

Abstract
We combine experiment and computer simulation to show how macromolecular crowding dramatically affects the structure, function, and folding landscape of phosphoglycerate kinase (PGK). Fluorescence labeling shows that compact states of yeast PGK are populated as the amount of crowding agents (Ficoll 70) increases. Coarse-grained molecular simulations reveal three compact ensembles: C (crystal structure), CC (collapsed crystal), and Sph (spherical compact). With an adjustment for viscosity, crowded wild-type PGK and fluorescent PGK are about 15 times or more active in 200 mg/ml Ficoll than in aqueous solution. Our results suggest a previously undescribed solution to the classic problem of how the ADP and diphosphoglycerate binding sites of PGK come together to make ATP: Rather than undergoing a hinge motion, the ADP and substrate sites are already located in proximity under crowded conditions that mimic the in vivo conditions under which the enzyme actually operates. We also examine T-jump unfolding of PGK as a function of crowding experimentally. We uncover a nonmonotonic folding relaxation time vs. Ficoll concentration. Theory and modeling explain why an optimum concentration exists for fastest folding. Below the optimum, folding slows down because the unfolded state is stabilized relative to the transition state. Above the optimum, folding slows down because of increased viscosity.
Keywords: enzymatic activity, FRET, folding kinetics, thermal denaturation, protein conformational changes
Phosphoglycerate kinase (PGK) is a 415-residue metabolic enzyme that produces ATP and is composed of two roughly equally sized subunits connected by a flexible hinge (1). In the crystal structure, the ADP and diphosphoglycerate binding sites, each located at an N and C subunit, are separated. It has been suggested that a large-scale conformational change (2) is necessary to bring the two subunits together when the phosphoryl group is catalytically transferred, and a hinge-bending mechanism has been postulated (3), bringing together both substrates at the inner surfaces of the C and N subdomains (4, 5).
It is still unclear how the conformational and folding dynamics of PGK is affected by the interior of a cell, which is heavily crowded by macromolecules (6, 7). Various computational and theoretical studies have been developed to address the effect of volume exclusion exerted by surrounding macromolecules on protein activity inside cells, called the “macromolecular crowding effect” (8). This effect, in addition to weak chemical interactions between proteins and crowders (9), can stabilize the folded states of a protein relative to the unfolded state (10), perturb folding barriers (11, 12), and alter folding rates (13) and folding routes (14).
Macromolecular crowding could selectively stabilize one folded protein structure over another (8, 15–17), particularly for proteins that are structurally malleable so their domains aligned in different orientations would have similar free energies (18). Thus, what we regard as the native structural ensemble depends on whether the protein is in aqueous buffer, in a solution crowded by macromolecules, or packed in a crystal.
In our work, structural changes of PGK are revealed by FRET experiments where the N and C termini are labeled with AcGFP1 and mCherry as fluorescent markers (Fig. 1). A crowded environment is achieved by the addition of inert Ficoll 70 carbohydrate to the samples. When the crowding content is increased, we observe changes in FRET that point to new compact structures being populated. Our simulations provide a high-resolution interpretation of the structural changes induced in PGK by crowding. Upon crowding, two other compact conformations [collapsed crystal (CC) and spherical (Sph)] exceed the stability of the crystal structure.
Fig. 1.

Programmable T-jump setup. A computer-controlled diode laser delivers a shaped heating pulse that induces a sustained step function in temperature for the duration of the measurement. A blue LED excites the donor label (green barrel on the PGK model). A microscope objective images donor and acceptor (red barrel) fluorescence from the very center of the heated area onto a single CCD camera, which collects successive snapshots of green and red fluorescence.
Strikingly, these structural changes of PGK upon crowding have a major functional consequence. An enzymatic activity assay shows that ATP production is enhanced over 25-fold in the crowded wild-type enzyme, when taking into account increased solvent viscosity. The donor/acceptor labeled PGK also becomes much more enzymatically active. We suggest that in vivo, PGK is already structurally primed to catalyze by forming in a collapsed compact form (e.g., CC and/or Sph structural ensembles) rather than requiring hinge motions.
The effects of crowding on PGK folding kinetics are also very pronounced. We measured the folding/unfolding relaxation time τ near the thermal denaturation midpoint of PGK. A plot of ln τ vs. Ficoll concentration has a “v” or chevron shape, indicating that there is an optimal crowder concentration for fastest folding. This observation is in accord with theory and computational models (13). High concentrations of sucrose do not show similar structural or kinetic effects, so the connectivity of Ficoll and hence crowding are needed to affect the large amplitude motions of the two-subunit protein PGK, be it functional dynamics involving enzymatic activity (a new compact native state) or folding dynamics (crowding-viscosity competition).
Results
Protein Donor–Acceptor Distance Decreases with Increased Crowding.
