

Abstract
The molecular mechanism underpinning regulation of eukaryotic translation initiation factor eIF4E by 4E-BP1 has remained unclear. We use isothermal calorimetry, circular dichroism, NMR, and computational modeling to analyze how the structure of the eIF4E-binding domain of 4E-BP1 determines its affinity for the dorsal face of eIF4E and thus the ability of this regulator to act as a competitive inhibitor. This work identifies the key role of solvent-facing amino acids in 4E-BP1 that are not directly engaged in interactions with eIF4E. These amino acid residues influence the propensity of the natively unfolded binding motif to fold into a conformation, including a stretch of α-helix, that is required for tight binding to eIF4E. In so doing, they contribute to a free energy landscape for 4E-BP1 folding that is poised so that phosphorylation of S65 at the C-terminal end of the helical region can modulate the propensity of folding, and thus regulate the overall free energy of 4E-BP1 binding to eIF4E, over a physiologically significant range. Thus, phosphorylation acts as an intramolecular structural modulator that biases the free energy landscape for the disorder–order transition of 4E-BP1 by destabilizing the α-helix to favor the unfolded form that cannot bind eIF4E. This type of order–disorder regulatory mechanism is likely to be relevant to other intermolecular regulatory phenomena in the cell.
Keywords: posttranscriptional control, conformational change, mRNA cap binding, intrinsically unstructured proteins
A critical step in cap-dependent translation initiation in eukaryotes involves association of eIF4G with the cap-binding protein eIF4E, because the bound eIF4G recruits the ribosomal 43S complex to the 5′end of mRNA (1). The eIF4G-eIF4E interaction can be blocked by small (∼12 kDa) heat-stable regulatory proteins called the 4E-binding proteins (4E-BPs) that act by competing for the same eIF4E dorsal site to which eIF4G binds (2, 3). The 4E-BPs and eIF4G factors share the Y(X)4Lϕ motif (Tyr; X = variable; Leu; ϕ = hydrophobic) (3) as a major part of the binding domain. Kinase-mediated phosphorylation of the 4E-BPs reduces their affinity for eIF4E, thus providing the cell with a regulatory mechanism for translation initiation that responds to growth factors, hormones, mitogens, and cytokines (3, 4). Phosphorylation of 4E-BP1 is thought to be at least a two-stage process, in which the FRAP/mTOR pathway phosphorylates T37 and T46 on 4E-BP1, and this serves as a priming event for phosphorylation of other sites (by the FRAP/mTOR or PI3 kinase/Akt pathways), including S65, T70, S83, and S112 (5). There are three, closely related, 4E-BPs that show somewhat different distributions in mammalian tissues (6, 7), but all act via the same mechanism. A second group of regulators, which contain the really interesting new gene (RING) domain, modulates eIF4E function via a partially overlapping dorsal binding site and via a different mechanism of conformational (allosteric) change (8).
The 4E-BPs belong to the large family of intrinsically unstructured proteins, an as yet poorly understood molecular family that performs a wide range of roles in the cell (9–12). Such proteins can undergo transitions between different conformational states and do not simply exist as random coils, although it has generally proved difficult to characterize the various states that any chosen protein can assume (12). Previous biophysical studies have suggested that there is little or no structure in 4E-BP1 in solution (13, 14). However, the binding regions of both eIF4G and 4E-BP1 become more structured (approximately 50% helical) upon forming complexes with eIF4E (15–19), whereby eIF4G also undergoes extensive folding in other parts of its structure (16). It has been shown that a peptide corresponding to 4E-BP1 residues 49–68 is sufficient to inhibit eIF4E function (13) and that a 17mer peptide copy of the same eIF4E-binding site manifests a very similar affinity for eIF4E to that of full-length 4E-BP1 (14), indicating that the major determinants of specific binding lie in the binding domain itself. But what predisposes the 4E-BP1 binding motif to bind in the correct conformation to the dorsal face of eIF4E? X-ray crystallographic analysis has identified interactions between the highly conserved 4E-BP1 residues Y54, F58, L59, the hydrophobic position 60 (M or L), and the basic position 63 (R or K), all of which lie in the helical region of the bound protein, and both the N-terminal region and the H1 and H2 helices of eIF4E (14, 17). Additionally, surface plasmon resonance (SPR) binding analysis demonstrated the importance of L39, W73, G139, and V69 on eIF4E (18), all of which are predicted to interact with the above 4E-BP1 contact residues (17). However, this has left unexplained the role of a number of residues (including D55, K57, E61, and N64) in the binding motif of 4E-BP1 that are not involved in direct interactions with eIF4E, yet are highly conserved across the mammalian 4E-BPs.
