

Abstract
Recent studies have revealed that accumulation of prion protein (PrP) in the cytoplasm results in the production of aggregates that are insoluble in non-ionic detergents and partially resistant to proteinase K. Transgenic mice expressing PrP in the cytoplasm develop severe ataxia with cerebellar degeneration and gliosis, suggesting that cytoplasmic PrP may play a role in the pathogenesis of prion diseases. The mechanism of cytoplasmic PrP neurotoxicity is not known. In this report, we determined the molecular morphology of cytoplasmic PrP aggregates by immunofluorescence and electron microscopy, in neuronal and non-neuronal cells. Transient expression of cytoplasmic PrP produced juxtanuclear aggregates reminiscent of aggresomes in human embryonic kidney 293 cells, human neuroblastoma BE(2)-M17 cells and mouse neuroblastoma N2a cells. Time course studies revealed that discrete aggregates form first throughout the cytoplasm, and then coalesce to form an aggresome. Aggresomes containing cytoplasmic PrP were 1–5-μm inclusion bodies and were filled with electron-dense particles. Cytoplasmic PrP aggregates induced mitochondrial clustering, reorganization of intermediate filaments, prevented the secretion of wild-type PrP molecules and diverted these molecules to the cytoplasm. Cytoplasmic PrP decreased the viability of neuronal and non-neuronal cells. We conclude that any event leading to accumulation of PrP in the cytoplasm is likely to result in cell death.
Keywords: aggresome, prion diseases, prion protein, protein aggregation
Prion diseases or transmissible spongiform encephalopathies are fatal neurodegenerative disorders characterized by spongiform degeneration of the brain, neurodegeneration and astrogliosis. These diseases can appear in three forms, sporadic, genetic and infectious. All forms involve modification of the normal prion protein (PrP), a secreted glycosylphosphatidylinositol (GPI)-anchored sialoglycoprotein, into a disease-causing prion protein (PrPSc) (Prusiner 1998). Although it is clearly established that PrPSc plays a key role in the origin and transmission of prion diseases, there is uncertainty about whether this molecule is neurotoxic by itself and causes neurodegeneration (Brandner et al. 1996). High levels of PrPSc in the brain of conditional PrP knockout mice are not toxic to these mice following PrP depletion (Mallucci et al. 2003). Alternative pathogenic PrP forms include a transmembrane form termed CtmPrP (Hegde et al. 1998; Hegde et al. 1999), cytoplasmic PrP (CyPrP) (Ma et al. 2002) and cross-linked PrP (Solforosi et al. 2004). Further investigation of these three alternative forms of PrP is required to determine their mechanisms of toxicity and whether they are involved in the pathogenesis of all or specific prion diseases.
CyPrP accumulates at high levels in cells treated with proteasome inhibitors and may have two different origins. First, PrP N-terminal signal peptide is relatively inefficient and generates CyPrP molecules if the degradative capacity of the proteasome is slightly impaired (Drisaldi et al. 2003; Rane et al. 2004). A second mechanism involves retrotrans-location. Like many secreted proteins, PrP can be subjected to the endoplasmic reticulum (ER)-associated degradation pathway, whereby unfolded molecules are retrotranslocated from the ER into the cytosol and degraded by the proteasome (Yedidia et al. 2001; Ma and Lindquist 2002) or/and other cytoplasmic proteases including calpain (Wang et al. 2005). These two mechanisms are not mutually exclusive and their contribution to CyPrP accumulation may vary depending on experimental conditions and cell types.
CyPrP has biochemical characteristics similar to PrPSc, including partial insolubility in non-ionic detergents and resistance to proteinase K (Ma and Lindquist 2002; Drisaldi et al. 2003). Interestingly, CyPrP molecules generated upon transient treatment with proteasome inhibitors accumulate at high levels over time, indicative of their self-perpetuating character (Ma et al. 2002; Drisaldi et al. 2003). The toxicity of CyPrP in cultured cells is controversial. Overexpression of wild-type PrP rendered neuroblastoma N2a cells susceptible to apoptosis induced by proteasome inhibitors (Ma and Lindquist 2001; Rane et al. 2004). However, other investigators have observed that proteasome inhibitor-mediated apoptosis is independent of PrP expression (Fioriti et al. 2005). In vivo, CyPrP is toxic, and mice expressing a transgene encoding PrP without both N- and C-terminal signal peptides develop a neurological illness characterized by a massive loss of granule cerebellar neurons and gliosis (Ma et al. 2002).
In an attempt to elucidate the mechanism of CyPrP toxicity and self-perpetuation, we transiently transfected cells with a PrP construct that did not contain the N- and C-terminal sequences, and analyzed the distribution of the protein by fluorescence microscopy of single live cells. CyPrP formed particles that coalesce in perinuclear aggregates in neuronal and non-neuronal cell lines. These aggregates displayed the main characteristics of aggresomes (Kopito 2000), and induced a major rearrangement of intermediate filament and mitochondrial networks. CyPrP aggregates also prevented wild-type PrP molecules from being secreted to the plasma membrane. Expression of CyPrP decreased the viability of all cell lines tested. Taken together, these data indicate that accumulation of cytoplasmic PrP molecules leads to the formation of toxic intracellular aggregates.
Experimental procedures
Antibodies and clones
Monoclonal anti-prion protein (clone 3F4) and anti-mitochondrial heat-shock protein 70 (mtHSP70)(clone MA3-028) antibodies were purchased from Chemicon International (Temecula, CA, USA) and Affinity Bioreagents (CedarLane laboratories, Hornby, ON, Canada) respectively. Monoclonal anti-γ-tubulin (clone GTU-88) and anti-vimentin (clone V9) were purchased from Sigma (Oakville, ON, Canada). Polyclonal anti-20S proteasome α/β subunits was purchased from Biomol International (CedarLane laboratories). Alexa Fluor 488 and 568 F(ab′)2 fragment of goat anti-mouse IgG and Alexa Fluor 568 F(ab′)2 fragment of goat anti-rabbit IgG were purchased from Molecular Probes (Invitrogen, Burlington, ON, Canada). Peroxidase-linked anti-mouse IgG from sheep was purchased from Amersham Biosciences (Baie d’Urfe, QC, Canada). GPIEGFP and CD4EGFP were provided by Dr Ben J. Nichols (MRC Laboratory of Molecular Biology, Cambridge, UK) and Dr Jana Stankova (Faculty of Medicine, University of Sherbrooke, Sherbrooke, QC, Canada), respectively.
