

Abstract
Phosphonate natural products possess a range of biological activities as a consequence of their ability to mimic phosphate esters or tetrahedral intermediates formed in enzymatic reactions involved in carboxyl group metabolism. The dianionic form of these compounds at pH 7 poses a drawback with respect to their ability to mimic carboxylates and tetrahedral intermediates. Microorganisms producing phosphonates have evolved two solutions to overcome this hurdle: biosynthesis of monoanionic phosphinates containing two P-C bonds or esterification of the phosphonate group. The latter solution was first discovered for the antibiotic dehydrophos that contains a methyl ester of a phosphonodehydroalanine group. We report here the expression, purification, substrate scope, and structure of the O-methyltransferase from the dehydrophos biosynthetic gene cluster. The enzyme utilizes S-adenosylmethionine to methylate a variety of phosphonates including 1-hydroxyethylphosphonate, 1,2-dihydroxyethylphosphonate, and acetyl-1-aminoethylphosphonate. Kinetic analysis showed that the best substrates are tripeptides containing as C-terminal residue a phosphonate analog of alanine suggesting the enzyme acts late in the biosynthesis of dehydrophos. These conclusions are corroborated by the X-ray structure that reveals an active site that can accommodate a tripeptide substrate. Furthermore, the structural studies demonstrate a conformational change brought about by substrate or product binding. Interestingly, the enzyme has low substrate specificity and was used to methylate the clinical antibiotic fosfomycin and the antimalaria clinical candidate fosmidomycin, showing its promise for applications in bioengineering.
Keywords: antibiotics, bioengineering, conformational change, X-ray crystallography, domain-swap
Natural products containing a phosphonate or phosphinate group have drawn increasing interest in recent years as a result of their use in agriculture and medicine (1). The biological activities of some members of this class of compounds stem from their structural similarity to phosphate esters combined with the hydrolytically stable P-C bond. Other members mimic carboxyl groups or the tetrahedral intermediates formed during enzymatic metabolism of these moieties. Recent studies have started to reveal nature’s biosynthetic strategies toward several of these compounds, including the clinically used antibiotic fosfomycin (2–4), the antimalaria clinical candidate FR900098 (5, 6), the antifungal tripeptide rhizocticin (7), and the widely used herbicide phosphinothricin (8–12) (Fig. 1A).
Fig. 1.

Examples of phosphonate natural products. (A) Chemical structures of fosfomycin, fosmidomycin, FR900098, rhizocticin A, and phosphinothricin. (B) Proposed biosynthetic pathway of dehydrophos (19). Biosynthetic steps that have been confirmed either genetically or biochemically are represented in solid arrows and proposed steps are shown in dashed arrows.
A general drawback of phosphonates is their dianionic charge state, which renders them less than ideal mimics of carboxyl groups and tetrahedral intermediates derived from them and limits their bioavailability. Two different strategies are found in nature to overcome these limitations. Phosphinothricin mimics the tetrahedral intermediate generated in Gln synthase (13, 14) by using a monoanionic phosphinate group containing two P-C bonds (Fig. 1A). In an alternative solution, the phosphonate group is esterified in dehydrophos (Fig. 1B), the first such example in a natural product phosphonate (15). The target of dehydrophos is currently not known, but analogous to other peptide antibiotics, it is anticipated that after uptake by a peptide transporter, a peptidase will release the active species (16, 17).
Dehydrophos, originally designated A53868, is produced by Streptomyces luridus and exhibits broad spectrum antibiotic activity against both Gram-negative and Gram-positive bacteria (18). The chemical structure of dehydrophos was recently revised to reveal a vinyl aminophosphonate moiety linked to a Gly-Leu dipeptide (15). The minimal contiguous gene cluster for dehydrophos biosynthesis in S. luridus has been identified (19), showing that the first three steps in the pathway are similar to those for fosfomycin and phosphinothricin, resulting in the formation of 2-hydroxyethyl phosphonate (2-HEP) (Fig. 1B). The remaining steps were proposed based on sequence homology of the gene products to other enzymes of known function and on the observation of accumulated intermediates in pathway mutants (19).
One key transformation in the pathway is the O-methylation of the phosphonic acid group that results in the phosphonate monomethyl ester that is characteristic for dehydrophos not found in any other natural product. This transformation was hypothesized to be carried out by an S-adenosyl-L-methionine (SAM) dependent methyltransferase encoded by dhpI (19). The exact timing of the methyl transfer reaction was less clear because any of the intermediates in Fig. 1B could be the natural substrate for DhpI. Here, we report in vitro characterization of DhpI using synthetic substrate candidates and provide support that the methylation takes place in the final step of the biosynthesis. In order to further elucidate the determinants of substrate recognition, we solved two structures of binary complexes of DhpI, one with the substrate SAM (2.3 Å resolution) and one with the product S-adenosyl-L-homocysteine (1.5 Å resolution). We also present the ternary complex with both SAM and 2-HEP (2.3 Å resolution). The structures reveal an interesting ligand-dependent conformational change that may facilitate product release following methyl transfer. Furthermore, we demonstrate the potential of using DhpI as a versatile catalyst for bioengineering studies because it has broad substrate tolerance and can be used to esterify other biologically active phosphonates.
Results
In Vitro Reconstitution of DhpI Activity.