The PGK construct fluorescence signature indicates the presence of at least two conformational states. The end-to-end size of the AcGFP1-PGK-mCherry construct was monitored as a function of temperature and Ficoll concentration. Monitoring the steady-state donor–acceptor (D/A) ratio of the construct at 22.3 ± 0.5 °C, we observed a small but reproducible lag upon going from 0 mg/ml Ficoll to 50 mg/ml Ficoll. As the concentration of Ficoll was further increased, D/A decreased continuously (Fig. 2A). The highest Ficoll concentration of 300 mg/ml corresponds to an experimental crowder volume fraction of 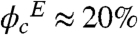 (see Experimental and Computational Methods for details on calculation of
(see Experimental and Computational Methods for details on calculation of 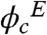 ). At room temperature, crowding conditions result in conformational changes of the protein that eventually decrease the donor–acceptor distance when more crowder is added.
). At room temperature, crowding conditions result in conformational changes of the protein that eventually decrease the donor–acceptor distance when more crowder is added.
Fig. 2.

Conformational, thermal stability and kinetics trends of the PGK construct as a function of Ficoll concentration. (A) Donor–acceptor ratio at 22.3 ± 0.5 °C (filled circles). Open triangles show the intrinsic fluorophore response of AcGFP1 and mCherry as a function of Ficoll concentration. (B) “Inverse chevron” plot of the observed T-jump relaxation time. Red squares show the kinetic trend obtained from simulations in ref. 13 at volume fraction 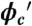 .
. 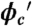 was computed using the size of the crowding agents equal to the native state of the protein. The simulations were scaled to match up the fastest folding rate observed in our experiments. (C) Melting temperature. (D) Cooperativity parameter Δg1 (free energy derivative) from Eq. 1. All error bars are ± 1 standard deviation.
was computed using the size of the crowding agents equal to the native state of the protein. The simulations were scaled to match up the fastest folding rate observed in our experiments. (C) Melting temperature. (D) Cooperativity parameter Δg1 (free energy derivative) from Eq. 1. All error bars are ± 1 standard deviation.
As a first control, we measured the D/A ratio as a function of sucrose concentration. We observed that D/A was almost constant over the entire range from 0–300 mg/ml (see SI Appendix), thus showing that Ficoll indeed induces compact states of PGK through macromolecular crowding and not through protein–carbohydrate nonspecific interactions. As a second control, we measured the intrinsic response of the AcGFP1 and mCherry fluorophores (open triangles in Fig. 2A) as a function of Ficoll concentration. For this purpose, we excited donor (green) fluorescence of an AcGFP1-PGK construct and acceptor (red) fluorescence of the AcGFP1-PGK-mCherry construct by exciting mCherry directly. D/A is nearly crowding-independent (Fig. 2A) and cannot account for the compact states of PGK observed at high crowder concentration.
Protein Stability as a Function of Crowding.
Upon Ficoll crowding, the stability of PGK construct increases, in agreement with predictions made previously (13) and also subsequent experiments on other proteins (19). In aqueous buffer, the construct had a melting temperature of Tm = 39 °C (see Experimental and Computational Methods). Tm increased slightly with crowder concentration (Fig. 2C). The maximum crowder concentration for the higher temperature experiments was 200 mg/ml (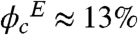 volume fraction) to avoid aggregation of the construct. However, addition of sucrose also results in significant thermodynamic stabilization (see Fig. S1), indicating that thermodynamic stabilization caused by macromolecular crowding is modest. Thermal titrations up to 50 °C were > 90% reversible to the room temperature signal in aqueous, sucrose, and Ficoll solutions.
volume fraction) to avoid aggregation of the construct. However, addition of sucrose also results in significant thermodynamic stabilization (see Fig. S1), indicating that thermodynamic stabilization caused by macromolecular crowding is modest. Thermal titrations up to 50 °C were > 90% reversible to the room temperature signal in aqueous, sucrose, and Ficoll solutions.
Enzymatic Activity Increases with Crowding.
As seen in Table 1, enzymatic activity of wild-type PGK increases about fivefold when the Ficoll concentration is raised to 100 mg/ml, and even more at 200 mg/ml. Ordinarily, the increased viscosity of the Ficoll solution should slow down the catalytic rate. If the viscosity increase is taken into account (Table 1), the net increase in activity is over 12-fold at 100 mg/ml, and even higher at 200 mg/ml of Ficoll (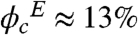 ).
).
Table 1.
Activity assays of wild-type PGK and the FRET-labeled mutant (PGK–FRET) construct at different crowding concentrations
| Mutant | Ficoll 70 concentration (mg/ml) | Enzyme activity (units/liter) | Viscosity 20 °CmPas |
| Wild-type PGK | 0 | 24 ± 1 | 1.0 |
| Wild-type PGK | 100 | 129 ± 33 | 2.3 |
| Wild-type PGK | 200 | 319 ± 199 | 5.0 |
| PGK–FRET | 0 | 18 ± 2 | 1.0 |
| PGK–FRET | 100 | 52 ± 4 | 2.3 |
| PGK–FRET | 200 | 58 ± 6 | 5.0 |
The FRET-labeled PGK yields similar results. Enzymatic activity is similar compared to the wild type (Table 1) in aqueous solution. At Ficoll concentrations ≥100 mg/ml, PGK activity increased about threefold. If one includes the effect of slower substrate diffusion in the viscous crowding matrix (Table 1), the activity increase is actually enhanced over sixfold in 100 mg/ml Ficoll, and over 15-fold in 200 mg/ml Ficoll. These increases in activity are much greater than the < 2-fold changes usually observed upon addition of osmolytes or crowders (20) to enzyme solutions, another indication of a larger structural change.