Cloning of PrP and CyPrP in pCEP4β (Invitrogen) has ben described previously (Bounhar et al. 2001; Roucou et al. 2003). PrP chimera with enhanced green fluorescent protein (PrPEGFP) in pCEP4β was a generous gift from Dr Neena Singh (Case Western Reserve University, Cleveland, OH, USA). CyPrPEGFP was amplified from the PrPEGFP-pCEP4β construct by PCR using primers CyPrPEGFP upstream (5′-CCCAAGCTTGTAATGTGCAAGAAGC-GCCCGAAGCCTGG-3′) and CyPrPEGFP downstream (5′-CGCGG-ATCCTCACGATCCTCTCTGGTAATAGGCCTG-3′). The PCR product was introduced in the HindIII and BamHI sites of pCEP4β. CyPrPEGFPΔOR, lacking the octapeptide region (OR), was cloned as described previously (Bounhar et al. 2001). CyPrPEGFP124stop and CyPrPEGFP157stop were amplified by PCR from the CyPrPEGFP-pCEP4β construct using primer CyPrPEGFPupstream, and primers CyPrPEGFP124 downstream (5′-CGCGGATCCTCAGCCCCCCAC-CACTGCCCCAGC-3′) and CyPrPEGFP157 downstream (5′-CGCGGATCCTCAGTAACGGTGCATGTTTTCACG-3′) respectively. The PCR products were introduced into the HindIII and BamHI sites of pCEP4β. Construct 124–230 was amplified from CyPrP-pCEP4β using primers CyPrPEGFP124–230 upstream (5′-CCCAAG-CTTTGGGCGGCTACATGCTGGGAAGTGC-3′) and CyPrPEGFP downstream. Construct 124–157 was amplified from CyPrP-pCEP4β using primers CyPrPEGFP124–157 upstream (5′-GGAAGCTT-CGGGCCTTGGCGGCTACATGCTG-3′) and CyPrPEGFP 157down-stream. The PCR products were introduced into the HindIII and BamHI restriction sites of EGFP-pCEP4β (Roucou et al. 2003) to generate CyPrPEGFP124–230 and CyPrPEGFP124–157 respectively. To construct PrPDsRed2, a PrP chimera with Discosoma sp. red fluorescent protein2 (DsRed2), DsRed2 was amplified from pDs-Red2-Bid (Clontech, Mississauga, ON, Canada), using primers DsRed2 upstream (5′-GGGTATGGCCTCCTCCGAGAACGTC-3′) and DsRed2 downstream (5′-GGGAGGAACAGGTGGTGGCGG-CCCTC-3′). The PCR product was introduced in the SmaI restriction site of PrP at residues 38–39. A clone containing DsRed2 in the right orientation was selected. All constructs were sequenced.
Cell culture and transfections
Human embryonic kidney (HEK) 293 and N2a mouse neuroblastoma cells were maintained in Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum (Wisent, Saint-Bruno, QC, Canada). Human neuroblastoma BE(2)-M17 cells were cultured in OptiMEM (Invitrogen) plus 10% fetal bovine serum.
Transfections were carried out using ExGen 500 according to the manufacturer’s protocol (MBI Fermentas, Burlington, ON, Canada).
Immunofluorescence and microscopy
Cells were fixed and processed for immunofluorescence as described previously (Roucou et al. 2005). Primary antibody dilutions were as follows: anti-PrP 1 : 200, anti-γ-tubulin 1 : 100, anti-vimentin 1 : 50, anti-mtHSP70 1 : 50, and anti-proteasome 1 : 500. Secondary antibodies were diluted 1 : 1000. Cells were examined with an Axioscop 2 phase-contrast/epifluorescence microscope (Carl Zeiss, Inc., Thornwood, NY, USA) equipped with band pass filters for fluorescence of Hoechst (excitation D360/40; emission D460/50), FITC (excitation D480/30; emission D535/40) and TRITC (excitation D560/40; emission D630/60) (all from Chroma Technology Corp., Rockingham, VT, USA). Photomicrographs of 1315 × 1033 pixels were captured using a 100 × oil immersion objective or a 40 × objective, and Spot cooled color digital camera (Diagnostic Instruments Inc., St Sterling Heights, MI, USA). Images were processed using SPOT software (Diagnostic Instruments).
For ultrastructural studies, transfected cells in six-well plates were prefixed for 30 min at room temperature (20°C) in fresh 1.4% glutaraldehyde diluted in 0.1 M cacodylate buffer, fixed overnight in 2.5% glutaraldehyde diluted in 0.1 M cacodylate, and post-fixed in 2% osmium tetroxide diluted in 0.1 M cacodylate for 1 h. They were dehydrated and covered with a 3-mm layer of Epon 812 resin. After polymerization for 48 h at 60°C, the plastic substratum was detached and specimens were inverted and re-embedded. Thin sections were visualized on a H-2500 electron microscope (Hitachi, Mississauga, ON, Canada).
Western blot
Protein expression was determined in lysates from cells grown in six-well plates as described previously (Roucou et al. 2003). Anti-PrP and anti-EGFP primary antibodies were diluted 1 : 10 000 and 1 : 1000 respectively. Secondary antibodies were diluted 1 : 5000.
Assay of phospholipase sensitivity
The procedure of Lehmann and Harris (1996) was used to test the ability of phosphatidylinositol-specific phospholipase C (PIPLC) to release PrPEGFP from the surface of intact N2a cells. Cell supernatants and pellets were further treated with N-glycosidase F (0.01 units/mL) for 12 h at 37°C before western blot analysis to produce a single band of deglycosylated PrPEGFP that could more easily be quantitated.