Phosphorylation of S65 on 4E-BP1 causes the greatest reduction in eIF4E-binding affinity of all single phosphoylation events on this inhibitor protein (20) and occurs at the site that is closest to the binding motif sequence of this protein. In vivo, interference with the 4E-BP1:eIF4E interaction is thought to be enhanced by phosphorylation of S65 combined with phosphorylation of at least one other site (see above; 20, 21). Given the major impact of S65 phosphorylation on the ability of 4E-BP1 to bind to the dorsal face recognition motif on eIF4E, it is essential to understand how this specific modification event influences binding between the two proteins. It has been suggested that phosphorylation of S65 gives rise to a repulsive electrostatic effect between this residue on 4E-BP1 and the E70 residue on eIF4E (14), but this hypothesis has remained experimentally untested. We now report on investigations that have generated a disorder–order transition model for regulation of 4E-BP binding. This model is likely to be of relevance to a number of intermolecular regulatory events in the cell and is in contrast to previously proposed phosphorylation-related mechanisms (22–24).
Results
Solvent-Oriented 4E-BP1 Amino Acids Promote the Correct Folding Transition.
We investigated the potential role of amino acid residues in the eIF4E-binding region that could influence folding into the conformation required for binding. We focused initially on residues that lie on the solvent face of the motif (referred to here as “solvent-oriented”) when 4E-BP1 is bound to eIF4E because their conservation across the 4E-BP species suggested to us that they could play a key role in determining the thermodynamics of the folding pathway. Of particular interest were the residues in the α-helical region adjacent to the S65 phosphorylation site, because proper folding of this region is required for tight binding to eIF4E (13–17; Fig. 1). We accordingly mutated solvent-oriented residues in this region in a series of derivative peptides corresponding to residues 51–67 of the human 4E-BP1 sequence (4E-BP151–67; Table 1) and used these derivatives in binding titration experiments assayed using isothermal titration calorimetry (ITC). The results revealed that single substitutions of D55, K57, E61, and N64 reduced the affinity (increased the KD) of the eIF4E-binding domain for eIF4E (Fig. 1A and Table 1). We wanted to put these effects in the context of the quantitative contributions of 4E-BP1 amino acids that X-ray crystallography has indicated are in direct contact with eIF4E. We therefore also performed ITC experiments with a further set of peptides containing substitutions at positions 60 and 63 (Fig. 1A and Table 1). Comparable increases in the KD value were observed as a consequence of mutations at these two positions. The majority of the 4E-BP151–67 variants manifested only a small amount of variation in the CD spectra relative to wild type (Fig. S1A), generally showing a strong negative band near 200 nm plus a weak shoulder in the region 209–222 nm. This type of spectrum is typical of proteins that are mostly unfolded in solution, but have a low level of α-helical structure. It is therefore evident that the primary effect of the respective solvent-side mutations manifests itself during the folding process and that the free energy of folding of the binding domain plays a major role in determining the affinity for eIF4E.
Fig. 1.

Affinities of 4E-BP151–67 variants for eIF4E. (A) Isothermal microcalorimetry was performed on titrations of 4E-BP151–67 peptides (as indicated) against human eIF4E. Typical results are shown that illustrate the breadth of affinities observed over the range of single and double amino acid substitutions (see also Table 1. (B) Electrostatic surface map for eIF4E, indicating the positions of key eIF4E residues, with 4E-BP151–67 superimposed (α-helical region highlighted). Acidic (negatively charged) regions are in red. (C) An enlarged (and labeled) version of the backbone structure of 4E-BP151–67 in the conformation determined by X-ray crystallography of the eIF4E:4E-BP1 complex (17). (D) The backbone for amino acids 54–65 of 4E-BP1 is shown, covering the helical segment, and aligned along the helix axis, viewing from the right in B and C. S65, the Y(X)4Lϕ motif and the side chains for Y54, L59, M60 (Y, L, ϕ, facing eIF4E) and for K57, E61 (solvent-facing) are highlighted. The binding domain sequences for mammalian eIF4GI and 4E-BP1 are compared at Bottom.
Table 1.