The dhpI gene was cloned from S. luridus genomic DNA and inserted into the expression vector pET15b. DhpI was heterologously expressed in Escherichia coli with an N-terminal hexahistidine tag (His6-DhpI). Successful purification by immobilized metal affinity chromatography afforded 10–15 mg of purified protein per liter of culture. With purified His6-DhpI in hand, the substrate scope of the enzyme was investigated. As reported for other SAM-dependent methyltransferases (20), the product S-adenosyl-L-homocysteine (SAH or AdoHcy) acted as a product inhibitor. Therefore AdoHcy nucleosidase, which converts SAH to adenine and S-ribosylhomocysteine, was overexpressed and isolated from E. coli (21) and added to the DhpI assays (Scheme S1). The catalytic efficacy of AdoHcy nucleosidase from E. coli (11.6 × 106 M-1 s-1) (21) is much greater than that of most known methyltransferases including DhpI (vide infra) allowing relief of product inhibition.
The phosphonate methyl ester of 2-HEP (2-HEP-OMe) accumulates in the spent growth medium of some of the genetic mutants generated in the heterologous host S. lividans (19). Therefore, we first investigated the ability of DhpI to convert the early biosynthetic intermediates 2-HEP and 1,2-dihydroxyethylphosphonic acid (1,2-DHEP, Fig. 1B) to their monomethyl phosphonate esters. 2-HEP and 1,2-DHEP were chemically synthesized and the rate of formation of the methyl ester products was determined by direct detection using HPLC coupled to atmospheric pressure chemical ionization mass spectrometry (APCI/MS) with synthetic compounds as authentic standards (Fig. S1). Although both 2-HEP and 1,2-DHEP were methylated by DhpI, they proved relatively poor substrates that were unable to saturate the enzyme even at high concentrations. N-Acetyl-1-aminoethylphosphonic acid (Ac-1-AEP or AcAlaP; Fig. 2A) was also converted to the corresponding methyl ester whereas little or no conversion was observed for 1-AEP and 2-AEP. Surprisingly, 1-hydroxyethylphosphonic acid (1-HEP) was a much better substrate (Table S1), and its methyl ester (1-HEP-OMe) was also produced by some of the mutants in S. lividans (19). However, it is difficult to envision a biosynthetic pathway that would include this compound as an intermediate, and 1-HEP-OMe could be a breakdown product of dehydrophos rather than a biosynthetic intermediate. Collectively, these results suggested that a positive charge attached to either C1 or C2 is not tolerated for DhpI activity and that DhpI may favor an amide group and an aliphatic substituent at the α-carbon. Desmethyl dehydrophos 2 satisfies both structural conditions, suggesting methylation might be the last step in the biosynthetic pathway (Fig. 1B).
Fig. 2.

Phosphonates tested as substrates for methylation by DhpI. (A) Chemical structures of various substrate candidates. (B) Formation of (±)-Gly-Leu-AlaP-OMe analyzed by HPLC coupled to APCI/MS. (C) Michaelis–Menten curve obtained by using variable concentrations of Gly-Leu-L-AlaP (open circles) and Gly-Leu-D-AlaP (closed circles) and a fixed SAM concentration of 3 mM.
Desmethyl dehydrophos is not readily available using our previously described synthetic route to dehydrophos (15). Instead, both diastereomers of the hydrogenated analog of desmethyl dehydrophos (Gly-Leu-AlaP, 3, Fig. 2A) were prepared using previously described methodology (15, 22). These peptides incorporate the well known phosphonate analog of alanine, AlaP (23). LC-MS analysis demonstrated that DhpI catalyzed the complete conversion of each of the diastereomeric peptides to their corresponding methylated derivatives (Fig. 2B). Additional transformation to dimethyl phosphonate esters was not observed even after a prolonged reaction time. The kcat/Km values of both diastereomers of Gly-L-Leu-AlaP (3) (14 and 5.9 × 102M-1s-1) were approximately 100-fold larger than that of 1-HEP 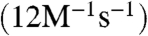 , which displays the highest kcat/Km among the other phosphonic acids tested (Fig. 2C and Table S1), but the enzyme did not strongly differentiate between the two diastereomers of 3. On the other hand, Gly-L-Leu-SerP (4, Fig. 2A), another potential substrate in the biosynthetic pathway as a precursor to the alkene of dehydrophos, was a very poor substrate for DhpI.
, which displays the highest kcat/Km among the other phosphonic acids tested (Fig. 2C and Table S1), but the enzyme did not strongly differentiate between the two diastereomers of 3. On the other hand, Gly-L-Leu-SerP (4, Fig. 2A), another potential substrate in the biosynthetic pathway as a precursor to the alkene of dehydrophos, was a very poor substrate for DhpI.
Cocrystal Structures of DhpI.