Fluorescence-activated cell sorting and viability assays
Cells were transiently transfected with EGFP, PrPEGFP or CyPrPEGFP. After 24 h, 3 × 106 cells were sorted with a FACS Vantage flow cytometer/CellQuest (Becton Dickinson, San Jose, CA, USA). Before each experiment the instrument was calibrated for optimization of alignment and sensitivity with FITC- and phycoerithrine (PE)-fluorescent CaliBRITE beads (Becton-Dickinson). The fluorescence was excited at 488 nm by ion laser and the intensity of emitted light was measured with a 530/30-nm optical filter. The viability of sorted untransfected (mock) and transfected (EGFP, PrPEGFP and CyPrPEGFP) cells measured by trypan blue exclusion was more than 95%. The viability of sorted N2a cells transfected with CyPrPEGFP was 80–85%. Sorted EGFP-positive (transfected) and EGFP-negative (mock) cells were plated at a density of 6000 viable cells per well in 96-well plates in a final volume of 100 μL, and incubated for 48 h at 37°C. Cell proliferation was measured with the reagent 4-[3-(4-lodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST-1) according to the manufacturer’s protocol (Roche, Mississauga, ON, Canada). WST-1 metabolizing activities were expressed as a percentage of control (WST-1 metabolizing activity of mock-transfected cells).
Apoptosis assay
Cells were transiently transfected with EGFP or CyPrPEGFP. After 24 h, cells were incubated in the presence of dimethylsulfoxide or staurosporine (STS) dissolved in dimethylsulfoxide for 6 h. Cells were then fixed and processed for nuclear staining as described previously (Roucou et al. 2003). Apoptosis was measured by counting EGFP-positive cells displaying apoptotic nuclei compared with the total number of EGFP-positive cells.
Statistical evaluation
Statistical significance was determined with ANOVA followed by Scheffé’s test post hoc using StatView (SAS Institute Inc., Cary, NC, USA); p < 0.05 was taken as a statistically significant.
Results
CyPrP molecules form aggregates upon transient transfection
Previous biochemical analyses showed that proteasome inhibitors induced the accumulation of CyPrP aggregates insoluble in non-ionic detergents (Ma and Lindquist 2001,Ma and Lindquist 2002; Yedidia et al. 2001; Drisaldi et al. 2003; Fioriti et al. 2005; Wang et al. 2005). In order to reconstitute specific cytoplasmic accumulation of PrP in cultured cells without using proteasome inhibitors, we transiently transfected cells with a construct that did not contain the N- and C-terminal sequences. This strategy also allowed us to avoid pleiotropic effects of proteasome inhibitors, including induction of cell death (Fioriti et al. 2005). We characterized CyPrP aggregate morphology by immunofluorescence of transfected single cells from three cell lines, mouse neuroblastoma N2a, human neuroblastoma BE(2)-M17 and HEK293. Expression of the different constructs used in the following experiments was analyzed by western blotting (Fig. 1a). CyPrP accumulated in aggregates consisting of bright fluorescent foci located in a juxtanuclear area, in over 80% of transfected cells (Figs 1b–d). CyPrP aggregates displayed a dense appearance in phase-contrast images (Figs 1b–d). Some 15–20% of N2a cells producing CyPrP aggregates underwent apoptosis and displayed fragmented nuclei (Fig. 1e). To determine the intracellular distribution of CyPrP by direct fluorescence, we engineered an EGFP-tagged CyPrP construct (CyPrPEGFP). In control experiments, PrPEGFP, like PrP, was mainly located at the plasma membrane and in the secretory pathway, as described previously (Fig. 1f) (Lee et al. 2001; Gu et al. 2003). CyPrPEGFP formed juxtanuclear aggregates similar to CyP-rP, indicating that the presence of the EGFP tag did not prevent the ability of CyPrP to aggregate next to the nucleus (Fig. 1g). The formation of these aggregates was not a consequence of cell fixation as they were also observed in live cells (not shown).
Fig. 1.
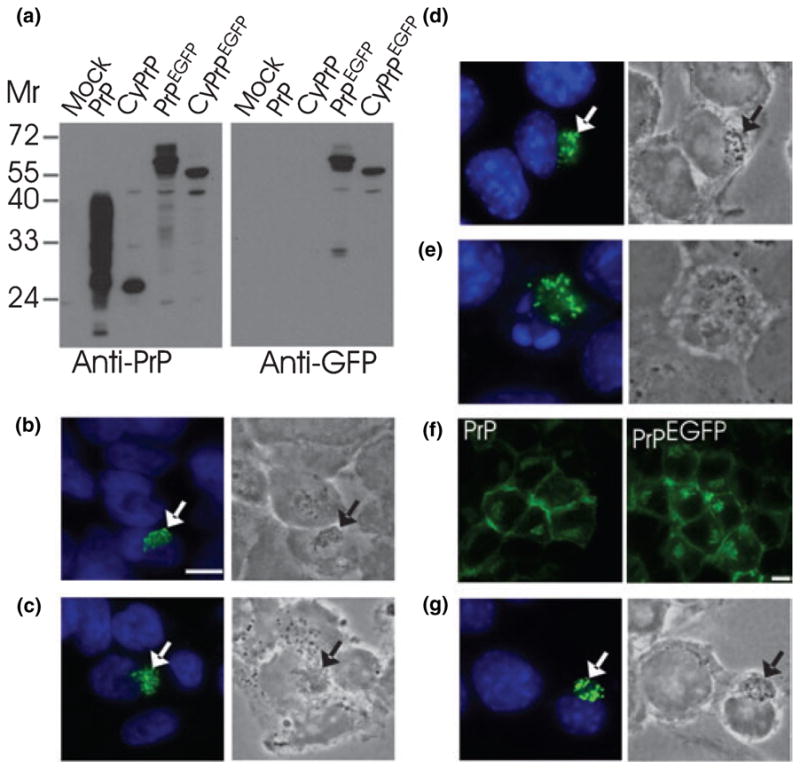
CyPrP forms aggresomes in transiently transfected non-neuronal and neuronal cells. (a) Western blot of PrP and EGFP in protein extracts (100 μg protein) from HEK293 cells expressing constructs indicated above the blots. Molecular masses (Mr) are indicated. Expected molecular masses are 27–40 for PrP, 27 for CyPrP, 54–67 for PrPEGFP and 54 for CyPrPEGFP. (b–d) HEK293 (b), BE(2)-M17 (c) and N2a (d) cells were transfected with CyPrP. PrP distribution was examined by immunofluorescence (3F4 mAb, green; left panels). Right panels show corresponding phase-contrast images. Nuclei were stained with Hoechst. The green and blue channels are shown merged. Arrows indicate juxtanuclear aggregates. Scale bar 10 μm. Original magnification × 100. (e) Some 15–20% of N2a cells expressing CyPrP underwent apoptosis. Apoptosis was assessed by nuclei fragmentation. (f) N2a cells were transfected with PrP or PrPEGFP, and examined by immunofluorescence with 3F4 mAb (left panel) or fluorescence of EGFP (right panel). (g) N2a cells were transfected with CyPrPEGFP and examined by fluorescence of EGFP.