Summary of KD data for all variants of 4E-BP151–67*
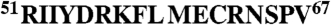 |
KD [eIF4E] (nM) |
Factor increase in KD |
| Wild type | 21 | — |
| D55E | 29 | 1.4 |
| D55S | 100 | 4.8 |
| K57E | 550 | 26 |
| K57R | 18 | 0.86 |
| K57E/E61G | 520 | 25 |
| K57E/E61K | 21 | 1.0 |
| E61K | 52 | 2.5 |
| M60E | 230 | 11 |
| M60T | 230 | 11 |
| M60A | 480 | 23 |
| R63Q | 67 | 3.2 |
| N64S | 67 | 3.2 |
| S65+P (pH 6.6) | 120 | 5.7 |
| S65+P (pH 6.9) | 160 | 7.6 |
| S65+P (pH 7.4) | 240 | 11 |
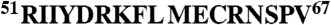 |
KD [eIF4E (E70A)] (nM) |
Factor increase in KD |
| Wild type | 15 | — |
| S65+P (pH 7.0) | 1,600 | Not comparable to WT |
*KD values are averages calculated on the basis of data from at least two independent ITC titrations performed at pH 7.0 (or at pH specified).
The K57E peptide showed some exceptional behavior in that it manifested a CD spectrum indicative of a distinctive mixture of disordered as well as β-type structure (with a more pronounced shoulder at 210–220 nm; Fig. S1B) and was also found to be of reduced solubility in aqueous solution. The increased tendency to aggregate may be attributable to an increased component of β-sheet structure (which is difficult to quantitate accurately in such a spectrum). In order to test the hypothesis that this change in detectable secondary structure was due to a shift in the thermodynamics of the folding landscape, we examined whether the presence of a known promoter of α-helix formation, trifluoroethanol (TFE) (25), would modify the folding behavior so as to reverse the partial conversion to β-sheet structure. We found that the secondary structure of the K57E peptide could indeed be converted to a more α-helical form (as illustrated for 40% TFE in Fig. S1C), whereby the transition was progressively dependent on TFE concentration (Fig. S1D), showing that the β-form is only marginally stable.
If the disorder–order transition in the binding pathway of 4E-BP1 to eIF4E exerts a controlling influence over binding, any sequence change that inhibits formation of the folded structure should, in turn, reduce binding. The K57E mutation results in a large reduction in eIF4E-binding affinity (Table 1), although the absolute KD cannot be compared in direct quantitative terms with the other KD values because measurements with this peptide had to be performed in the presence of a low concentration of DMSO to counteract its enhanced tendency to aggregate at the high concentrations needed for the ITC experiments. In contrast, the control mutation K57R results in only a very minor change in KD (Table 1). The residues K57 and E61 lie on the solvent side of the α-helical region of the eIF4E-binding motif of 4E-BP1 at a distance that is amenable to salt-bridge formation. We tested whether a second mutation that reverses the destabilizing effect of K57E could at least partially restore wild-type binding by studying the effect of engineering the “charge-compensatory” substitution E61K. The K57E/E61K double-mutated version of the binding motif manifested a similar binding affinity for eIF4E to that of the wild-type peptide (Table 1), as well as an enhanced shoulder region around 222 nm (Fig. S1E), indicating an increased tendency to form α-helical structure. No such restoration of the binding affinity was achieved with the peptide bearing the K57E/E61G double mutation, in which no compensatory interaction can be formed (Table 1). This is further evidence that the solvent-oriented amino acid residues act to guide the folding pathway of the 4E-BP1 binding motif (upon binding eIF4E), so that its minimal free energy conformation coincides with folding that promotes binding to eIF4E. We note that the estimated effect of the K57E mutation on the affinity of 4E-BP1 for eIF4E may have been partially attributable to the generally altered physical properties of this derivative, and thus the absolute value of the estimated KD value is less significant than the observed reversal of the effect by the second mutation at E61.
The Folding Propensity of 4E-BP1 in Solution Captured by NMR.
