JL, Sipols AJ, Baskin DG, Woods SC, Kahn SE, Porte Jr D 1991 Central insulin administration reduces neuropeptide Y mRNA expression in the arcuate nucleus of food-deprived lean (Fa/Fa) but not obese (fa/fa) Zucker rats. Endocrinology 128:2645–2647 [DOI] [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E 1996 A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature 380:243–247 [DOI] [PubMed] [Google Scholar]
- Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV 1998 Rapid communication: hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and in ob/ob and db/db mice, but is stimulated by leptin. Diabetes 47:294–297 [DOI] [PubMed] [Google Scholar]
- Mizuno TM, Makimura H, Silverstein J, Roberts JL, Lopingco T, Mobbs CV 1999 Fasting regulates hypothalamic neuropeptide Y, agouti-related peptide, and proopiomelanocortin in diabetic mice independent of changes in leptin or insulin. Endocrinology 140:4551–4557 [DOI] [PubMed] [Google Scholar]
- Makimura H, Mizuno TM, Isoda F, Beasley J, Silverstein JH, Mobbs CV 2003 Role of glucocorticoids in mediating effects of fasting and diabetes on hypothalamic gene expression. BMC Physiol 3:5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert M, Magnan C, Turban S, André J, Guerre-Millo M 2003 Leptin receptor-deficient obese Zucker rats reduce their food intake in response to a systemic supply of calories from glucose. Diabetes 52:277–282 [DOI] [PubMed] [Google Scholar]
- Bady I, Marty N, Dallaporta M, Emery M, Gyger J, Tarussio D, Foretz M, Thorens B 2006 Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes 55:988–995 [DOI] [PubMed] [Google Scholar]
- Yang XJ, Mastaitis J, Mizuno T, Mobbs CV 2007 Glucokinase regulates reproductive function, glucocorticoid secretion, food intake, and hypothalamic gene expression. Endocrinology 148:1928–1932 [DOI] [PubMed] [Google Scholar]
- Stolarczyk E, Guissard C, Michau A, Even PC, Grosfeld A, Serradas P, Lorsignol A, Pénicaud L, Brot-Laroche E, Leturque A, Le Gall M 2010 Detection of extracellular glucose by GLUT2 contributes to hypothalamic control of food intake. Am J Physiol Endocrinol Metab 298:E1078–E1087 [DOI] [PubMed] [Google Scholar]
- Mounien L, Marty N, Tarussio D, Metref S, Genoux D, Preitner F, Foretz M, Thorens B 2010 Glut2-dependent glucose-sensing controls thermoregulation by enhancing the leptin sensitivity of NPY and POMC neurons. FASEB J 24:1747–1758 [DOI] [PubMed] [Google Scholar]
- Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP 2000 Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 288:2379–2381 [DOI] [PubMed] [Google Scholar]
- Shu IW, Lindenberg DL, Mizuno TM, Roberts JL, Mobbs CV 2003 The fatty acid synthase inhibitor cerulenin and feeding, like leptin, activate hypothalamic pro-opiomelanocortin (POMC) neurons. Brain Res 985:1–12 [DOI] [PubMed] [Google Scholar]
- He W, Lam TK, Obici S, Rossetti L 2006 Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat Neurosci 9:227–233 [DOI] [PubMed] [Google Scholar]
- López M, Lelliott CJ, Tovar S, Kimber W, Gallego R, Virtue S, Blount M, Vázquez MJ, Finer N, Powles TJ, O'Rahilly S, Saha AK, Diéguez C, Vidal-Puig AJ 2006 Tamoxifen-induced anorexia is associated with fatty acid synthase inhibition in the ventromedial nucleus of the hypothalamus and accumulation of malonyl-CoA. Diabetes 55:1327–1336 [DOI] [PubMed] [Google Scholar]
- López M, Lage R, Saha AK, Pérez-Tilve D, Vázquez MJ, Varela L, Sangiao-Alvarellos S, Tovar S, Raghay K, Rodríguez-Cuenca S, Deoliveira RM, Castañeda T, Datta R, Dong JZ, Culler M, Sleeman MW, Alvarez CV, Gallego R, Lelliott CJ, Carling D, Tschöp MH, Diéguez C, Vidal-Puig A 2008 Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 7:389–399 [DOI] [PubMed] [Google Scholar]
- Wolfgang MJ, Cha SH, Millington DS, Cline G, Shulman GI, Suwa A, Asaumi M, Kurama T, Shimokawa T, Lane MD 2008 Brain-specific carnitine palmitoyl-transferase-1c: role in CNS fatty acid metabolism, food intake, and body weight. J Neurochem 105:1550–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortman MD, Clegg DJ, D'Alessio D, Woods SC, Seeley RJ 2003 C75 inhibits food intake by increasing CNS glucose metabolism. Nat Med 9:483–485 [DOI] [PubMed] [Google Scholar]
- Mobbs CV, Kow LM, Yang XJ 2001 Brain glucose-sensing mechanisms: ubiquitous silencing by aglycemia vs. hypothalamic neuroendocrine responses. Am J Physiol Endocrinol Metab 281:E649–E654 [DOI] [PubMed] [Google Scholar]
- Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH 2001 Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 50:2673–2681 [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn-Meynell AA, Routh VH 2002 CNS sensing and regulation of peripheral glucose levels. Int Rev Neurobiol 51:219–258 [DOI] [PubMed] [Google Scholar]
- Mobbs CV, Yen K, Mastaitis J, Nguyen H, Watson E, Wurmbach E, Sealfon SC, Brooks A, Salton SR 2004 Mining microarrays for metabolic meaning: nutritional regulation of hypothalamic gene expression. Neurochem Res 29:1093–1103 [DOI] [PubMed] [Google Scholar]