Such intracellular localization is reminiscent of structures called inclusion bodies or aggresomes that consist of highly concentrated protein aggregates (Kopito 2000). Aggresomes are formed around the centrosome and are surrounded by the intermediate filament protein vimentin (Johnston et al. 1998). CyPrPEGFP aggregates also localized near γ-tubulin, a component of the centrosome, and were surrounded by a cage composed of vimentin protein (Figs 2a and b). Proteasomes were recruited to the aggregation sites, whereas they are normally diffuse in the cytoplasm and the nucleus (Fig. 2c). Proteasome clustering did not result from overexpression of a recombinant protein as it did not occur in PrP-overexpressing cells (Fig. 2c). PrP aggregates also immunostained for ubiquitin (not shown).
Fig. 2.
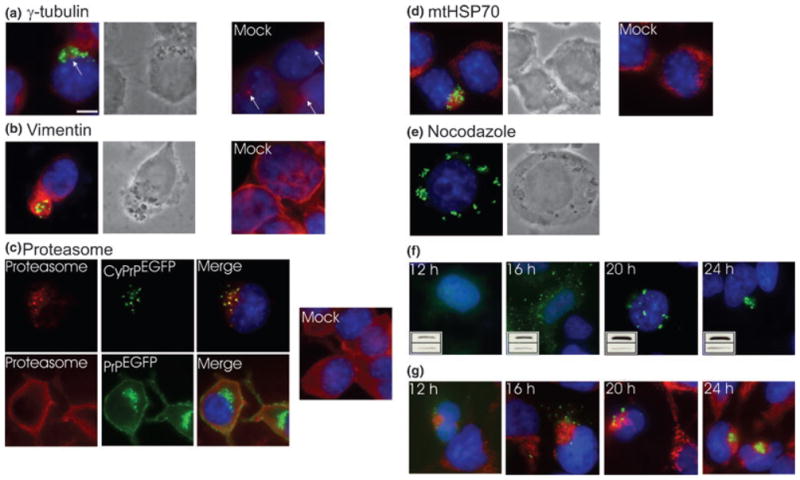
Characterization of PrP aggregates in N2a cells. (a–d) Immunofluorescence analysis of (a) γ-tubulin, (b) vimentin, (c) proteasome and (d) mtHSP70 (red channel) in cells transfected with CyPrPEGFP (green channel). Nuclei were stained with Hoechst (blue channel). Left panels represent an overlay of the three channels and phase-contrast images of cells transfected with CyPrPEGFP (a, b, d). Right panels represent mock-transfected cells (a–d). Arrow in (a) indicates the centrosome. (e) Distribution of CyPrPEGFP aggregates in cells incubated for 12 h in the presence of nocodazole (10 μg/mL). Left panel is an overlay of the green and blue channels and right panel shows phase-contrast image. (f) Aggregate distribution at different times after transfection determined by direct fluorescence of CyPrPEGFP. Insets show western blot analysis of CyPrPEGFP (upper blot). Equal loading was verified by western blot analysis of α-tubulin (lower blot). (g) Mitochondrial network at different times after transfection. Panels represent an overlay of the green (CyPrPEGFP), red (mtHSP70) and blue (Hoechst) channels. Scale bar 10 μm. Original magnification × 100. Similar results were obtained in HEK293 and BE(2)-M17 cells (not shown).
We noted a major rearrangement of the mitochondrial network in cells containing CyPrP aggregates. Mitochondria, which are normally distributed throughout the cytoplasm, were clustered around the aggresomes (Fig. 2d), leaving no detectable mitochondria in other regions of the cytoplasm. Aggresome formation requires microtubules (Johnston et al. 1998), and we tested the effect of the microtubule-depolymerizing agent nocodazole on CyPrP aggregation. Nocodazole did not prevent the formation of CyPrP aggregates, but abrogated their coalescence around the centrosome (Fig. 2e). Thus, in addition to having similar cytological features, CyPrP aggregates also share functional features with aggresomes.
To determine whether CyPrP has to accumulate at high levels to induce the formation of aggresomes, CyPrP aggregates were analyzed by fluorescence at different times after transfection (Fig. 2f). Discrete cytoplasmic aggregates could be detected as early as 12 h after transfection. We observed a correlation between the molecular morphology of these aggregates and the levels of CyPrP (Fig. 2f). Aggresomes were formed after CyPrP levels reached a peak, at 24 h after transfection (Fig. 2f). We further characterized the re-modelling of the mitochondrial network by determining whether it preceded or followed the formation of CyPrP aggresomes (Fig. 2g). Mitochondrial clustering occurred as early as 12 h after transfection. Mitochondria were distributed throughout the cytoplasm in mock-transfected cells (Fig. 2d).
To examine the ultrastructure of CyPrP aggregates, cells were examined by transmission electron microscopy. After 16 h, cells displayed several electron-dense particles, distributed in the cytoplasm (Fig. 3b). Twenty-four hours after transfection, cells contained an amorphous electron-dense structure of several microns in the vicinity of the nucleus (Fig. 3c). These aggregates were similar to those observed by fluorescence, and probably represent aggregates before they coalesce next to the nucleus and aggresomes respectively. Mitochondria were detected surrounding the particulate structures (Figs 3b–d). These structures were consistently observed in CyPrP-transfected cells, but never in mock-transfected cells (Fig. 3d).
Fig. 3.

Transmission electron microscopy of CyPrP aggregates. N2a cells were transiently transfected with CyPrP and processed for electron microscopy. (a, b) Electron-dense particles in CyPrP-expressing cells at 16 h (a) and 24 h (b) after transfection. (c) Higher magnification of section indicated by white box in (b) showing mitochondria clustering around an aggresome. (d) Mock-transfected cells. n, nucleus; m, mitochondria. Scale bar 500 nm.