A number of 4E-BP1 residues that contact eIF4E are also involved in α-helical structure formation. We therefore studied the 1H-15N heteronuclear sequential quantum correlation (HSQC) spectra of 13C,15N-labeled wild-type, R63Q and M60E versions of 4E-BP151–67 (overlaid in Fig. 2A). The chemical shift changes encountered upon mutation are shown in Fig. 2B. The N termini of the mutant peptides do not show any change in chemical environment upon point mutation at M60 and R63. On the other hand, the R63Q mutation causes the HSQC peaks of residues C62, Q63, and S65 to move (Fig. 2B, Middle), whereas the M60E mutation causes significant chemical shift changes in residues R56, F58, E60, E61, C62, R63, and N64 (Fig. 2B, Bottom). The overall chemical environment of the residues in the R63Q variant is quite similar to WT in contrast to the significant chemical shift observed in the M60E variant (compare Fig. 2B, Bottom and Middle). This change in chemical shift can be correlated with the increase in KD for eIF4E binding measured for these two mutant peptides: R63Q shows a ∼3-fold increase in KD, whereas the M60E variant shows an ∼11-fold increase in the KD value (Table 1). As noted above, the eIF4E-binding domain of the 4E-BP protein shows a propensity to form α-helix (R56—E61) upon binding to its partner eIF4E, and it is interesting that the residues that are at the same side of this helix show significant chemical shift perturbations (R56, E60, and R63). In the crystal structure of the eIF4E:4E-BP1 complex [Protein Data Bank (PDB) ID code 1wkw)], the side chain of R56 of wild-type 4E-BP1 has direct contact with E132, L135, and R186 of eIF4E. The arginine ϵ proton (in R56) is very labile and is detectable only at low pH and/or when the proton is hydrogen-bonded. Thus the appearance of an arginine ϵ amide peak in the M60E variant suggests that the side chain of R56 may form a hydrogen bond with E60, thus reducing the ability of R56 to interact with eIF4E and providing a potential explanation for the observed increase in KD. In addition, the  coupling constant values were calculated on the basis of 3JHNHα coupling constant (HNHA) measurements for each residue of the WT, M60E, and R63Q peptides. In all three peptides, the residues that form an α-helix upon binding to eIF4E (positions R56—E61) have smaller J values compared to the residues at the N and C termini (Table S1). The typical range of J values for residues within α-helical structure is 3–6 Hz, whereas it is 6–8 for random coil and > 8 for β-sheet, with some dependence on sequence (26). Overall, the NMR data are consistent with a propensity to form α-helix in the noncomplexed form and also indicate the involvement of eIF4E-interacting residues in determining the stability of the helical conformation. This is also evident from the J values obtained for wild-type 4E-BP151–67 in 40% TFE (Table S1).
coupling constant values were calculated on the basis of 3JHNHα coupling constant (HNHA) measurements for each residue of the WT, M60E, and R63Q peptides. In all three peptides, the residues that form an α-helix upon binding to eIF4E (positions R56—E61) have smaller J values compared to the residues at the N and C termini (Table S1). The typical range of J values for residues within α-helical structure is 3–6 Hz, whereas it is 6–8 for random coil and > 8 for β-sheet, with some dependence on sequence (26). Overall, the NMR data are consistent with a propensity to form α-helix in the noncomplexed form and also indicate the involvement of eIF4E-interacting residues in determining the stability of the helical conformation. This is also evident from the J values obtained for wild-type 4E-BP151–67 in 40% TFE (Table S1).
Fig. 2.

NMR spectra of single site mutant forms of 4E-BP151–67. A shows the overlaid 1H-15N HSQC spectra of wild-type (black), R63Q (green), and M60E (red) 4E-BP151–67 peptides (see amino acid sequence in the bottom left-hand corner; M60 and R63 are highlighted red and green, respectively). The arginine ϵ proton (RHNε) evident in the M60E peptide (7.2 ppm/120.4 ppm) is very labile and is detectable only at low pH and/or when the proton is H bonded. (B) The chemical shift differences between the WT and M60E (Bottom), R63Q (Middle), and phosphorylated WT (Top) peptides are plotted against the residue number. The horizontal black line denotes the nominal significance threshold of 0.08 ppm. The red and green colored bars represent mutated residues. In the phosphorylated WT peptide the S65 is not 15N-labeled and is denoted by an asterisk.
Phosphorylation Modulates the Propensity to Fold upon Binding eIF4E.