Initial crystallographic phases for the DhpI-SAM complex were determined by single wavelength anomalous diffraction data collected on crystals grown from selenomethionine labeled protein (24). The structures of the DhpI-SAH and DhpI-SAM-2-HEP complexes were solved by the molecular replacement method (25). In the DhpI-SAM-2-HEP complex structure, electron density corresponding to the substrate is visible in only one of the four complexes in the crystallographic asymmetric unit. The identity of the ligand as 2-HEP was firmly established through structural analysis of multiple datasets collected from different crystals. Relevant data collection and refinement statistics are given in Table S2. The overall structure of DhpI consists of a core Rossman-fold domain composed of seven β-strands surrounded by six α-helices, with an overall architecture similar to those of other methyltransferases (Fig. 3A) (26). Two augmentations to this core scaffold distinguish DhpI, an amino-terminal extension of 30 residues (Thr3 through Met30) that covers across the SAM-binding site, and an unusual insertion between strand β5 and helix α6 of the Rossman fold (residues Val147 through Ser184) that covers the active site. This latter insertion consists of a short helix (which we refer to as the “capping helix”) followed by two β strands, and residues within this insertion appear to play critical roles in modulating substrate and product binding. While insertions to the core Rossman scaffold are commonly found in the structures of methyltransferases that work on small molecules (27), such insertions typically occur between strands β6 and β7.
Fig. 3.

(A) Ribbon diagram for the overall structure of DhpI monomer with SAM and a sulfate anion. The core methyltransferase domain is shown in pink and the DhpI-specific insertions, consisting of the capping helix and β-hairpin insert, are highlighted in blue. The SAM cofactor is shown in green. (B) Structure of the DhpI dimer showing the domain-swapped interactions between the SAM-binding site of one monomer (shown in pink and blue) and the capping helix and β-hairpin insertion from another monomer (shown in gray and green). (C) Close-up view of the composite active site in the DhpI-SAM-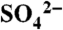 structure. The sulfate anion is shown in orange, residues that contact the sulfate are shown in yellow, and the intersubunit interaction between Tyr29 and Glu155 is shown in yellow and white, respectively.
structure. The sulfate anion is shown in orange, residues that contact the sulfate are shown in yellow, and the intersubunit interaction between Tyr29 and Glu155 is shown in yellow and white, respectively.
One molecule of SAM is bound within the interior of DhpI with the adenine ring enclosed in a hydrophobic cavity defined by Leu12, Tyr15, Leu99, Phe100, and Trp116. Strong electron density corresponding to a sulfate molecule, originating from the crystallization buffer, can be observed adjacent to the methyl group of SAM (Fig. 3C). The sulfate makes contacts with Tyr15, His119, Arg168, and Lys180 (Fig. 3C). These interactions with substrate are recapitulated in the DhpI-SAM-2-HEP cocrystal structure, establishing a role for these residues in substrate binding.
Within the crystal, DhpI forms a homodimer in the presence of SAM and sulfate or phosphonate (Fig. 3B). In the DhpI-SAM-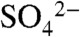 and DhpI-SAM-2-HEP cocrystal structures, the active site of one monomer is enclosed by the capping helix from an adjacent monomer, forming a domain-swapped dimer. Extensive interactions between the 30-residue amino-terminal extension with the capping helix of the adjacent monomer creates a pocket for the methyl group of SAM defined by Tyr15A, Arg18A, and Tyr22A (superscript indicates this residue is from monomer A). Lastly, the side chain of Arg168A, which engages the sulfate in a bidentate fashion, is fixed by the loop formed between residues Gln157B through Ser160B from the adjacent capping helix (Fig. 3C). Structure-based DALI searches (28) against the structural database identified several methyltransferases that are architecturally similar to DhpI (Z-score greater than 15), but none of these structures bears insertions between β5 and α6 that harbor the residues involved in domain swapping, further highlighting the functional role of this structural element. To our knowledge, this is a unique example of a methyltransferase harboring a composite active site composed of residues from multiple subunits.
and DhpI-SAM-2-HEP cocrystal structures, the active site of one monomer is enclosed by the capping helix from an adjacent monomer, forming a domain-swapped dimer. Extensive interactions between the 30-residue amino-terminal extension with the capping helix of the adjacent monomer creates a pocket for the methyl group of SAM defined by Tyr15A, Arg18A, and Tyr22A (superscript indicates this residue is from monomer A). Lastly, the side chain of Arg168A, which engages the sulfate in a bidentate fashion, is fixed by the loop formed between residues Gln157B through Ser160B from the adjacent capping helix (Fig. 3C). Structure-based DALI searches (28) against the structural database identified several methyltransferases that are architecturally similar to DhpI (Z-score greater than 15), but none of these structures bears insertions between β5 and α6 that harbor the residues involved in domain swapping, further highlighting the functional role of this structural element. To our knowledge, this is a unique example of a methyltransferase harboring a composite active site composed of residues from multiple subunits.