The aggregate determinant of CyPrP lies in the C-terminal globular domain
We took advantage of the chimeric CyPrPEGFP construct to identify by direct fluorescence a domain of PrP responsible for its intracellular aggregation. Based on the observation that genetic fusion of the OR of PrP (residues 51–91) to myoglobin results in the formation of amyloid fibrils in vitro, it was proposed that this domain is an aggregation-inducing motif (Tanaka et al. 2002). To test whether the OR motif is also an aggregate determinant in vivo, cells were transiently transfected with CyPrPEGFPΔOR, a mutant with the OR domain deleted. As shown in Fig. 4, the absence of the OR domain did not prevent the formation of PrP aggregates.
Fig. 4.

The aggregation determinant of CyPrP includes β-sheet 1 and α-helix 1. (a) Diagrams of the of CyPrPEGFP and several deletion mutants engineered in this study are shown. Numbers indicate residues at the junction of different structural domains (adapted from Zahn et al. 2000). The black box represents the EGFP coding sequence. Aggregation was evaluated by fluorescence microscopy of transiently transfected N2a cells (original magnification ×40). (b) Western blot analysis of the constructs described in (a); extracts containing 100 μg protein from N2a cells were immunostained using antibodies directed against EGFP; mock-transfected cells (lanes 1), CyP-rPEGFP (lanes 2, expected Mr 54), CyP-rPEGFPΔOR (lanes 3, expected Mr 49.5), CyPrPEGFP124stop (lanes 4, expected Mr 38, CyPrPEGFP124–230 (lanes 5, expected Mr 39), CyPrPEGFP157stop (lanes 6, expected Mr 42) and CyPrPEGFP124–157 (lanes 7, expected Mr 33.5). Mr values are indicated on the left. S, supernatant; P, pellet.
The N-terminal domain of PrP (residues 23–121) is unstructured (Riek et al. 1997), and we hypothesized that it might be the determinant of CyPrP aggregate formation. However, CyPrPEGFP124stop was unable to form aggregates, suggesting that the domain responsible for CyPrP aggregation is in the structured C-terminal region of the protein (Fig. 4). Indeed, CyPrPEGFP124–230, representing a chimeric protein between EGFP and the C-terminal region of CyPrP, formed aggregates (Fig. 4).
Next, we engineered several C-terminal PrP mutants with various structural domains deleted in order to delineate more precisely the domain responsible for the aggregation of CyPrP. Cells expressing CyPrPEGFP157stop were still able to produce aggregates (Fig. 4). This observation indicated that α-helices 2 and 3 and β-sheet 2 are not essential for the formation of cytoplasmic PrP aggregates. We concluded that the 33-amino acid region located between residues 124–157 and containing β-sheet 1 and α-helix 1 is an essential determinant for intracellular aggregation of PrP. When genetically fused to EGFP, this region did not induce the aggregation of EGFP by itself (Fig. 4). This indicated that, although β-sheet 1 and α-helix 1 are essential for the aggregation of CyPrP, they probably interact with other domains of PrP in order to induce aggregation of the protein.
CyPrP aggregates prevent the trafficking of wild-type PrP to the plasma membrane
Conversion to a PrPSc-like conformation in the cytoplasm can be initiated using proteasome inhibitors (Ma and Lindquist 2002). This conversion process is sustained even after removal of the inhibitors and restoration of proteasomal activity. This indicates that aggregated CyPrP molecules may influence newly PrP molecules entering the cytoplasm to change conformation (Ma and Lindquist 2002). The impact of PrP aggregation in the cytoplasm on the efficiency of secretion of normal PrP molecules was not determined. To address this issue, cells were co-transfected with PrPEGFP and CyPrP (Fig. 5). In contrast to cells expressing PrPEGFP (Fig. 1f), almost no EGFP fluorescence was detected at the plasma membrane of cells co-transfected with PrPEGFP and CyPrP (Fig. 5a). This suggested that PrPEGFP accumulated in the secretory pathway and did not reach the plasma membrane. Indeed, PrPEGFP fluorescence co-localized with immunostaining of protein disulfide isomerase (PDI), an enzyme located in the ER (Fig. 5b). In approximately 30% of cells co-transfected with PrPEGFP and CyPrP, the fluorescence displayed a punctuate pattern similar to that of cytoplasmic aggregates described previously (Fig. 5a, right panel).
Fig. 5.

CyPrP aggregates prevent the secretion of wild-type PrP molecules. (a, b) N2a cells were co-transfected with PrPEGFP and CyPrP. Cells were analyzed by direct fluorescence of EGFP (a; numbers indicate percentage of cells with the same pattern of EGFP fluorescence) or immunostained with PDI antibodies (b). (c) Expression and localization of PrPDsRed2 in N2a cells determined by direct fluorescence of DsRed2. (d) N2a cells were co-transfected with PrPDsRed2 and CyPrPEGFP. (e) N2a cells were co-transfected with PrPDsRed2 and CyPrPEGFP124stop. (f) Western blot analysis of PrPEGFP in the supernatant (lanes 1–3) or in the pellet (lanes 4–6) of N2a cells expressing PrPEGFP (lanes 1 and 4), N2a cells expressing PrPEGFP and CyPrP (lanes 2 and 5), or mock-transfected N2a cells (lanes 3 and 6) after treatment with PIPLC. Protein extracts were further treated with peptide N-glycosidase F. Equal loading in fractions from the cell pellet was verified by western blot analysis of α-tubulin. (g) N2a cells were transfected with CD4EGFP, CD4EGFP and CyPrP, GPIEGFP, or GPIEGFP and CyPrP. Scale bar 10 μm. Original magnification ×100. Similar results were obtained with HEK293 cells (not shown).