- Yang XJ, Kow LM, Funabashi T, Mobbs CV 1999 Hypothalamic glucose sensor: similarities to and differences from pancreatic β-cell mechanisms. Diabetes 48:1763–1672 [DOI] [PubMed] [Google Scholar]
- Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ 2005 Diverging regulation of pyruvate dehydrogenase kinase isoform gene expression in cultured human muscle cells. FEBS J 272:3004–3014 [DOI] [PubMed] [Google Scholar]
- MacDonald MJ 1995 Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J Biol Chem 270:20051–20058 [PubMed] [Google Scholar]
- Yang XJ, Kow LM, Pfaff DW, Mobbs CV 2004 Metabolic pathways that mediate inhibition of hypothalamic neurons by glucose. Diabetes 53:67–73 [DOI] [PubMed] [Google Scholar]
- Arden C, Hampson LJ, Huang GC, Shaw JA, Aldibbiat A, Holliman G, Manas D, Khan S, Lange AJ, Agius L 2008 A role for PFK-2/FBPase-2, as distinct from fructose 2,6-bisphosphate, in regulation of insulin secretion in pancreatic β-cells. Biochem J 411:41–51 [DOI] [PubMed] [Google Scholar]
- Agius L, Alberti KG 1985 Regulation of flux through pyruvate dehydrogenase and pyruvate carboxylase in rat hepatocytes. Effects of fatty acids and glucagon. Eur J Biochem 152:699–707 [DOI] [PubMed] [Google Scholar]
- Perdomo G, Kim DH, Zhang T, Qu S, Thomas EA, Toledo FG, Slusher S, Fan Y, Kelley DE, Dong HH 2010 A role of apolipoprotein D in triglyceride metabolism. J Lipid Res 51:1298–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokka A, Antonenkov VD, Soininen R, Immonen HL, Pirilä PL, Bergmann U, Sormunen RT, Weckström M, Benz R, Hiltunen JK 2009 Pxmp2 is a channel-forming protein in mammalian peroxisomal membrane. PLoS One 4:e5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal NS, Patel RT, Qi Y, Lee YS, Ahima RS 2008 Loss of resistin ameliorates hyperlipidemia and hepatic steatosis in leptin-deficient mice. Am J Physiol Endocrinol Metab 295:E331–E338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Horikawa Y, Iizuka K, Sakurai N, Tanaka T, Shihara N, Oshima A, Takeda J, Mikuni M 2008 Large-scale analysis of glucocorticoid target genes in rat hypothalamus. J Neurochem 106:805–814 [DOI] [PubMed] [Google Scholar]
- Koliwad SK, Kuo T, Shipp LE, Gray NE, Backhed F, So AY, Farese Jr RV, Wang JC 2009 Angiopoietin-like 4 (ANGPTL4, fasting-induced adipose factor) is a direct glucocorticoid receptor target and participates in glucocorticoid-regulated triglyceride metabolism. J Biol Chem 284:25593–25601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S 2008 UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 454:846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage R, Vázquez MJ, Varela L, Saha AK, Vidal-Puig A, Nogueiras R, Dieguez C, Lopez M 2010 Ghrelin effects on neuropeptides in the rat hypothalamus depend on fatty acid metabolism actions on BSX but not on gender. FASEB J 24:2670–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Isoda F, Belsham DD, Mobbs CV 2008 Inhibition of agouti-related peptide expression by glucose in a clonal hypothalamic neuronal cell line is mediated by glycolysis, not oxidative phosphorylation. Endocrinology 149:703–710 [DOI] [PubMed] [Google Scholar]
- Randle PJ 1998 Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 14:263–283 [DOI] [PubMed] [Google Scholar]
- Yang SY, He XY, Schulz H 1987 Fatty acid oxidation in rat brain is limited by the low activity of 3-ketoacyl-coenzyme A thiolase. J Biol Chem 262:13027–13032 [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Vavvas D, Witters LA 1999 Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol Endocrinol Metab 276:E1–E18 [DOI] [PubMed] [Google Scholar]
- Pocai A, Lam TK, Obici S, Gutierrez-Juarez R, Muse ED, Arduini A, Rossetti L 2006 Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest 116:1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell M, Gerdin AK, Jönsson M, Surve VV, Svensson L, Huang XF, Törnell J, Bohlooly YM 2006 G protein-coupled receptor 12 deficiency results in dyslipidemia and obesity in mice. Biochem Biophys Res Commun 348:359–366 [DOI] [PubMed] [Google Scholar]
- Mandard S, Zandbergen F, van Straten E, Wahli W, Kuipers F, Müller M, Kersten S 2006 The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J Biol Chem 281:934–944 [DOI] [PubMed] [Google Scholar]
- Staiger H, Haas C, Machann J, Werner R, Weisser M, Schick F, Machicao F, Stefan N, Fritsche A, Häring HU 2009 Muscle-derived angiopoietin-like protein 4 is induced by fatty acids via peroxisome proliferator-activated receptor (PPAR)-δ and is of metabolic relevance in humans. Diabetes 58:579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola A, Liu ZW, Andrews Z, Paradis E, Roy M-C, Friedman JM, Ricquier D, Richard D, Horvath TL, Gao X-B, Diano S 2007 A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab 5:21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB 2007 Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449:228–232 [DOI] [PubMed] [Google Scholar]