A control experiment in 100 mg/ml sucrose simply resulted in a reduction of enzymatic activity (see Fig. S2), as expected at the higher viscosity. Thus, the enhancement of activity in Ficoll goes hand-in-hand with the more compact structure detected by FRET upon macromolecular crowding, in contrast to sucrose (Fig. S1), and detected by simulations below.
Chevron Plot of Relaxation Time vs. Crowder Concentration.
The PGK relaxation rate is maximized at a well-defined crowder concentration. The AcGFP1-PGK-mCherry construct was laser temperature jumped from 39.8 ± 0.5 to 44.0 ± 0.5 °C at several crowder concentrations. When relaxation is measured by temperature jumps to 44 °C, the unfolding rate is weighted slightly more than the folding rate in the observed relaxation rate because Tm ranged from 39 to 40.5 °C. The relaxation time vs. crowder concentration is a “chevron” plot (Fig. 2B). The fastest relaxation is observed at 100 mg/ml Ficoll concentration. In contrast, the relaxation rate in sucrose decreases monotonically with increasing concentration (see SI Appendix). Addition of sucrose only results in increased viscosity, not in any crowding effect.
Folding Energy Landscape of PGK in Silico Changes upon Crowding.
In order to investigate the stability and structure of PGK, we calculated the free energy landscapes of PGK. We identified four states: C (crystal structure), CC (collapsed crystal structure), Sph (spherical compact state), and U (thermally denatured state). The structures of these states are discussed in detail in the next subsection. In Fig. 3 we plot the ensemble populations as a function of volume fraction of crowders (ϕc = 0–40%) and temperature (0.9 to 1.47kBT/ε) using structural similarity χ (χ equals 0 for the crystal structure, 1 for highly dissimilar structures) (21) and radius of gyration Rg as the order parameters characterizing the energy landscape of PGK.
Fig. 3.

Two-dimensional folding free energy landscape of PGK in the bulk (A–C) and under the crowded conditions at ϕc = 25% (D–F) and ϕc = 40% (G–I) as a function of the radius of gyration Rg and the overlap function χ at different temperatures (in units of kBT/ε). The color is scaled by kBT where kB is the Boltzmann constant and ε is 0.6 kcal/mol. The crystal structure is labeled C, the collapsed crystal structure CC, the spherical state Sph, and the unfolded state U.
At low temperature and in the absence of crowders, the C and CC states are populated (Fig. 3A). As ϕc increases, the population of the C states reduces. At ϕc = 25%, CC and Sph are both populated, while the population of C is significantly destabilized by approximately 4kBT (Fig. 3D). Finally, at the highest crowding (ϕc = 40%), the Sph structure prevails. (Fig. 3G).
When the temperature is increased from kBT/ε = 0.9 to kBT/ε = 1.17 (ε is the solvent-mediated interaction; see SI Appendix for details), the CC and Sph structures become extensively populated even in the absence of crowding (Fig. 3B), and the Sph structure dominates under crowded conditions (Fig. 3 E and H). Finally, at the highest temperature, the unfolded state dominates under all crowding conditions.
Structural Characteristics of Compact PGK States in Silico.
We characterized the coarse-grained structural properties of PGK with four parameters (see Experimental and Computational Methods): structural similarity χ, radius of gyration Rg, asphericity Δ (Δ = 0 is sphere, Δ = 1 is rod) and shape parameter S (S = -0.25 is oblate, S = 2 is prolate). The crystal structure ensemble C had 0.05 ≤ χ ≤ 0.18 and 22.0 Å ≤ Rg ≤ 24.0 Å. The collapsed crystal structure CC state had higher χ values but smaller Rg (0.16 ≤ χ ≤ 0.26 and 20.0 Å ≤ Rg ≤ 21.5 Å) than the C state. The calculation of shape and asphericity parameters for the CC structure (S = 0.05 and Δ = 0.1) demonstrated a dramatic shape change to a closed compact state from the open crystal state (S = 0.26 and Δ = 0.26). Furthermore, at a higher temperature or crowding level the Sph state appears with greater values of χ (0.3 ≤ χ ≤ 0.36 and 20.0 Å ≤ Rg ≤ 21.5 Å). The ensemble structures from the Sph state are characterized by S = 0.01 and Δ = 0.02, revealing a spherical nature of this state.
To better illustrate the structural differences among ensemble conformations of PGK in atomistic detail, we reconstructed the coarse-grained protein models of the C, CC, and Sph states into all-atomistic protein models using the reconstruction algorithm SCAAL (side chain-Cα to all-atom) (15, 22) (Fig. 4 A–C). In the C state, the two subunits are split wide apart as in an open “pacman” form. In the CC state, the cleft between the two subunits, a space accommodating the two substrates of PGK, closes, resembling a closed “pacman.” The Sph state is formed by a major rearrangement of the two subunits over the hinge that involves twisting one of the two subunits with respect to the other, leaving at least one of the binding sites exposed to solvent.