In order to determine whether CyPrP aggregates could induce PrPEGFP to aggregate in the cytoplasm, we engineered a chimeric protein between PrP and the DsRed2 fluorescent protein (PrPDsRed2). PrPDsRed2 had the same distribution as PrP and PrPEGFP (Fig. 1f), and was mainly located in the golgi and at the plasma membrane (Fig. 5c). In cells co-expressing CyPrPEGFP and PrPDsRed2, we observed the presence of cytoplasmic aggregates displaying both green and red fluorescence (Fig. 5d). Mutant CyPrPEGFP124stop, which was unable to form intracellular aggregates, did not prevent trafficking of PrPDsRed2 to the plasma membrane (Fig. 5e). The interference of PrP secretion by CyPrP was confirmed by estimating the amount of PrP at the cell surface with a phosphatidylinositol-specific phospholipase (Fig. 5f). We found that the amount of PrP released by the phospholipase was decreased by at least 50% in cells expressing CyPrP. The specificity of the effect of CyPrP on PrP trafficking was tested with two other plasma membrane proteins, CD4EGFP and GPIEGFP. CD4 is a transmembrane protein and GPIEGFP, like PrPEGFP, is a GPI-anchored EGFP protein (Nichols et al. 2001). CyPrP did not interfere with CD4EGFP and GPIEGFP localization at the plasma membrane (Fig. 5g).
Altogether, these results demonstrate that CyPrP aggregates efficiently prevent PrP molecules from being trafficked to the plasma membrane.
Toxicity of CyPrP aggregates
It was reported initially that CyPrP specifically promotes apoptosis in neuronal cells (Ma et al. 2002). Further work showed that CyPrP-mediated apoptosis is restricted to N2a cells. CyPrP does not induce apoptosis in human neuroblastoma cell lines BE(2)-M17 and SK-N-SH, and in human primary neurons (Roucou et al. 2003). In order to resolve this controversy, we reasoned that cells producing CyPrP aggresomes may not be as healthy as control cells, and that N2a cells would be more sensitive and undergo apoptosis.
In a first set of experiments, we determined the effect of the presence of CyPrP aggregates on the proliferation of HEK293 and N2a cells by measuring their metabolic activity (Fig. 6a). EGFP expression slightly decreased cell proliferation of HEK293 and N2a cells. In agreement with previous work, HEK293 cells expressing PrPEGFP displayed a reduction of their metabolic activity compared with mock-transfected cells (Paitel et al. 2002). This effect of PrP on cell proliferation was not detected in N2a cells (Fig. 6a). Cells expressing CyPrP displayed a reduction in proliferation of between 70 and 80% (Fig. 6a). This might result either from a cytostatic or a cytotoxic effect of CyPrP aggregates. To distinguish between these possibilities, cells were transfected with EGFP, PrPEGFP or CyPrPEGFP, and the cell cycle was analyzed by flow cytometry. Cells producing CyPrPEGFP aggregates had a cell cycle similar to cells expressing PrPEGFP, EGFP or mock-transfected cells (not shown). Expression of CyPrPEGFP124stop, a mutant unable to form aggregates, did not have any effect on cell proliferation (Fig. 6a). Thus, CyPrP aggregates are cytotoxic in HEK293 and N2a cells.
Fig. 6.

Toxicity of CyPrP aggregates. (a) Cell proliferation of HEK293 and N2a cells transfected with EGFP, PrPEGFP, CyPrPEGFP or CyP-rPEGFP124stop. Activities are expressed as a percentage of the WST-1 metabolizing activity of mock-transfected cells. Values are mean ± SD of three independent experiments. Activity in EGFP-expressing N2a cells was significantly different from that in mock-transfected N2a cells (*p < 0.05). Activity in PrPEGFP-expressing HEK293 and N2a cells was not significantly different from that in EGFP-transfected cells. Activity in CyPrPEGFP-expressing HEK293 and N2a cells was significantly different from that in mock-transfected cells, EGFP-expressing cells and PrPEGFP-expressing cells (†p < 0.01). (b) Cells producing CyPrP aggresomes were more sensitive to STS-induced apoptosis. Cells were transfected with CyP-rPEGFP. Twenty-four hours after transfection, cells were treated with STS at the indicated concentrations for 6 h. The percentage cell death was determined by examining condensed chromatin and fragmented nuclei with Hoechst staining. Values are mean ± SD of three independent experiments. More than 200 cells were counted for each condition. In the absence of STS, cell death in CyPrPEGFP-expressing N2a cells was significantly different from that in CyPrPEGFP-expressing HEK293 and BE(2)-M17 cells (*p < 0.01). Cell death in CyPrPEGFP-expressing N2a cells treated with 0.25 μM STS was significantly different from that in CyPrPEGFP-expressing HEK293 and BE(2)-M17 cells treated with 0.25 μM STS (# p < 0.05). Statistical significance was determined by ANOVA followed by post-hoc Scheffe’s analysis.
To determine whether cells producing CyPrP aggregates are more sensitive to apoptotic insults, we treated control cells and CyPrPEGFP-expressing cells with different concentrations of STS, an inducer of apoptosis (Fig. 6b). In the absence of STS, N2a cells expressing CyPrPEGFP displayed between 15 and 20% more cell death than HEK293 and BE(2)-M17 cells. In the presence of 0.25 μM staurosporine, N2a cells expressing CyPrPEGFP displayed twice as many apoptotic nuclei as BE(2)-M17 and HEK293 cells (Fig. 6b). At a high STS concentration, the percentage cell death was similar in all cell lines.
Discussion
We have shown here that PrP forms cytoplasmic aggregates in transiently transfected cells. These intracellular aggregates decrease the viability of neuronal and non-neuronal cells, and self-sustain by preventing normal PrP molecules from being expressed at the plasma membrane.
A previous report indicated that accumulation of PrP in the cytoplasm of neuronal cells is not toxic (Fioriti et al. 2005), in contrast to previous work from other laboratories (Ma and Lindquist 2002; Ma et al. 2002; Rane et al. 2004; present paper). The reason for this difference is not clear. However, in this report the authors induced CyPrP accumulation by using different proteasome inhibitors. As the proteasome is responsible for most non-lysosomal intracellular protein degradation, its inhibition results in the cytoplasmic accumulation of many different proteins. The proteasome plays a critical role in the maintenance of cellular homeostasis by removing short-lived proteins that regulate the cell cycle, cell growth and differentiation. Depending on experimental conditions (e.g. cell line, inhibitor concentration, duration of treatment in the presence of inhibitors), some proteins that accumulate or cellular pathways that are altered might inhibit, mediate or enhance CyPrP toxicity directly or indirectly. In order to avoid these potential drawbacks, we used a transgene encoding a cytoplasmic form of PrP that allows specific accumulation of PrP in the cytoplasm.