Despite the fact that cocrystals with SAM and SAH can be grown under identical conditions, crystals of the DhpI-SAH complex occupy a different space group and diffract to a significantly higher resolution. While the overall structures of the two complexes are similar, the DhpI-SAH complex is monomeric in the crystal, and no electron density can be observed for sulfate in the active site (Fig. 4A). A superposition of the structure of the DhpI-SAH complex with that of DhpI-SAM-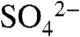 reveals a number of significant rearrangements near the substrate-binding site and at the capping helix (Fig. 4 B and C). While His5 through Leu41 form a continuous helix in the SAH complex (Fig. 4A), in the DhpI-SAM-
reveals a number of significant rearrangements near the substrate-binding site and at the capping helix (Fig. 4 B and C). While His5 through Leu41 form a continuous helix in the SAH complex (Fig. 4A), in the DhpI-SAM-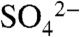 structure residues Val27 through Tyr29 protrude into the active site and adopt a notably different conformation, resulting in a break in this helix (Fig. 3A). In the latter structure, Val27 makes van der Waals contacts with the methyl group of SAM and this interaction facilitates the helix breakage, which is further facilitated by the presence of an adjacent proline (Pro28A). As a consequence, in the DhpI-SAM-
structure residues Val27 through Tyr29 protrude into the active site and adopt a notably different conformation, resulting in a break in this helix (Fig. 3A). In the latter structure, Val27 makes van der Waals contacts with the methyl group of SAM and this interaction facilitates the helix breakage, which is further facilitated by the presence of an adjacent proline (Pro28A). As a consequence, in the DhpI-SAM-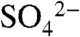 structure, several residues in this region protrude away from the active site to engage in intersubunit interactions, most notably Tyr29A with Glu155B (Fig. 4B) and Ser32A with Arg152B. These interactions facilitate additional intersubunit hydrogen bond interactions that result in a second notable local conformational difference between the two structures. A β-hairpin encompassing residues Val166 through Val179 moves 6 Å toward the active site in the DhpI-SAM-
structure, several residues in this region protrude away from the active site to engage in intersubunit interactions, most notably Tyr29A with Glu155B (Fig. 4B) and Ser32A with Arg152B. These interactions facilitate additional intersubunit hydrogen bond interactions that result in a second notable local conformational difference between the two structures. A β-hairpin encompassing residues Val166 through Val179 moves 6 Å toward the active site in the DhpI-SAM-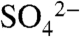 structure, resulting in hydrogen bonding interactions between Arg153A and Asp149B, the main chain oxygens of Glu157B and Asp158B with Arg168A (Fig. 4B), and Glu156A with the main chain oxygen of Gly207B. The last significant difference between the two structures is that in the SAH complex, residues Glu151 through Pro162 that encompass the capping helix are disordered and cannot be unambiguously modeled. In this structure, the continuous amino-terminal helix orients Tyr29A toward the monomer (Fig. 4C), thereby disrupting interactions with Glu155B that would stabilize the capping helix.
structure, resulting in hydrogen bonding interactions between Arg153A and Asp149B, the main chain oxygens of Glu157B and Asp158B with Arg168A (Fig. 4B), and Glu156A with the main chain oxygen of Gly207B. The last significant difference between the two structures is that in the SAH complex, residues Glu151 through Pro162 that encompass the capping helix are disordered and cannot be unambiguously modeled. In this structure, the continuous amino-terminal helix orients Tyr29A toward the monomer (Fig. 4C), thereby disrupting interactions with Glu155B that would stabilize the capping helix.
Fig. 4.

Comparison of cocrystal structures with substrates and product. (A) Ribbon diagram for the overall structure of DhpI-SAH complex. Regions of the polypeptide that undergo a conformational shift, relative to the DhpI-SAM-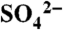 structure are shown in pink. (B) Close-up view of the composite active site in the DhpI-SAM-
structure are shown in pink. (B) Close-up view of the composite active site in the DhpI-SAM-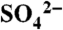 structure showing the interaction between two monomers, colored in pink and green. Active site residues from the pink monomer are shown in yellow and those from the capping helix from the green monomer are shown in green. (C) A close-up view of the equivalent region in the DhpI-SAH structure, showing the reorganization of the active site residues Tyr29 and Arg168.
structure showing the interaction between two monomers, colored in pink and green. Active site residues from the pink monomer are shown in yellow and those from the capping helix from the green monomer are shown in green. (C) A close-up view of the equivalent region in the DhpI-SAH structure, showing the reorganization of the active site residues Tyr29 and Arg168.
Site-Directed Mutagenesis.
In order to investigate the role of putative catalytic residues, several single point mutations were generated of residues that were in close proximity to the sulfate in the SAM cocrystal structure. Substitution of His119, Lys180, and Arg168 with Ala resulted in loss of methylation activity with the tripeptide substrates within the detection limits of our assays. Mutation of Tyr15 to Phe did not abolish activity but did greatly decrease kcat and increased Km, resulting in overall 1,000-fold decrease in catalytic efficiency (Table S1). Thus, all four residues are likely important for binding of the phosphonic acid moiety of the tripeptide substrate. In addition, the importance of two residues that appear to be critical for the disruption of the N-terminal helix in the SAM cocrystal structure and for setting up the sulfate binding pocket was investigated. Mutation of Val27 that makes van der Waals contacts to the methyl group of SAM to Ala had a modest effect, resulting in a fivefold decrease in kcat/Km. On the other hand, mutation of Tyr29, which makes a contact with Glu155 of the other subunit in the SAM and SAM-2-HEP structures, to Phe had a more pronounced effect. The kcat/Km value for the tripeptide substrate decreased about 500-fold compared to the wild-type enzyme (Table S1).
Methylation of Phosphonate Natural Products.
Given the improved pharmacokinetic properties of phosphonate esters compared to phosphonic acids, the substrate scope of DhpI was further investigated. The enzyme was able to fully convert the antimalaria clinical candidate fosmidomycin to its methyl ester (Fig. 5A and Fig. S2). Similarly, the clinically used antibiotic fosfomycin was transformed to its methyl ester by DhpI (Fig. 5B and Fig. S2). The substrate promiscuity of the enzyme also extended to L-(+)-2-amino-4-phosphonobutyric acid (L-AP4, Fig. 2A), an often used nonhydrolizable analog of phosphoSer. Interestingly, phosphoserine itself was also methylated by DhpI, although somewhat less effeciently than L-AP4 (Fig. S3). Hence, the enzyme does not strongly differentiate between a phosphonate and the corresponding phosphate ester. Collectively, these experiments demonstrate the promise of using DhpI or in vitro evolved analogs for bioengineering of esterified phosphonates.