Fig. 4.

Structural characteristics of the dominant compact ensemble structures of PGK in cartoon representation. (A) Crystal state C, (B) collapsed crystal state CC, and (C) spherical state Sph. The coloring of each protein model ranges from N terminus (red) to C terminus (blue). The N and C termini are represented with van der Waals spheres. The schematic representation at the bottom left of each panel is to address a simplistic view of the structural arrangement of the N- and C-lobes in each conformation. (D) The probability distribution of the distance between N and C termini (DN-C) of the three dominant structures of PGK under the condition in which each prevails in the simulations. C state (solid black), CC (dashed red), and Sph (dotted blue). Atomistic structures were reconstructed from coarse-grained protein models using SCAAL.
In order to investigate the domain rearrangement in the Sph state in detail, we calculated the orientation between the N and C domains of PGK by measuring an angle (θ) formed by two representative vectors. Two vectors were defined, one on each domain, that are parallel in the crystal structure, and these were used to assess the relative orientation of the two domains (see SI Appendix for details). The calculation reveals that in the ensemble of the crystal state (C) θ is 0° (cos θ° = 1 represents that the two vectors are in parallel) as shown in Fig. S3 A and C. However, in the ensemble of the Sph state, θ between the two vectors is centered around 180° (cos θ = -1) as shown in Fig. S3 B and D, representing the alignment of two domains is nearly antiparallel. This result suggests a twist bending motion in the Sph state of PGK involving a significant rearrangement of the protein’s lobes under high crowding conditions.
The domain rearrangement can also be demonstrated by the gain or loss of contact formation, in the form of a contact map, in the C, CC, and Sph states characterized from the respective ensembles in Fig. S4. The formation of new nonnative contacts (in green boxes) and the loss of native contacts (in orange boxes) across subdomains in the CC and Sph states compared to the crystal state (C) suggest a large-scale structural rearrangement in PGK.
In Silico Investigation of the N–C Termini Distance.
The D/A ratios from the FRET-labeled construct correlate with the N–C termini distribution. The experiments demonstrated a strong decrease of D/A with increasing Ficoll concentration, indicating the existence of at least two types of PGK conformations in the presence of crowders. These observations prompted us to compute the distance between the N and C termini (DN-C) for the three populated states (C, CC, and Sph) of PGK from computer simulations. DN-C is defined as the distance between the Cα beads of residues Ser-1 and Lys-415.
Fig. 4D reveals the distinctive profile of the N–C distance (DN-C) distribution for each compact structure taken from the crowding/temperature ensembles where it dominates. The CC state has the highest average DN-C, followed by the C and Sph states. In addition, the DN-C distribution of the CC state is by far the broadest, ranging from 5 Å to 50 Å. The reason for this wide distribution profile as well as high average is the greater mobility of the N and C termini, which are outwardly protruded when the protein’s hinge is bent. In contrast, in the Sph state in which the two domains are twisted against each other, the N and C termini residues are found in the interior of the protein, thus significantly reducing the distribution and the average of DN-C.
In Silico Investigation of the Mg-ATP/3PGA Binding Sites.
The effect of crowding on the structure of PGK pertinent to its enzymatic activity can be indicated by the spatial distance Db between the ATP and 3PGA binding sites. The two binding sites are located separately at the inner surfaces of the N and C subunits, and it is postulated that the closure of this cleft, bringing the two sites together, is necessary for an active enzymatic reaction (3, 23). Fig. 5 shows the Db distribution of the three ensemble structures, C, CC, and Sph. For the crystal structure ensemble (C), which dominates the population only in the absence of Ficoll 70 (ϕc = 0), Db is centered at 21.5 Å. For the CC state ensemble, which is prominent at ϕc = 25%, the most probable Db is drastically reduced to 12.54 Å. Lastly, for the spherical state ensemble Sph ϕc = 40%, the Db distribution is centered at 16.41 Å, still shorter than the most probable Db in the C state.
Fig. 5.

The probability distribution of the distance between the centers of mass of the Mg-ATP and 3PG binding sites (Db) calculated for the three dominant structures of PGK under the condition in which each prevails in the simulations. (Db) of C state (solid black), CC (dotted red), and Sph (dotted blue) ensembles are plotted. The purple solid line represents the convolution of the two profiles from CC and Sph states. The three characteristic structures are illustrated in cartoon representation. The coloring of each protein model ranges from N terminus (red) to C terminus (blue). The Mg-ATP and 3PG binding sites are colored in yellow and green representations of van der Waals spheres, respectively. Atomistic structures were reconstructed from coarse-grained protein models using SCAAL.