The above results indicate that the solvent-oriented residues in the eIF4E binding motif, in comparable measure to the eIF4E-contacting amino acid residues, make a key contribution to the free energy landscape for folding of the eIF4E-binding motif in 4E-BP1. We therefore asked the question whether S65 phosphorylation might also influence the propensity of the motif sequence to fold, because this would provide a mechanistic explanation of the inhibitory effect of this modification. We first investigated whether the magnitude of the effect of S65 phosphorylation on binding is pH-dependent, because this is relevant to our understanding of the role of charge at the C-terminal end of the α-helical stretch of the motif. ITC experiments with eIF4E and the phosphorylated 4E-BP1 peptide at pH values of 6.6, 6.9, and 7.4 (Table 1 and Fig. S2) revealed that the KD values for binding of the phosphopeptide to eIF4E increased at higher values of pH, consistent with a reduction in affinity as the phosphate group is titrated from -1 to -2, and with a more positive free energy of formation of α-helical structure in the eIF4E-binding domain. Phosphoserine is highly destabilizing to an alpha-helix when at its C terminus, particularly when it has a -2 charge, primarily due to an unfavorable repulsion to the negative end of the helix dipole (27). We then used NMR to investigate the effect of phosphorylation on the folding and binding characteristics of 4E-BP151–67. The HSQC spectra of the wild-type peptide and of its counterpart phosphorylated at S65 reveal that phosphorylation affects most strongly the local residues (R63, N64, and V67) in solution (Table S1, Fig. 2B, Top, and Fig. 3A). The S65 in the phosphorylated 4E-BP151–67 was not 15N-labeled so that no resonances were seen for this residue in the spectrum (denoted by an asterisk in Fig. 2B). It is again evident that the chemical shift changes between the wild-type and phosphorylated 4E-BP151–67 correlate with the changes in KD for eIF4E binding. Calculation of single residue secondary structure propensity scores (28) highlights the effect of phosphorylation on helical propensity, whereby this effect is amplified in the presence of TFE (Fig. S3; compare Fig. S4 and Table S1).
Fig. 3.

NMR spectra reveal conformational changes. (A) Overlaid 1H-15N HSQC spectra of wild-type (black) and S65-phosphorylated (pink) versions of the 4E-BP151–67 peptide (see sequence in lower left-hand corner; S65 is highlighted pink). (B) Overlaid 1H-15N HSQC spectra of wild-type 4E-BP151–67 peptide complexed with human eIF4E (black), and of noncomplexed wild-type 4E-BP151–67 peptide (blue).
To examine the influence of S65 phosphorylation on interactions between 4E-BP1 and eIF4E, we compared the HSQC spectra of wild-type 4E-BP151–67 in the free form and bound to eIF4E (overlaid in Fig. 3B) and of phosphorylated wild-type 4E-BP151–67 in the free and complexed forms (overlaid in Fig. S5A). In both cases, the data (see black and green peaks in Fig. S5B) are consistent with binding inducing peptide folding, consistent with earlier X-ray crystallography studies (14, 17). The well-dispersed peaks of both forms of 4E-BP151–67 complexed with eIF4E suggest that these peptides form a folded structure in the complex. However, comparison of the HSQC spectra reveals changes that are consistent with altered binding interactions for 4E-BP151–67 caused by phosphorylation (Fig. S5B). We have identified residues that have similar chemical environments (circled red) and others with slightly different chemical environments (circled blue). N64, S65, and V67, however, show significant chemical shift changes that could be due to distinct binding modes for the phosphorylated and nonphosphorylated peptides. These changes are likely to be directly attributable to phosphorylation, because they correlate with comparably large shifts seen with 4E-BP151–67 in solution (Fig. 3A). In conclusion, NMR reveals that S65 phosphorylation affects the folding state of the C-terminal region of 4E-BP151–67 complexed with eIF4E.
The Role of Electrostatic Repulsion in Molecular Regulation.
It has been suggested that E70 in eIF4E will influence the affinity of phosphorylated 4E-BP1 by means of electrostatic repulsion of the phosphate group on S65 (14). Modeling of the electrostatic surface charge of the wild-type and mutant forms of eIF4E shows how E70 can be expected to influence charge distribution on the dorsal binding face of this protein (Fig. 1B). Using a previously developed procedure (29), we estimated a 0.5 to 1 kJ mol-1 increase in energy (depending on the continuum calculation method used) for the addition of a phosphate group (-2 charge form) to S65. For the interaction between the phospho-S65 and E70 on eIF4E, this would translate into only a 1.3–1.6 factor change in the KD between the two proteins. For comparison, the equivalent calculation for the interaction between 4E-BP1 S65 and eIF4E D144 estimates a change in interaction energy of 0.0 kJ mol-1. We also measured the binding behavior of the eIF4E mutant E70A. Previous yeast two-hybrid results suggested that the E70A mutant form of eIF4E still binds strongly to 4E-BP1 (18), and here the ITC results reveal that eIF4E E70A shows an affinity for 4E-BP1 that is comparable to wild type (Table 1). Phosphorylation of S65 in 4E-BP151–67 still results in inhibition of binding to this mutant form of eIF4E; indeed the effect is even greater than that observed with wild-type eIF4E, possibly because the phosphopeptide folded state is less compatible with the eIF4E E70A binding interface. In conclusion, both the ITC results and the theoretical calculations indicate that charge repulsion between eIF4E E70 and 4E-BP1 S65 does not play a major role in phosphorylation-dependent modulation of binding between these two proteins.
