

Abstract
Characterizing the ionic distribution around chromatin is important for understanding the electrostatic forces governing chromatin structure and function. Here we develop an electrostatic model to handle multivalent ions and compute the ionic distribution around a mesoscale chromatin model as a function of conformation, number of nucleosome cores, and ionic strength and species using Poisson-Boltzmann theory. This approach enables us to visualize and measure the complex patterns of counterion condensation around chromatin by examining ionic densities, free energies, shielding charges, and correlations of shielding charges around the nucleosome core and various oligonucleosome conformations. We show that: counterions, especially divalent cations, predominantly condense around the nucleosomal and linker DNA, unburied regions of histone tails, and exposed chromatin surfaces; ionic screening is sensitively influenced by local and global conformations, with a wide ranging net nucleosome core screening charge (56–100e); and screening charge correlations reveal conformational flexibility and interactions among chromatin subunits, especially between the histone tails and parental nucleosome cores. These results provide complementary and detailed views of ionic effects on chromatin structure for modest computational resources. The electrostatic model developed here is applicable to other coarse-grained macromolecular complexes.
Introduction
Understanding chromatin organization and its control of gene expression represents a fundamental open biological challenge. Eukaryotic transcription can only proceed when the DNA is accessible, which is influenced by physiological salts and regulated by a complex network of chemical modifications, chromatin remodeling, and interactions with regulatory protein factors. Because chromatin structure is highly charged, electrostatic interactions play a major role in chromatin folding. For example, acetylation of all H4 histone tails at lysine 16, a prevalent epigenetic modification in eukaryotes, causes charge reduction and, in turn, inhibits the formation of compact 30-nm chromatin fibers and hampers the formation of cross-fiber interactions (1). Such covalent modifications can dictate the higher-order chromatin structure and orchestrate the recruitment of enzyme complexes to access the DNA (2,3).
Many experimental studies have shown that chromatin folding states are profoundly influenced by its ionic environment. In vitro studies of oligonucleosome folding have demonstrated that the compact 30-nm chromatin fiber can form in the presence of ∼0.001 M divalent salt, whereas even elevated levels of monovalent salt (∼0.1 M) attain less compact fibers (4–6). Moreover, higher concentrations of divalent salt (>0.002 M) can induce self-association of nucleosome arrays (7) and stacking of nucleosome core particles without DNA linkers to form dense lamellar and hexagonal phases (8). These phenomena underscore the strong dependence of chromatin organization on the concentration and valence of ionic species. Despite the importance of ionic screening for chromatin organization, no systematic theoretical research has been performed to characterize the relative effectiveness of the charge shielding patterns around chromatin structures. This situation is partly due to the absence of atomic-resolution chromatin structures needed for analysis of electrostatics using molecular dynamics or mean-field theories, such as the Poisson-Boltzmann equation (PBE).
Computational studies of chromatin folding have used different strategies to treat ionic interactions. Korolev et al. (9) employed Langevin dynamics with explicit ions to simulate aggregation of isolated nucleosomes under monovalent and multivalent salt conditions. Other studies using coarse-grained chromatin models include Brownian dynamics simulations of DNA unrolling from the nucleosome (10); Monte Carlo (MC) simulations of the dependence on the nucleosome repeat length (11) and stretching of a single chromatin fiber (12); molecular dynamics simulations of the nucleosome core using a coarse-grained force field (13); and Langevin dynamics simulations of the effects of histone tail modifications on nucleosome core interactions (14).
We have been developing a mean-field approach to treat chromatin charge screening by monovalent ions. Specifically, we represent screened interactions between chromatin sites using the Debye-Hückel (DH) approximation with salt-dependent charges, which are optimized to match the atomistic electrostatic potential derived from the nonlinear PBE (15–17). This approach allows efficient sampling of chromatin conformations using physically detailed models that incorporate irregular nucleosome surface (16,17), flexible histone tails (18,19), and variable linker DNA lengths (20,21). Other groups have also used the DH approximation to treat ionic interactions in chromatin folding simulations (11,22). Recently, we applied a first-order approximation approach to treat divalent ion effects by reducing the repulsive interactions among DNA linkers and the persistence length of the linker DNA sequences (23) based on experimental data on DNA (24,25). Applications of this model, in combination with cross-linking experiments that measure internucleosome interaction patterns, suggest that chromatin fibers under physiological conditions can stabilize a hybrid zigzag and solenoid fiber, where bent linker DNAs accent a mostly zigzag conformation with straight linkers (23).