Discussion
Despite numerous studies of PGK’s folding stability and dynamics (24–26) and the motion of its flexible hinge, by experiment (27) as well as by computation (28, 29), it remains an open question to what extent the highly crowded environment can affect the PGK folding and structure and, ultimately, its enzymatic activity involving large-scale structural movement inside a cell. Although experimental studies using synthetic crowding agents have shown that the folding properties and enzymatic activity of simple proteins can be perturbed (30), our study shows that crowding leads to major conformational changes of PGK, with a major impact on its enzymatic activity. The tethers labeling PGK are too short to allow the donor–acceptor fluorescence ratio D/A to change by nearly a factor of 2 while PGK remains open, and the individual fluorophore controls reveal no intrinsic crowding dependence of D/A due to changing quantum yields (Fig. 2A). The D/A ratio observed in our experiments decreases rapidly with increasing Ficoll concentration, showing that the two subunits of PGK close in on each other upon being placed in a crowded environment. The observed D/A values correspond to the existence of at least two states of PGK and possibly a third state (resulting in a lag of D/A below 50 mg/ml).
Our experimental interpretation is entirely consistent with the coarse-grained molecular simulations, which reveal three compact ensemble structures of PGK below its folding temperature. The lag and decrease of the D/A ratio with increased crowding may be explained by a progression of PGK through C, CC, and Sph states. In Fig. 3 A, D, and G the most probable PGK state is the C state at ϕc = 0, the CC state at ϕc = 25%, and the Sph state at ϕc = 40%. The corresponding average N- to C-terminal distance of the C, CC, and Sph states is DN-C ∼ 19 Å, ∼25 Å, and ∼14 Å, respectively. This nonmonotonic trend of DN-C is in qualitative agreement with the FRET results: a lag phase (or slight increase) of D/A from 0–50 mg/ml Ficoll, then a strong decrease of D/A.
Crowding dramatically increases PGK activity, as demonstrated by an over 15-fold increase in viscosity-adjusted enzymatic activity of the construct. The increase is even more dramatic for wild type yeast PGK (without FRET labels) (Table 1). Complementary structural analysis of the compact PGK ensembles observed in the computer simulations reveals that the CC and Sph states indeed bring the two catalytic sites closer together (Fig. 5). The binding sites of the CC ensemble are intact after PGK undergoes large structural changes while the binding sites in the Sph ensemble are rearranged. We suggest that, although the distance between the binding sites is reduced in both CC and Sph states, the CC states whose binding sites are structurally intact may be the most biochemically active. The additional activity of wild-type unlabeled PGK may be due to stabilization of CC compared to Sph, relative to the fluorescence-labeled triple mutant.
Our folding kinetics experiments show that domain collapse is not the only large amplitude motion affected by crowding. The measured folding kinetics vary nonmonotonically with crowder concentration, leading to a chevron-like plot with a maximal folding rate observed at a Ficoll concentration of 100 mg/ml (Fig. 2B). Below and above this concentration, the folding/unfolding relaxation time increases. Models of macromolecular crowding have predicted an initial increase in the relaxation rate as crowder concentration is increased, followed by a decrease at even higher crowder concentrations. Based on predictions made by computer simulations on the macromolecular crowding effects on WW folding (13) (Fig. 2B), we rationalize the observed kinetic behavior as follows: Below the optimal crowder concentration, [Ficoll]*, where the folding rate is maximum (i.e., [Ficoll]∗ = 100 mg/ml), the unfolded state becomes conformationally more stable, slowing down the refolding rate; above the optimal crowder concentration, the viscosity increases rapidly, slowing down both the unfolding and refolding reactions. Chemical and crowding effects can obviously compete with one another, but in the case of PGK, simple crowding can explain all the observations (31).
In summary, we have shown that macromolecular crowding profoundly affects the structure of PGK, resulting in the formation of more compact and enzymatically more active structures. This has far-reaching implications, despite Ficoll 70 still being a cytomimetic material and lacking the polydisperse nature of a cell (32)—it suggests that in the crowded interior of a living cell, hinge-bending motions may not be necessary for PGK to carry out its function. Instead, the nucleotide and 1,3-DPG binding domains may already be in close proximity to each other for efficient catalysis to occur. Finally, we showed that macromolecular crowding and viscosity compete with each other, resulting in a uniquely defined concentration at which PGK folding is fastest. A speedup of folding upon crowding was also observed for lysozyme’s “prompt” folding phase (33) (the one relevant here because PGK has no disulfide bridges).
Our combined experimental and computational effort highlights the importance of in vivo studies (34) in addition to crystallographic structure-function relationships and in vitro studies. The states in which PGK protein exists under crowding conditions have structural and kinetic characteristics very different from the state in dilute aqueous buffer (35) or in the crystal. This could turn out to be a general phenomenon for multisubunit (36–38) or partly unstructured proteins: the structural characteristics of the so-called “native state(s)” and how rapidly they fold or unfold will depend sensitively on the nature of their surrounding crowding conditions.
Experimental and Computational Methods
Protein Design and Expression.
We designed a fusion protein consisting of a low-melting mutant (Y122W/W308F/W333F) of phosphoglycerate kinase (PGK) (24), fused between a Green Fluorescent Protein (AcGFP1) donor (D) and an mCherry acceptor (A). AcGFP1 and mCherry are connected to the N and C termini by amino acid linkers consisting of two amino acids each. Unfolding of this protein leads to an increase in the N–C terminal distance, resulting in an increase in donor to acceptor fluorescence ratio (D/A) due to decrease in energy transfer by Förster resonance energy transfer. We expressed the fusion protein in E. coli and purified the construct on a Ni-NTA column as described previously (35). A fusion protein consisting of AcGFP1 tagged to the N terminus of PGK was expressed and purified as described above.