Our observations show that CyPrP aggregates are distributed throughout the cytoplasm and can coalesce in a juxtanuclear area to form aggresomes. An intact microtubule network is required for the coalescence of PrP aggregates in the centrosomal region. Interestingly, a direct interaction between PrP and tubulin was recently reported (Nieznanski et al. 2005). Aggresomes form by the deposition of aggregated proteins in a large structure surrounding the centrosome, and have been proposed to form as a general cellular response to aggregated proteins (Johnston et al. 1998). Cells producing CyPrP aggresomes undergo major intracellular rearrangements, including the formation of a cage-like structure composed of vimentin protein surrounding the aggregates, accumulation of proteasomes, and clustering of mitochondria at the site of CyPrP aggregate deposition. The formation of PrP-containing aggresomes is not specific to CyPrP. PrP mutants V203I and E211Q, associated with Creutzfeldt–Jakob disease, and PrP Q212P, associated with Gerstmann–Straussler–Scheinker syndrome, all form aggresomes in response to proteasomal inhibition (Mishra et al. 2003). Inhibition of peptidylprolyl cis–trans isomerases by cyclosporine A also results in the production of PrP-containing aggresomes (Cohen and Taraboulos 2003). However, the formation of CyPrP aggresomes does not require inhibition of either the proteasome or peptidylprolyl cis–trans isomerases.
Retrotranslocation of PrP from the ER to the cytosol for proteasomal degradation was recently challenged (Drisaldi et al. 2003). The authors reported that PrP molecules that accumulated in cells treated with proteasome inhibitors still contained the N-terminal signal peptide. It was proposed that the hydrophobic N-terminal signal peptide induced the aggregation of PrP in the cytoplasm (Harris et al. 2004). Our results obtained with a form of CyPrP lacking the N-terminal signal peptide demonstrate that the signal peptide is not essential for intracellular aggregation of PrP. Therefore, all PrP molecules have the potential to aggregate when diverted into the cytoplasm. The intracellular aggregation determinant of PrP lies in the C-terminal globular domain and includes the structural domains α-helix 1 and β-sheet 1.
Transient proteasome inhibition induced the self-perpetuation of PrPSc-like molecules in the cytoplasm, but the mechanism was unclear (Ma et al. 2002). Our data show that wild-type PrP molecules are not normally secreted in cells producing CyPrP aggregates. Instead, PrP accumulates in the ER and in the cytosol. This is a clear indication that CyPrP aggregates recruit wild-type PrP and may use this mechanism to self-perpetuate. This phenomenon seems to be specific for PrP because CyPrP aggregates did not prevent trafficking of two other plasma membrane proteins, CD4EGFP and GPIEGFP. At present, we do not know at what stage of its trafficking wild-type PrP is diverted to the cytosol. CyPrP may interfere with PrP translocation in the ER. Alternatively, aggresomes induce fragmentation of the golgi (Garcia-Mata et al. 2002), and PrP may be diverted in the cytoplasm during its transport through the secretory pathway, before reaching the plasma membrane. This requires further investigation.
Previous studies investigating the toxicity of CyPrP reported that it promotes apoptosis in N2a cells (Ma et al. 2002; Roucou et al. 2003). These studies relied on the assumption that toxicity occurred exclusively by apoptosis. Based on metabolic activity measurements, we have shown here that CyPrP impedes normal proliferation not only in N2a cells, but also in non-neuronal HEK293 cells. Moreover, CyPrP expression sensitizes these cells to apoptotic insults. Toxicity may result from disruption of the mitochondrial network and clustering of mitochondria that occurs before the formation of aggresomes. Such mitochondrial clustering has been observed previously in cells containing aggresomes composed of mutant huntingtin fragments or intracellular clusterin (Waelter et al. 2001; Debure et al. 2003). Disruption of the mitochondrial network is likely to perturb the cellular metabolism, including localized energy requirements and calcium homeostasis. In experiments using the phenol red pH indicator, we have observed that the pH of the culture medium becomes more rapidly acidic in N2a cell cultures than in other neuronal and non-neuronal cell cultures (not shown). We propose that N2a cells have a very active metabolism and are more sensitive to major mitochondrial perturbations resulting from the formation of CyPrP aggregates.
What is the function of CyPrP and is CyPrP responsible for the neurodegeneration observed in prion diseases? In some neurons, PrP is relatively abundant in the cytoplasm (Mironov et al. 2003), and may have a beneficial effect (Roucou et al. 2003). However, when the ability of the proteasome to degrade PrP or when PrP translocation into the ER is compromised, PrP levels increase in the cytoplasm and the protein forms aggregates. PrP aggresomes are not a pathological feature of prion disease and are unlikely to be responsible for the neurodegeneration. However, if in vivo CyPrP levels do not reach levels high enough to produce aggresomes similar to those observed in cultured cells, intracellular aggregates might still be produced. Interestingly, intracellular PrP aggregates are observed in some forms of prion diseases including sporadic Creutzfeld–Jakob disease and in some Gertsmann–Sträussler–Scheinker syndromes (Ghetti et al. 1994; Kovács et al. 2005). It was shown recently that small aggregates composed of 14–28 molecules are the most infectious PrP particles (Silveira et al. 2005). Small intracellular PrP aggregates may also be very toxic. As a growing body of evidence indicates that PrP molecules different from PrPSc, including CyPrP, are neurotoxic intermediates in prion diseases, it is essential to further characterize the molecular activity of these molecules.
Acknowledgments
We are grateful to Dr Neena Singh (Case Western Reserve University, Cleveland, OH, USA), Dr Jana Stankova (University of Sherbrooke, Sherbrooke, QC, Canada) and Dr Ben Nichols (Medical Research Council Laboratory of Molecular Biology, Cambridge, UK) for providing the plasmids encoding PrPEGFP, CD4EGFP and GPIEGFP respectively. We thank the people from the electron microscopy facility of the University of Sherbrooke for expert assistance with transmission electron microscopy. This work was supported by a grant from Canadian Institutes for Health Research. XR is a Junior 2 research scholar from the Fonds de la Recherche en Santé du Québec.