Fig. 5.

Methylation of useful phosphonates. Extracted ion chromatograms showing the complete methylation of fosmidomycin and fosfomycin by DhpI. See Fig. S2 for MS spectra. (A) Fosmidomycin (RT = 5.9, m/z 184) was fully converted to fosmidomycin-OMe (RT = 9.4 min, m/z 198). (B) Fosfomycin (RT = 6.1 min, m/z 139) was fully converted to fosfomycin-OMe (RT = 9.1 min, m/z 153). N-Acetyl aspartic acid (AcAsp) was used as internal standard (IS).
Discussion
PEP mutase (PEPM) installs the P-C bond in all phosphonates for which the biosynthetic gene clusters are currently known. As part of a multidisciplinary program focused on naturally occurring phosphonates we have recently demonstrated that the gene clusters of these compounds can be readily identified using the pepM gene as a marker (3–10). In addition, these studies have shown that phosphonate biosynthesis is widespread and that new clusters that encode for as yet structurally unidentified phosphonates are readily found (1). Given the range of biological activities that known phosphonates display and given their current applications in medicine and agriculture, these unidentified phosphonates may provide a reservoir for new compounds with useful activities. As noted, the bisanionic charge state of phosphonates provides a potential hurdle for their use, a drawback that in synthetic phosphonates is circumvented by esterification. Dehydrophos is currently the only known esterified naturally occurring phosphonate. O-Methylation appears also relatively rare for phosphate esters (29–32), and only very few phosphate methyl transferases have been characterized (33–35). The only phosphate O-methyltransferase with sequence information, a member of the isoprenylcysteine carboxyl methyltransferase (ICMT) superfamily that is involved in the biosynthesis of a modified form of lipopolysaccharide containing a methylated phosphate, does not show homology with DhpI. However, DhpI is shown here to methylate a phosphate with close structural similarity to a phosphonate substrate. Interestingly, the hits with the highest homology with DhpI in a search of the current protein databases are from genomes that do not contain a pepM gene. Hence, the function of these homologs may be to methylate phosphate groups.
In this study we identified tripeptides closely resembling desmethyl dehydrophos (2) as the preferred substrates of the SAM-dependent O-methyltransferase DhpI, strongly suggesting it acts as the last step in the biosynthetic pathway. This observation argues against one alternative role of the methyl ester: as a protective measure to prevent the biosynthetic intermediates from inhibiting endogenous enzymatic reactions in the producing strain. Instead, introduction of the methyl group in the last step suggests that the methyl group is important for biological activity on surrounding organisms. Acetylphosphonate methyl ester, the expected product after hydrolysis of the amide bonds of dehydrophos within the targeted organism, is an 80-fold better inhibitor of pyruvate dehydrogenase than acetylphosphonate itself at pH 7 (36) and has about 1,000-fold higher affinity for the enzyme than the pyruvate substrate (37). Thus, our current hypothesis for methylation is to provide a better mimic of the monoanionic carboxylate group of the pyruvate substrate of pyruvate dehydrogenase. DhpI displayed broad substrate specificity and methylated two clinically important phosphonate natural products. As such, it may prove to be a very useful biocatalyst to prepare methylated analogs of naturally occurring phosphonates by fermentation in engineered organisms.
An examination of the composite active site within DhpI provides a molecular rationale for the tolerance of tripeptides as well as a broad range of natural product substrates. Within the structure, the orientation of Arg168 is stabilized through interactions with residues Gln157B through Ser160B from the capping helix of the adjacent monomer. This crossover interaction creates a binding groove where extended substrates may bind. The DhpI-HEP-SAM cocrystal structure suggests that larger substrates would direct away from the active site to bind along the trajectory of the capping helix. Because nearly all of the residues within this helix are charged (Glu151-RRLEQQDDS-Glu161) (Fig. S4), these residues may also provide an electrostatic interface for the stabilization of the N terminus of the natural tripeptide substrate.
In addition to providing information regarding the biosynthesis of dehydrophos and showing promise for use as a biocatalyst, our work also uncovered mechanistic insights into DhpI catalysis. The three cocrystal structures illustrate a conformational change that is triggered by the presence of both the methyl group of SAM and bound sulfate/phosphonate. Although crystals of both DhpI-SAM and DhpI-SAH complexes could be grown under identical conditions, they exhibit distinct morphologies, occupy different space groups, and demonstrate drastically different diffraction properties. Both crystal morphologies can be observed in crystallization drops that were stored for prolonged period, consistent with the hydrolysis of SAM to SAH in the crystallization buffer after several weeks. Because both the SAM and SAH cocrystals were grown under identical conditions, the presence of sulfate in only the SAM complex argues that the methyl group of SAM instigates the structural rearrangements that are further stabilized by substrate binding. The movement of Val27 in toward the active site in the DhpI-SAM-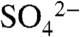 structure provides a solvent-excluded binding pocket for the methyl group and phosphonate that orients the substrates in an optimal orientation for methyl transfer. This movement disrupts the continuous helix containing Val27 that is seen when SAH is present. The energetic penalty for disrupting the helix is compensated by four new intersubunit interactions that stabilize the capping helix. Mutation of Val27 to Ala resulted in only a fivefold decrease in the catalytic efficiency over the wild type, indicating that the van der Waals interaction between Val27 and the methyl group of SAM provides only a limited energetic driving force for the structural rearrangement. It is likely that additional energy may be provided by the electrostatic interaction between the negatively charged sulfate/phosphonate group and the positively charged sulfonium group of SAM. In the SAH structure, interactions between the sulfate/phosphonate and SAH are likely destabilizing, favoring the continuous helix, disrupting the extensive intersubunit interactions, and preventing the stabilization of the sulfate/phosphonate binding pocket, thereby facilitating product release. Additional biochemical studies to further probe this model are in progress.