Stability and Kinetics of Crowded Phosphoglycerate Kinase.
For both thermodynamic and kinetic measurements, the protein solution was sandwiched between a cover slip and microscope slide by a 100 μm spacer, resulting in a column density of about 1,000 proteins/μm2. This low density avoids protein–protein interactions while providing sufficient fluorescence for good detection sensitivity with a N/A 0.65 objective in an inverted fluorescence microscope.
Equilibrium titrations were performed by resistive heating of the microscope stage. Temperature was detected by mCherry fluorescence excited at 532 nm, which decreases as temperature increases, and independently calibrated by thermocouples below and above the sample slide. Measuring the equilibrium D/A ratio as a function of temperature yielded protein unfolding curves that were fitted to a sigmoidal model with a linear temperature dependence:
| [1] |
The equilibrium constant is an effective equilibrium constant because PGK is known not to be a two-state folder. We analyzed the melting temperature Tm and the cooperativity parameterΔg1 (negentropy, or first derivative of the free energy at Tm) of the unfolding transition in aqueous solution up to Ficoll concentrations of 200 mg/ml. Δg1 from Eq. 1 was fitted to 1.07 ± 0.16 kJ/mole/°C. The measurement uncertainty was too large to discern a trend (Fig. 2D). The AcGFP1 and mCherry melting points exceed 70 °C, so the observed transition corresponds to melting of the PGK within the construct. Concentrations greater than 200 mg/ml resulted in PGK aggregation at the higher temperatures. 200 mg/ml corresponds to a volume fraction of approximately 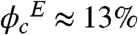 . Thermodynamic measurements in sucrose (Fisher Scientific) were possible up to a concentration of 150 mg/ml; at higher concentrations, the protein started aggregating before yielding a denatured baseline, thus precluding determination of the melting temperature and effective cooperativity.
. Thermodynamic measurements in sucrose (Fisher Scientific) were possible up to a concentration of 150 mg/ml; at higher concentrations, the protein started aggregating before yielding a denatured baseline, thus precluding determination of the melting temperature and effective cooperativity.
Experimental volume fractions of Ficoll 70 (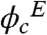 ) were calculated using the partial specific volume of Ficoll, which is 0.67 ml/g (39). We used Ficoll with an average molecular mass of 70 kDa from Sigma-Aldrich. The highest volume fraction achievable in our experiments was
) were calculated using the partial specific volume of Ficoll, which is 0.67 ml/g (39). We used Ficoll with an average molecular mass of 70 kDa from Sigma-Aldrich. The highest volume fraction achievable in our experiments was 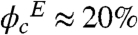 , while the simulations (see below) were carried out at ϕc = 0%, 25%, and 40%, thus allowing us to qualitatively compare the experimental results with the computer simulations.
, while the simulations (see below) were carried out at ϕc = 0%, 25%, and 40%, thus allowing us to qualitatively compare the experimental results with the computer simulations.
Relaxation kinetics were induced by temperature jumps and monitored with 17 ms time-resolution for 15 s. A tailored mid-IR pulse (λ = 2,200 nm) emitted by a diode laser jumped up the temperature of the protein solution from 39 °C to 43 °C, across the thermal unfolding transition. The key advantage of using a programmable diode laser is that the temperature profile can be adjusted to a step function, so the temperature after the jump is constant to within ± 0.25 °C. No sample recooling and backward relaxation occurs. The kinetics were described by a stretched-exponential signal of the form S(t) ∼ e-(t/τ)β, which has been used previously to describe folding kinetics of several PGK mutants (24, 40). The hierarchical stretched-exponential description or “strange kinetics” provides a convenient way of summarizing the intermediate stages of folding with a single parameter β. The precision of the stretched-exponential fits for the time constant τ was ± 0.01 s, while the error bar in Fig. 2B shows the accuracy of our kinetics measurements based on repeated measurement.
PGK Activity Assay.
We measured PGK activity using the coupled reaction with glyceraldehyde-3-phosphate dehydrogenase from the previous step of the glycolytic pathway, as described in detail earlier. NADH consumption was monitored as a function of time by following the absorbance at 340 nm. The rate of NADH consumption was measured at various concentrations of the substrate 3-phosphoglycerate and plotted as a function of substrate concentration to extract the enzyme activity. For purposes of comparison, we assayed the activity of wild-type PGK without any labels. Wild-type PGK and all chemicals required for the assay were purchased from Sigma-Aldrich.
Simulation Details.
The coarse-grained models of PGK proteins, as well as the simulation methods, are provided in the SI Appendix. The thermodynamic properties of PGK were studied at the volume fractions of crowder Ficoll 70 ϕc = 0%, ϕc = 25%, and ϕc = 40% by coarse-grained molecular simulation (by convention, the volume fraction of crowders is presented as a percentage of total volume; i.e., ϕc = 25% corresponds to 0.25 of the total volume). Several represented coarse-grained structures were reconstructed to all-atomistic protein models for illustration by using SCAAL (15, 22).