Discussion
We have characterized a mechanistic principle underpinning the regulation of eukaryotic translation initiation. Amino acids in the eIF4E-binding domain of 4E-BP1 that are not directly involved in contacts with the dorsal face of eIF4E play a key role in the mechanism of regulation by virtue of their influence on the free energy of folding of the binding domain (ΔGfolding). This term, which will have a considerable entropic component, contributes to the free energy of binding (ΔGbinding) between 4E-BP1 and eIF4E through the relationship
| [1] |
which also includes a free energy term associated with the interactions between the two proteins (ΔGinteractions; this term includes any changes in the solvation states of the participating molecules).
In our model, noncomplexed 4E-BP1 does not fold to any great degree into secondary structure because ΔGfolding is comparatively small and positive (Fig. 4A). Upon binding to eIF4E, a comparatively large value for ΔGinteractions generates a favorable value for ΔGbinding. This free energy balance places the unbound eIF4E-binding motif at a point on the overall free energy landscape for binding that results in a physiologically appropriate affinity between 4E-BP1 and eIF4E. ITC and SPR studies have indicated that the respective affinities of 4E-BP1 and eIF4GI/II for eIF4E fall in the range 5–30 nM (14, 16, 18). Moreover, the relative intracellular abundance of 4E-BP1 is likely to be greater than that of the eIF4G factors (30). These thermodynamic parameters allow effective inhibition of translation when 4E-BP1 is hypophosphorylated and also enable a good dynamic range of up-regulation in response to phosphorylation, especially when combined with the additional effects of phosphorylation at other sites (see below). S65 phosphorylation swings the value of ΔGfolding significantly positive (see Eq. 1), thus shifting the thermodynamic balance away from a favorable affinity between the two proteins, allowing eIF4E to interact with eIF4G to promote translation initiation (Fig. 4B).
Fig. 4.

Model of disorder–order transition. (A) Our data indicate that a major part of the effect of phosphorylation on binding affinity is due to a reduced propensity to fold into a binding-compatible conformation. Free energy diagram for the nonphosphorylated eIF4E-binding domain of 4E-BP1 (in blue), showing the order–disorder transition of the noncomplexed protein in solution on the left-hand side and binding of the folded protein to eIF4E on the right-hand side (eIF4E in orange). Suggested relationships between the respective ΔG values are shown (these are not to scale). (B) As in A, but for the phosphorylated form, showing altered ΔG values (see text).
We have examined the role of charged amino acid side chains in the regulatory mechanism. Our evidence indicates that both acidic and basic residues are important here, but not according to a simple charge-repulsion mechanism between 4E-BP1 and eIF4E as previously suggested. E70 in eIF4E plays at most a minimal role in the modulatory effect of phosphorylation. In contrast, we find that the charged solvent-oriented amino acid residues K57 and E61 contribute in a significant way to the ΔGfolding component of Eq. 1, possibly by forming a salt bridge that stabilizes α-helical structure. The K57E mutation has a marked effect on secondary structure, apparently shifting it more toward a mixture of random coil/β-sheet structure with an increased tendency to aggregate, thus demonstrating the fine balance between the respective determinants of secondary structure in this domain. A possible explanation for a switch to increased β-sheet structure would be an anomalous salt bridge forming between the mutant glutamic acid residue at position 57 and R63 in the 4E-BP1 sequence, although we have not investigated this possibility further. We propose that the additional substitution of a lysine residue at position 61 in the K57E/E61K variant stabilizes formation of an alternative α-helix-promoting salt bridge, thus improving the free energy of formation of the fold suitable for tight binding to eIF4E.
D55, K57, and N64, the solvent-side amino acid residues that have been identified here as important for tight binding to eIF4E, are identical across all three 4E-BP isoforms, and E61 is substituted conservatively by D in 4E-BP2. The large degree of conservation of these residues is consistent with their key role in determining the free energy of folding of the 4E-BPs as these bind to eIF4E. Interestingly, the 4E-BP2 binding motif differs from that of 4E-BP1 only through the substitution of L60D61R62 for M60E61C62, and this results in a threefold increase in affinity for eIF4E (18). Positions 60 and 62 interact directly with residues on the dorsal face of eIF4E, and thus adjustments here may represent one of the only ways for the affinity for eIF4E to be increased beyond the already high eIF4E-binding affinity of 4E-BP1.