Abbreviations used
- CyPrP
cytoplasmic prion protein
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- GPI
glycosylphosphatidylinositol
- HEK
human embryonic kidney
- OR
octapeptide region
- PDI
protein disulfide isomerase
- PIPLC
phosphatidylinositol-specific phospholipase C
- PrP
prion protein
- PrPDsRed2
Discosoma sp. red fluorescent protein2-tagged PrP
- PrPEGFP
enhanced green fluorescent protein-tagged PrP
- PrPSc
disease-causing prion protein
- STS
staurosporine
- WST-1
4-[3-(4-lodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate
References
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- Cohen E, Taraboulos A. Scrapie-like prion protein accumulates in aggresomes of cyclosporin A-treated cells. EMBO J. 2003;22:404–417. doi: 10.1093/emboj/cdg045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debure L, Vayssiere JL, Rincheval V, Loison F, Le Drean Y, Michel D. Intracellular clusterin causes juxtanuclear aggregate formation and mitochondrial alteration. J Cell Sci. 2003;116:3109–3121. doi: 10.1242/jcs.00619. [DOI] [PubMed] [Google Scholar]
- Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–21743. doi: 10.1074/jbc.M213247200. [DOI] [PubMed] [Google Scholar]
- Fioriti L, Dossena S, Stewart LR, Stewart RS, Harris DA, Forloni G, Chiesa R. Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J Biol Chem. 2005;280:11320–11328. doi: 10.1074/jbc.M412441200. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R, Gao YS, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 2002;3:388–396. doi: 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- Ghetti B, Tagliavini F, Giaccone G, Bugiani O, Frangione B, Farlow MR, Dlouhy SR. Familial Gerstmann–Straussler–Scheinker disease with neurofibrillary tangles. Mol Neurobiol. 1994;8:41–48. doi: 10.1007/BF02778006. [DOI] [PubMed] [Google Scholar]
- Gu Y, Verghese S, Mishra RS, Xu X, Shi Y, Singh N. Mutant prion protein-mediated aggregation of normal prion protein in the endoplasmic reticulum: implications for prion propagation and neurotoxicity. J Neurochem. 2003;84:10–22. doi: 10.1046/j.1471-4159.2003.01255.x. [DOI] [PubMed] [Google Scholar]
- Harris DA, Peters PJ, Taraboulos A, Lingappa V, DeArmond SJ, Prusiner SB. Prion Biology and Diseases. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor: 2004. [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Tremblay P, Groth D, DeArmond SJ, Prusiner SB, Lingappa VR. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature. 1999;402:822–826. doi: 10.1038/45574. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Kovács GG, Preusser M, Strohschneider M, Budka H. Subcellular localization of disease-associated prion protein in the human brain. Am J Pathol. 2005;166:287–294. doi: 10.1016/S0002-9440(10)62252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Magalhaes AC, Zanata SM, Brentani RR, Martins VR, Prado MA. Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J Neurochem. 2001;79:79–87. doi: 10.1046/j.1471-4159.2001.00529.x. [DOI] [PubMed] [Google Scholar]
- Lehmann S, Harris DA. Mutant and infectious prion proteins display common biochemical properties in cultured cells. J Biol Chem. 1996;271:1633–1637. doi: 10.1074/jbc.271.3.1633. [DOI] [PubMed] [Google Scholar]
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA. 2001;98:14955–14960. doi: 10.1073/pnas.011578098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lindquist S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science. 2002;298:1785–1788. doi: 10.1126/science.1073619. [DOI] [PubMed] [Google Scholar]
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Mironov A, Jr, Latawiec D, Wille H, Bouzamondo-Bernstein E, Legname G, Williamson RA, Burton D, DeArmond SJ, Prusiner SB, Peters PJ. Cytosolic prion protein in neurons. J Neurosci. 2003;23:7183–7193. doi: 10.1523/JNEUROSCI.23-18-07183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra RS, Bose S, Gu Y, Li R, Singh N. Aggresome formation by mutant prion proteins: the unfolding role of proteasomes in familial prion disorders. J Alheimers Dis. 2003;5:15–23. doi: 10.3233/jad-2003-5103. [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Kenworthy AK, Polishchuk RS, Lodge R, Roberts TH, Hirschberg K, Phair RD, Lippincott-Schwart ZJ. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J Cell Biol. 2001;153:529–541. doi: 10.1083/jcb.153.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieznanski K, Nieznanska H, Skowronek KJ, Osiecka K, Stepkowski D. Direct interaction between prion protein and tubulin. Biochem Biophys Res Commun. 2005;334:403–411. doi: 10.1016/j.bbrc.2005.06.092. [DOI] [PubMed] [Google Scholar]
- Paitel E, Alves Da Costa C, Vilette D, Grassi J, Checler F. Overexpression of PrPc triggers caspase 3 activation: potentiation by proteasome inhibitors and blockade by anti-PrP antibodies. J Neurochem. 2002;83:1208–1214. doi: 10.1046/j.1471-4159.2002.01234.x. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane NS, Yonkovich JL, Hegde RS. Protection from cytosolic prion protein toxicity by modulation of protein translocation. EMBO J. 2004;23:4550–4559. doi: 10.1038/sj.emboj.7600462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP (23–231) FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, Leblanc A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005;12:783–795. doi: 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solforosi L, Criado JR, McGavern DB, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303:1514–1516. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Machida Y, Nishikawa Y, Akagi T, Morishima I, Hashikawa T, Fujisawa T, Nukina N. The effects of aggregation-inducing motifs on amyloid formation of model proteins related to neurodegenerative diseases. Biochemistry. 2002;41:10277–10286. doi: 10.1021/bi0258905. [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang F, Sy MS, Ma J. Calpain and other cytosolic proteases can contribute to the degradation of retro-translocated prion protein in the cytosol. J Biol Chem. 2005;280:317–325. doi: 10.1074/jbc.M410649200. [DOI] [PubMed] [Google Scholar]
- Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 2001;20:5383–5391. doi: 10.1093/emboj/20.19.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]