Structural Characterization of PGK.
To analyze the structural characteristics of PGK, we used four parameters: (i) Overlap function χ is to measure the structural similarity to the crystal structure (21). χ equals 0 for the crystal structure, 1 for highly dissimilar structures. (ii) The radius of gyration Rg is to measure the size of PGK. (iii) Asphericity S and (iv) shape parameters Δ are calculated from the inertia tensor (15). S lies between -0.25 (oblate ellipsoid) and 2 (prolate ellipsoid). Δ lies between 0 (sphere) and 1 (rod). For a sphere, S and Δ are equal to zero.
Binding Sites Distance Calculation.
To calculate the distance between the Mg-ATP and 3PGA binding sites in the characteristic structures of PGK we defined Db to be the distance between the centers of mass of the amino acid residues involved in the two substrate binding sites. The selection of Mg-ATP binding site residues were obtained from ref. 1), while the residues that bind 3PGA were taken from ref. 23).
Supplementary Material
Acknowledgments.
We thank Tripta Mishra for assistance with protein growth and purification. This work was supported by National Science Foundation grants MCB 1019958 (M.G.) and MCB 0919974 (M.S.C.). M.S.C. thanks the computational sources from the Texas Learning and Computation Center and the University of Houston. S.E. was the recipient of a von Humboldt Fellowship. A.D. and S.E. were supported by the NSF Center for the Physics of Living Cells.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 17457.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1006760107/-/DCSupplemental.
References
- 1.Watson HC, et al. Sequence and structure of yeast phosphoglycerate kinase. EMBO J. 1982;1:1635–1640. doi: 10.1002/j.1460-2075.1982.tb01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 3.Blake C. Glycolysis—phosphotransfer hinges in PGK. Nature. 1997;385:204–205. doi: 10.1038/385204a0. [DOI] [PubMed] [Google Scholar]
- 4.Haran G, Haas E, Szpikowska BK, Mas MT. Domain motions in phosphoglycerate kinase: Determination of interdomain distance distributions by site-specific labeling and time-resolved fluorescence energy transfer. Proc Natl Acad Sci USA. 1992;89:11764–11768. doi: 10.1073/pnas.89.24.11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein BE, Michels PAM, Hol WGJ. Synergistic effects of substrate-induced conformational changes in phosphoglycerate kinase activation. Nature. 1997;385:275–278. doi: 10.1038/385275a0. [DOI] [PubMed] [Google Scholar]
- 6.Ellis RJ. Macromolecular crowding: An important but neglected aspect of the intracellular environment. Curr Opin Struc Biol. 2001;11:114–119. doi: 10.1016/s0959-440x(00)00172-x. [DOI] [PubMed] [Google Scholar]
- 7.Zimmerman SB, Minton AP. Macromolecular crowding: Biochemical, biophysical, and physiological consequences. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- 8.Minton AP. Excluded volume as a determinant of macromolecular structure and reactivity. Biopolymers. 1981;20:2093–2120. [Google Scholar]
- 9.Winzor DJ, Wills PR. Molecular crowding effects of linear polymers in protein solutions. Biophys Chem. 2006;119:186–195. doi: 10.1016/j.bpc.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 11.Minton AP. Models for excluded volume interaction between an unfolded protein and rigid macromolecular cosolutes: Macromolecular crowding and protein stability revisited. Biophys J. 2005;88:971–985. doi: 10.1529/biophysj.104.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou HX. Loops, linkages, rings, catenanes, cages, and crowders: Entropy-based strategies for stabilizing proteins. Accounts Chem Res. 2004;37:123–130. doi: 10.1021/ar0302282. [DOI] [PubMed] [Google Scholar]
- 13.Cheung MS, Klimov D, Thirumalai D. Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc Natl Acad Sci USA. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Homouz D, Stagg L, Wittung-Stafshede P, Cheung MS. Macromolecular crowding modulates folding mechanism of alpha/beta protein apoflavodoxin. Biophys J. 2009;96:671–680. doi: 10.1016/j.bpj.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Homouz D, Perham M, Samiotakis A, Cheung MS, Wittung-Stafshede P. Crowded, cell-like environment induces shape changes in aspherical protein. Proc Natl Acad Sci USA. 2008;105:11754–11759. doi: 10.1073/pnas.0803672105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samiotakis A, Wittung-Stafshede P, Cheung MS. Folding, stability and shape of proteins in crowded environments: Experimental and computational approaches. Int J Mol Sci. 2009;10:572–588. doi: 10.3390/ijms10020572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kudlay A, Cheung MS, Thirumalai D. Crowding effects on the structural transitions in a flexible helical homopolymer. Phys Rev Lett. 2009;102:118101. doi: 10.1103/PhysRevLett.102.118101. [DOI] [PubMed] [Google Scholar]