structure provides a solvent-excluded binding pocket for the methyl group and phosphonate that orients the substrates in an optimal orientation for methyl transfer. This movement disrupts the continuous helix containing Val27 that is seen when SAH is present. The energetic penalty for disrupting the helix is compensated by four new intersubunit interactions that stabilize the capping helix. Mutation of Val27 to Ala resulted in only a fivefold decrease in the catalytic efficiency over the wild type, indicating that the van der Waals interaction between Val27 and the methyl group of SAM provides only a limited energetic driving force for the structural rearrangement. It is likely that additional energy may be provided by the electrostatic interaction between the negatively charged sulfate/phosphonate group and the positively charged sulfonium group of SAM. In the SAH structure, interactions between the sulfate/phosphonate and SAH are likely destabilizing, favoring the continuous helix, disrupting the extensive intersubunit interactions, and preventing the stabilization of the sulfate/phosphonate binding pocket, thereby facilitating product release. Additional biochemical studies to further probe this model are in progress.
Materials and Methods
Expression and Purification of DhpI.
E. coli Rosetta2 (DE3) cells harboring the DhpI-encoding plasmid were grown in a Luria–Bertani (LB) medium supplemented with antibiotic markers (100 μg/mL ampicillin and 20 μg/mL chloramphenicol) at 37 °C until OD600 reached 0.5–0.7. The expression of N-terminally His6-tagged DhpI was induced by addition of isopropyl-1-thio-β-D-galactoside (IPTG) to 0.5 mM and the culture was grown at 18 °C for 15 h. The protein was purified by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography (Qiagen, Valencia, CA). A detailed description of the protein purification and mutagenesis methods used in this study can be found in the SI Appendix and Table S3.
DhpI Activity Assay.
Enzyme kinetic assays were carried out at 30 °C in 50 mM Tris·HCl (pH 7.8) in 1.5 mL reaction vials. The reaction mixture contained 3 mM SAM, 2–24 μM DhpI, 1 μM AdoHcy nucleosidase, and 30–2000 μM phosphonate substrates in a final volume of 300 μL. After adding DhpI, the reaction mixture was preincubated at 30 °C for 3 min before the reaction was initiated by addition of SAM. For quantification, 30 μL aliquots of the assay mixture were removed at designated time points and quenched with an equal volume of 0.1% formic acid containing 200 μM N-acetyl aspartate (AcAsp) as internal standard for quantification during LC-MS analysis. The assay sample was injected to a Synergi C18 RP-fusion column (150 mm × 4.6 mm, Phenomenex) on an HPLC system (Agilent Technologies 1200 series) equipped with a multimode electrospray ionization/APCI spray chamber. HPLC parameters were as follows: column temperature 25 °C; solvent A, 0.1% formic acid in water; solvent B, methanol; for monomeric phosphonates: isocratic mobile phase of 100% solvent A, for tripeptide-like phosphonates: gradient from 100% A to 30% B over 13 min; flow rate, 0.5 mL/ min; detection by APCI/MS operating in positive-ion mode.
MS Data Processing and Kinetic Analysis.
For each sample, the eluting fractions from HPLC were ionized by APCI and the mass spectrum was detected in positive-ion mode. Relative intensities were determined by integrating the selected ion chromatogram for a specific ion across the entire elution profile. Quantification of each analyte was achieved by constructing a standard curve of chemically synthesized authentic sample. Each signal was normalized with a response factor (i.e., ratio of the integrated area of the analyte peak to the integrated area of the internal standard, AcAsp) to account for variations arising from the instrument and sampling conditions. The initial rates of the enzyme reactions were plotted for at least eight varying concentrations of substrates and fitted to the Michaelis–Menten equation using nonlinear regression (KaleidaGraph program, ver.3.5) to estimate the apparent steady-state rate constants.
All of the assays used for determination of kcat and Km were performed in duplicate.
X-ray Crystallization and Structure Determination.
Cocrystals of all complexes were grown by the hanging drop vapor diffusion method under similar conditions. Typically, 1 μL of the protein-ligand complex (25 mg/mL) was mixed with 1 μL of precipitant solution containing 1.6 M ammonium sulfate, 0.1 M Tris-HCl, pH = 8.0 and the drop was equilibrated over a well containing the same precipitant solution at 8 °C. Selenomethionine-incorporated DhpI was crystallized under the same condition except the protein concentration was decreased to 20 mg/mL. Cocrystals were step-wise equilibrated with incremental concentrations of glycerol up to a final concentration of 30% prior to vitrification in liquid nitrogen. For data collection and model building, see SI Appendix.