Many phosphorylation sites in proteins lie in intrinsically disordered regions (31), but the effect of this modification differs from system to system. For example, phosphorylation of the unstructured kinase-inducible domain (pKID) of the transcription factor cyclic-AMP-response-element-binding (CREB) protein is thought to promote electrostatic interactions that enhance binding to the KID-binding (KIX) domain of CREB-binding protein (CBP) (24, 32). Our results now demonstrate that, although electrostatic repulsion (particularly involving other 4E-BP1 phosphorylation sites) may be a factor in determining the overall affinity between 4E-BP1 and eIF4E (see below), the major regulatory function of S65 phosphorylation is to modulate the propensity of the 4E-BP1 binding motif to fold into a conformation that fits the binding surface on eIF4E (Fig. 4). This model of destabilizing an α-helical fold that is required for tight binding to another protein is likely to be relevant to a number of intracellular protein–protein interactions modulated by phosphorylation, particularly because helix formation is in general strongly affected by phosphorylation (27). A key advantage of this mechanism is that the dynamic range of achievable regulation can be tuned to suit physiological requirements through the selection of noninteracting amino acid residues in the region containing the binding motif. Amino acid residues that are not directly involved in contacts with the target protein exercise significant control over the ΔGfolding term. Thus the overall ΔGbinding term, and correspondingly the degree of inhibition exerted by the regulatory protein, can be adjusted over a wide range without having to change the residues that are directly involved in the intermolecular interactions.
T46 and T70 on 4E-BP1 are not only farther away than S65 from the eIF4E-binding motif, but they may also lie closer to E70 and D144 on eIF4E when complexed with the latter protein (16). As a result, phosphorylation at these additional sites on 4E-BP1 could theoretically modulate 4E-BP1:eIF4E binding via electrostatic (repulsive) effects, although direct experimental evidence for this has yet to be obtained. Here, we have shown that modulation of the free energy of a folding transition is a key principle for controlling and regulating the binding affinity between a 4E-BP and its target. This mechanism provides the basis for both coarse and fine tuning of binding affinities. Further work will be needed to characterize in equal detail the mode of action of the phosphorylation sites that are more remote from the eIF4E-binding-motif of 4E-BP1.
Materials and Methods
Preparation of Proteins and Peptides.
Human eIF4E was prepared as described previously (20). Peptides were either synthesized on an Advanced Chemtech APEX 396 synthesizer using Fmoc-protected amino acids or were obtained in HPLC-purified form from a commercial source (Peptide Protein Research Ltd.). Purity was checked using mass spectrometry. The K57E peptide was held as a 1% DMSO solution because of reduced solubility in water.
Biophysical Measurements and Modeling.
These are described in SI Text.
Supplementary Material
Acknowledgments.
We thank the Biotechnology and Biological Sciences Research Council for financial support through project Grant B17206 and a Professorial Fellowship (to J.E.G.M.). Research at NYSBC was supported by National Institutes of Health grants GM66354 and GM47021.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1008242107/-/DCSupplemental.