- 18.Homouz D, Sanabria H, Waxham MN, Cheung MS. Modulation of calmodulin plasticity by the effect of macromolecular crowding. J Mol Biol. 2009;391:933–943. doi: 10.1016/j.jmb.2009.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stagg L, Zhang S-Q, Cheung MS, Wittung-Stafshede P. Molecular crowding enhances native structure and stability of alpha/beta protein flavodoxin. Proc Natl Acad Sci USA. 2007;104:18976–18981. doi: 10.1073/pnas.0705127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lonhienne TGA, Reilly PEB, Winzor DJ. Further evidence for the reliance of catalysis by rabbit muscle pyruvate kinase upon isomerization of the ternary complex between enzyme and products. Biophys Chem. 2003;104:189–198. doi: 10.1016/s0301-4622(02)00366-6. [DOI] [PubMed] [Google Scholar]
- 21.Camacho CJ, Thirumalai D. Kinetics and thermodynamics of folding in moldel proteins. Proc Natl Acad Sci USA. 1993;90:6369–6372. doi: 10.1073/pnas.90.13.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samiotakis A, Homouz D, Cheung MS. Multiscale investigation of chemical interference in proteins. J Chem Phys. 2010;132:175101. doi: 10.1063/1.3404401. [DOI] [PubMed] [Google Scholar]
- 23.Harlos K, Vas M, Blake CF. Crystal structure of the binary complex of pig muscle phosphoglycerate kinase and its substrate 3-phospho-D-glycerate. Proteins. 1992;12:133–144. doi: 10.1002/prot.340120207. [DOI] [PubMed] [Google Scholar]
- 24.Osvath S, Sabelko JJ, Gruebele M. Tuning the heterogeneous early folding dynamics of phosphoglycerate kinase. J Mol Biol. 2003;333:187–199. doi: 10.1016/j.jmb.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Gruebele M, Sabelko JJ, Ballew R, Ervin J. Laser temperature jump induced protein refolding. Accounts Chem Res. 1998;31:699–707. [Google Scholar]
- 26.Damaschun G, Damaschun H, Gast K, Zirwer D. Proteins can adopt totally different folded conformations. J Mol Biol. 1999;291:715–725. doi: 10.1006/jmbi.1999.3009. [DOI] [PubMed] [Google Scholar]
- 27.Gast K, et al. Cold denaturation of yeast phosphoglycerate kinase: Which domain is more stable? FEBS Lett. 1995;358:247–250. doi: 10.1016/0014-5793(94)01437-6. [DOI] [PubMed] [Google Scholar]
- 28.Guilbert C, Pecorari F, Perahia D, Mouawad L. Low frequency motions in phosphoglycerate kinase. A normal mode analysis. Chem Phys. 1996;204:327–336. [Google Scholar]
- 29.Balog E, Laberge M, Fidy J. The influence of interdomain interactions on the intradomain motions in yeast phosphoglycerate kinase: A molecular dynamics study. Biophys J. 2007;92:1709–1716. doi: 10.1529/biophysj.106.093195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monterroso B, Minton AP. Effect of high concentration of inert cosolutes on the refolding of an enzyme. J Biol Chem. 2007;282:33452–33458. doi: 10.1074/jbc.M705157200. [DOI] [PubMed] [Google Scholar]
- 31.Ladurner AG, Fersht AR. Upper limit of the diffusion and chain collapse in chymotrypsin inhibitor 2. Nat Struct Biol. 1999;6:28–31. doi: 10.1038/4899. [DOI] [PubMed] [Google Scholar]
- 32.Elcock AH. Models of macromolecular crowding effects and the need for quantitative comparisons with experiment. Curr Opin Struc Biol. 2010;20:196–206. doi: 10.1016/j.sbi.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van den Berg B, Wain R, Dobson CM, Ellis RJ. Macromolecular crowding perturbs protein refolding kinetics: implications for folding inside the cell. EMBO J. 2000;19:3870–3875. doi: 10.1093/emboj/19.15.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dedmon MM, Patel CN, Young GB, Pielak GJ. FlgM gains structure in living cells. Proc Natl Acad Sci USA. 2002;99:12681–12684. doi: 10.1073/pnas.202331299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebbinghaus S, Dhar A, McDonald JD, Gruebele M. Protein folding stability and dynamics imaged in a single living cell. Nat Methods. 2010;7:319–323. doi: 10.1038/nmeth.1435. [DOI] [PubMed] [Google Scholar]
- 36.Arora K, Brooks CL. Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc Natl Acad Sci USA. 2007;104:18496–18501. doi: 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hyeon C, Jennings PA, Adams JA, Onuchic JN. Lingand-induced global transitions in the catalytic domain of protein kinase A. Proc Natl Acad Sci USA. 2009;106:3023–3028. doi: 10.1073/pnas.0813266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyashita O, Onuchic JN, Wolynes PG. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc Natl Acad Sci USA. 2003;100:12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavrenko PN, Mikriukova OI, Okatova OV. On the separation ability of various Ficoll gradient solutions in zonal centrifugation. Anal Biochem. 1987;166:287–297. doi: 10.1016/0003-2697(87)90577-x. [DOI] [PubMed] [Google Scholar]
- 40.Sabelko JJ, Ervin J, Gruebele M. Observation of strange kinetics in protein folding. Proc Natl Acad Sci USA. 1999;96:6031–6036. doi: 10.1073/pnas.96.11.6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.