The structure of the DhpI-SAH complex was determined to 1.5 Å resolution by molecular replacement using the refined coordinates of the DhpI structure as a search probe (25). Multiple rounds of manual model building using XtalView were interspersed with refinement using REFMAC5 to complete structure refinement. Despite extensive manual rebuilding and refinement, residues in the capping loop region, bridging Glu151 through Glu161, remain ill defined and have not been modeled in the structure. As a consequence of the mobility of the residues in the capping loop, their contribution to scattering is minimal as reflected by the fact that the free R factor of 22.1% for this structure is within the range of expectations for a 1.5 Å resolution structure. For each of the structures, stereochemistry of the model was monitored throughout the course of refinement using PROCHECK (38).
Preparation of Phosphonate Substrates.
1,2-DHEP, 1,2-DHEP-OMe, 1-HEP, 1-HEP-OMe, 2-HEP, and 2-HEP-OMe were synthesized according to literature procedures (19). The synthesis and characterization of Gly-Leu-AlaP and Gly-Leu-AlaP-OMe are described in the SI Appendix. Commercially available 1-AEP, 2-AEP, fosfomycin and fosmidomycin were obtained from Sigma-Aldrich (St. Louis, MO) and L-AP4 was purchased from Tocris Bioscience (Ellisville, MO); these compounds were used without further purification. 1-AEP was acetylated using acetic anhydride to prepare Ac-1-AEP.
Supplementary Material
Acknowledgments.
We thank John E. Cronan (Department of Microbiology, University of Illinois at Urbana-Champaign) for the SAH nucleosidase expressing E. coli strain, Amanda Brunner for help with purification of mutants of DhpI, and David Zhang for assistance in crystallization experiments. This work was supported by the National Institutes of Health (P01 GM077596 to W.W.M., S.K.N., and W.A.v.d.D.). M.K. was supported by a postdoctoral fellowship from the Swiss National Science Foundation. B.T.C. was supported by a National Institutes of Health Chemistry–Biology Interface Training Program (GM070421).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates for the refined structures have been deposited in the Protein Data Bank, www.rcsb.org [accession nos. 3OU2 (DhpI-SAH), 3OU6 (DhpI-SAM-sulfate), and 3OU7 (DhpI-SAM-2-HEP)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1006848107/-/DCSupplemental.
References
- 1.Metcalf WW, van der Donk WA. Biosynthesis of phosphonic and phosphinic acid natural products. Annu Rev Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seto H, Kuzuyama T. Bioactive natural products with carbon-phosphorus bonds and their biosynthesis. Nat Prod Rep. 1999;16:589–596. doi: 10.1039/a809398i. [DOI] [PubMed] [Google Scholar]
- 3.Woodyer RD, Li G, Zhao H, van der Donk WA. New insight into the mechanism of methyl transfer during the biosynthesis of fosfomycin. Chem Commun. 2007:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 4.Woodyer RD, et al. Heterologous production of fosfomycin and identification of the minimal biosynthetic cluster. Chem Biol. 2006;13:1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Eliot AC, et al. Cloning, expression, and biochemical characterization of Streptomyces rubellomurinus genes required for biosynthesis of antimalarial compound FR900098. Chem Biol. 2008;15:765–770. doi: 10.1016/j.chembiol.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johannes TW, et al. Deciphering the late biosynthetic steps of antimalarial compound FR-900098. Chem Biol. 2010;17:57–64. doi: 10.1016/j.chembiol.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Biosynthesis of rhizocticins, antifungal phosphonate oligopeptides produced by Bacillus subtilis ATCC6633. Chem Biol. 2010;17:28–37. doi: 10.1016/j.chembiol.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blodgett JA, et al. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat Chem Biol. 2007;3:480–485. doi: 10.1038/nchembio.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao Z, et al. Biosynthesis of 2-hydroxyethylphosphonate, an unexpected intermediate common to multiple phosphonate biosynthetic pathways. J Biol Chem. 2008;283:23161–23168. doi: 10.1074/jbc.M801788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cicchillo RM, et al. An unusual carbon-carbon bond cleavage reaction during phosphinothricin biosynthesis. Nature. 2009;459:871–874. doi: 10.1038/nature07972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitteck JT, Cicchillo RM, van der Donk WA. Hydroperoxylation by hydroxyethylphosphonate dioxygenase. J Am Chem Soc. 2009;131:16225–16232. doi: 10.1021/ja906238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JH, Evans BS, Li G, Kelleher NL, van der Donk WA. In vitro characterization of a heterologously expressed nonribosomal peptide synthetase involved in phosphinothricin tripeptide biosynthesis. Biochemistry. 2009;48:5054–5056. doi: 10.1021/bi900164d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leason M, Cunliffe D, Parkin D, Lea PJ, Miflin BJ. Inhibition of pea leaf glutamine synthetase by methionine sulfoximine, phosphinothricin, and other glutamate analogs. Phytochemistry. 1982;21:855–857. [Google Scholar]