References
- 1.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pause A, et al. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 3.Mader S, Lee H, Pause A, Sonenberg N. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4 gamma and the translational repressors 4E-binding proteins. Mol Cell Biol. 1995;15:4990–4997. doi: 10.1128/mcb.15.9.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin TA, et al. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science. 1994;266:653–6. doi: 10.1126/science.7939721. [DOI] [PubMed] [Google Scholar]
- 5.Gingras AC, et al. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsukiyama-Kohara K, et al. Tissue distribution, genomic structure, and chromosome mapping of mouse and human eukaryotic initiation factor 4E-binding proteins 1 and 2. Genomics. 1996;38:353–363. doi: 10.1006/geno.1996.0638. [DOI] [PubMed] [Google Scholar]
- 7.Poulin F, Gingras AC, Olsen H, Chevalier S, Sonenberg N. 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. J Biol Chem. 1998;273:14002–14007. doi: 10.1074/jbc.273.22.14002. [DOI] [PubMed] [Google Scholar]
- 8.Volpon L, Osborne MJ, Capul AA, de la Torre JC, Borden KL. Structural characterization of the Z RING-eIF4E complex reveals a distinct mode of control for eIF4E. Proc Natl Acad Sci USA. 2010;107:5441–5446. doi: 10.1073/pnas.0909877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 10.Uversky VN. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Gunasekaran K, Tsai CJ, Kumar S, Zanuy D, Nussinov R. Extended disordered proteins: Targeting function with less scaffold. Trends Biochem Sci. 2003;28:81–85. doi: 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 13.Fletcher CM, et al. 4E binding proteins inhibit the translation factor eIF4E without folded structure. Biochemistry. 1998;37:9–15. doi: 10.1021/bi972494r. [DOI] [PubMed] [Google Scholar]
- 14.Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol Cell. 1999;3:707–716. doi: 10.1016/s1097-2765(01)80003-4. [DOI] [PubMed] [Google Scholar]
- 15.Hershey PE, et al. The cap-binding protein eIF4E promotes folding of a functional domain of yeast translation initiation factor eIF4G1. J Biol Chem. 1999;274:21297–21304. doi: 10.1074/jbc.274.30.21297. [DOI] [PubMed] [Google Scholar]
- 16.Gross JD, et al. Ribosome loading onto the mRNA cap is driven by conformational coupling between eIF4G and eIF4E. Cell. 2003;115:739–750. doi: 10.1016/s0092-8674(03)00975-9. [DOI] [PubMed] [Google Scholar]
- 17.Tomoo K, Abiko F, Miyagawa H, Kitamura K, Ishida T. Effect of N-terminal region of eIF4E and Ser65-phosphorylation of 4E-BP1 on interaction between eIF4E and 4E-BP1 fragment peptide. J Biochem. 2006;140:237–246. doi: 10.1093/jb/mvj143. [DOI] [PubMed] [Google Scholar]
- 18.Ptushkina M, von der Haar T, Karim MM, Hughes JM, McCarthy JEG. Repressor binding to a dorsal regulatory site traps human eIF4E in a high cap-affinity state. EMBO J. 1999;18:4068–4075. doi: 10.1093/emboj/18.14.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomoo K, et al. Structural basis for mRNA cap-binding regulation of eukaryotic initiation factor 4E by 4E-binding protein, studied by spectroscopic, X-ray crystal structural, and molecular dynamics simulation methods. Biochim Biophys Acta. 2005;1753:191–208. doi: 10.1016/j.bbapap.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 20.Karim MM, et al. A quantitative molecular model for modulation of mammalian translation by the eIF4E-binding protein 1. J Biol Chem. 2001;276:20750–20757. doi: 10.1074/jbc.M011068200. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Li W, Parra JL, Beugnet A, Proud CG. The C terminus of initiation factor 4E-binding protein 1 contains multiple regulatory features that influence its function and phosphorylation. Mol Cell Biol. 2003;23:1546–1557. doi: 10.1128/MCB.23.5.1546-1557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pufall MA, et al. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005;309:142–145. doi: 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- 24.Zor T, Mayr BM, Dyson HJ, Montminy MR, Wright PE. Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators. J Biol Chem. 2002;277:42241–42248. doi: 10.1074/jbc.M207361200. [DOI] [PubMed] [Google Scholar]
- 25.Rohl CA, Chakrabartty A, Baldwin RL. Helix propagation and N-cap propensities of the amino acids measured in alanine-based peptides in 40 volume percent trifluoroethanol. Protein Sci. 1996;5:2623–2637. doi: 10.1002/pro.5560051225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penkett CJ, et al. NMR analysis of main-chain conformational preferences in an unfolded fibronectin-binding protein. J Mol Biol. 1997;274:152–159. doi: 10.1006/jmbi.1997.1369. [DOI] [PubMed] [Google Scholar]
- 27.Andrew CD, Warwicker J, Jones GR, Doig AJ. Effect of phosphorylation on α-helix stability as a function of position. Biochemistry. 2002;41:1897–1905. doi: 10.1021/bi0113216. [DOI] [PubMed] [Google Scholar]
- 28.Marsh J, Singh K, Zongchao J, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between α- and γ- synuclein: Implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitchen J, Saunders RE, Warwicker J. Charge environments around phosphorylation sites in proteins. BMC Struct Biol. 2008;8:19. doi: 10.1186/1472-6807-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von der Haar T, Gross JD, Wagner G, McCarthy JEG. The mRNA cap-binding protein in eIF4E in post-transcriptional gene expression. Nat Struct Mol Biol. 2004;11:503–511. doi: 10.1038/nsmb779. [DOI] [PubMed] [Google Scholar]
- 31.Iakoucheva LM, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radhakrishnan I, et al. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.