- 14.Tachibana K, Watanabe T, Sekizawa Y, Takematsu T. Action mechanism of bialaphos. I. Inhibition of glutamine synthetase and quantitative changes of free amino acids in shoots of bialaphos-treated Japanese barnyard millet. Nippon Noyaku Gakkaishi; J Pestic Sci. 1986;11:27–31. [Google Scholar]
- 15.Whitteck JT, et al. Reassignment of the structure of the antibiotic A53868 reveals an unusual amino dehydrophosphonic acid. Angew Chem Int Ed. 2007;46:9089–9092. doi: 10.1002/anie.200703810. [DOI] [PubMed] [Google Scholar]
- 16.Allen JG, Havas L, Leicht E, Lenox-Smith I, Nisbet LJ. Phosphonopeptides as antibacterial agents: Metabolism and pharmacokinetics of alafosfalin in animals and humans. Antimicrob Agents Chemother. 1979;16:306–313. doi: 10.1128/aac.16.3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atherton FR, et al. Phosphonopeptides as substrates for peptide transport systems and peptidases of Escherichia coli. Antimicrob Agents Chemother. 1983;24:522–528. doi: 10.1128/aac.24.4.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson RD, Kastner RM, Larsen SH, Ose EE. 4,463,092. US Pat. 1984
- 19.Circello BT, Eliot AC, Lee J-L, van der Donk WA, Metcalf WW. Molecular cloning and heterologous expression of the dehydrophos biosynthetic gene cluster. Chem Biol. 2010;17:402–411. doi: 10.1016/j.chembiol.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendricks CL, Ross JR, Pichersky E, Noel JP, Zhou ZS. An enzyme-coupled colorimetric assay for S-adenosylmethionine-dependent methyltransferases. Anal Biochem. 2004;326:100–105. doi: 10.1016/j.ab.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Choi-Rhee E, Cronan JE. A nucleosidase required for in vivo function of the S-adenosyl-L-methionine radical enzyme, biotin synthase. Chem Biol. 2005;12:589–593. doi: 10.1016/j.chembiol.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Soroka M, Zygmunt J. Tritylamine (Triphenylmethylamine) in organic-synthesis. 1. The synthesis of N-(Triphenylmethyl)alkanimines, 1-(Triphenylmethylamino)alkylphosphonic esters, and 1-Aminoalkylphosphonic acids and esters. Synthesis. 1988:370–375. [Google Scholar]
- 23.Hanson JE, Kaplan AP, Bartlett PA. Phosphonate analogues of carboxypeptidase A substrates are potent transition-state analogue inhibitors. Biochemistry. 1989;28:6294–6305. doi: 10.1021/bi00441a022. [DOI] [PubMed] [Google Scholar]
- 24.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin JL, McMillan FM. SAM (dependent) I AM: The S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 27.Singh S, et al. Structure and mechanism of the rebeccamycin sugar 4′-O-methyltransferase RebM. J Biol Chem. 2008;283:22628–22636. doi: 10.1074/jbc.M800503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holm L, Sander C. Dali: A network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 29.Gabel CA, Costello CE, Reinhold VN, Kurz L, Kornfeld S. Identification of methylphosphomannosyl residues as components of the high mannose oligosaccharides of Dictyostelium discoideum glycoproteins. J Biol Chem. 1984;259:13762–13769. [PubMed] [Google Scholar]
- 30.Kates M, Moldoveanu N, Stewart LC. On the revised structure of the major phospholipid of Halobacterium salinarium. Biochim Biophys Acta. 1993;1169:46–53. doi: 10.1016/0005-2760(93)90080-s. [DOI] [PubMed] [Google Scholar]
- 31.Singh R, Reddy R. Gamma-monomethyl phosphate: A cap structure in spliceosomal U6 small nuclear RNA. Proc Natl Acad Sci USA. 1989;86:8280–8283. doi: 10.1073/pnas.86.21.8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Que-Gewirth NL, et al. A methylated phosphate group and four amide-linked acyl chains in leptospira interrogans lipid A. The membrane anchor of an unusual lipopolysaccharide that activates TLR2. J Biol Chem. 2004;279:25420–25429. doi: 10.1074/jbc.M400598200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freeze HH, Hindsgaul O, Ichikawa M. A novel pathway for phosphorylated oligosaccharide biosynthesis. Identification of an oligosaccharide-specific phosphate methyltransferase in dictyostelium discoideum. J Biol Chem. 1992;267:4431–4439. [PubMed] [Google Scholar]
- 34.Shimba S, Reddy R. Purification of human U6 small nuclear RNA capping enzyme. Evidence for a common capping enzyme for gamma-monomethyl-capped small RNAs. J Biol Chem. 1994;269:12419–12423. [PubMed] [Google Scholar]
- 35.Boon Hinckley M, et al. A Leptospira interrogans enzyme with similarity to yeast Ste14p that methylates the 1-phosphate group of lipid A. J Biol Chem. 2005;280:30214–30224. doi: 10.1074/jbc.M506103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Brien TA, Kluger R, Pike DC, Gennis RB. Phosphonate analogues of pyruvate. Probes of substrate binding to pyruvate oxidase and other thiamin pyrophosphate-dependent decarboxylases. Biochim Biophys Acta. 1980;613:10–17. doi: 10.1016/0005-2744(80)90186-2. [DOI] [PubMed] [Google Scholar]
- 37.Kluger R, Pike DC. Active site generated analogues of reactive intermediates in enzymic reactions. Potent inhibition of pyruvate dehydrogenase by a phosphonate analogue of pyruvate. J Am Chem Soc. 1977;99:4504–4506. doi: 10.1021/ja00455a052. [DOI] [PubMed] [Google Scholar]
- 38.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.