Although experimental and theoretical studies have provided a global understanding of ionic effects on chromatin organization, the patterns of ionic screening in the vicinity of various chromatin subunits have not been analyzed systematically. Such an analysis can help characterize the condensation of ions around chromatin constituents and its dependence on conformation and multivalent ions. Significantly, an analysis of ionic shielding effects could be used to assess the accuracy of current approaches to ionic interactions.
Computational approaches with explicit and implicit ions have been used to compute the ionic atmosphere around chromatin components. A 200-ns all-atom simulation of a nucleosome core particle showed that monovalent cations cluster around the nucleosomal DNA and the acidic patch of the histone proteins (26). All-atom simulations of larger systems with multiple nucleosome cores, DNA linkers, and histone tails remain a challenge. A more computationally feasible approach for large systems is to calculate electrostatic potentials and ionic distributions around biomolecules using mean-field theories such as the PBE (27) and its extensions (28,29); recent applications include assessment of ionic clouds around tRNA (30) and DNA duplexes (28).
Our computation of chromatin's ionic atmosphere is based on a mesoscale oligonucleosome model, Poisson-Boltzmann theory, and a modified DH approximation relating ionic density to the electrostatic potential (27). Our mesoscale oligonucleosome model incorporates all the basic chromatin constituents, including the irregular nucleosome core, linker DNA, histone tails, and linker histone (15,17–19,23,31,32). In particular, we developed the discrete surface charge optimization (DiSCO) method to represent the irregular nucleosome core using surface beads and assigned salt-dependent site charges to reproduce the atomistic electrostatic field from the nonlinear PBE (15,16). Our chromatin model has been extensively tested and validated relative to experimental data as well as applied to investigate the roles of histone tails, linker histone, variable nucleosome repeat lengths, and divalent ions in chromatin folding using MC simulations (18–20,23,32). Full model details are available in the literature (18,19,23,32); the main features are summarized in Materials and Methods and in the Supporting Material.
In this work, we compute ionic densities and screening charges around oligonucleosomes generated from mesoscale MC simulations. The ionic densities are computed using a modified DH approximation relating ionic density to the electrostatic potential. We introduce an exclusion zone in the ionic density function to correct for finite ion size, which relies on the molecular theory of electrolytes (28,29). This modified DH approximation avoids unrealistically high density values in the vicinity of macromolecular charges, as previously noted (30). The mesoscale electrostatic model developed here can be generalized to other large, coarse-grained macromolecular complexes.
Using the developed mesoscale model, our analysis of the nucleosome core and oligonucleosome conformations shows that divalent ions provide an enhanced screening of chromatin structure compared with monovalent salt by forming a dense layer of counterions around nucleosomal and linker DNA, unburied regions of histone tails, and exposed chromatin surfaces (Figs. 1 and 2). For the nucleosome core, monovalent cations form a screening layer near the nucleosomal DNA which is similar to that obtained by more expensive atomistic simulations (26); in contrast, divalent cations form a continuous layer extending significantly beyond the nucleosome surface (Fig. 1). For oligonucleosome folds, we find that charge screening is most effective on the exposed chromatin surfaces and sensitively dependent on local and global chromatin conformations; for example, the net shielding charge near the nucleosome core in a 12-core fiber spans a large range, from 56e to 100e (Fig. 3). Interestingly, our screening charge correlations (or fluctuations) reflect conformational flexibility and interactions among chromatin components (Table S3); for example, the histone tails and nucleosome cores exhibit the largest and smallest charge fluctuations, respectively. In addition, our analysis of chromatin folding trajectories shows that the free energy due to ions fluctuates by ∼10% about the equilibrium value, and an addition of physiological levels (∼0.01 M) of divalent ions lowers the free energy by 20% (Fig. 4 and later in Fig. 6). These findings imply that ionic screening should be dynamically computed during chromatin simulations. Our analyses provide both qualitative and quantitative results of ionic screening of chromatin structure which are complementary to other chromatin and polyelectrolyte modeling studies.
Figure 1.
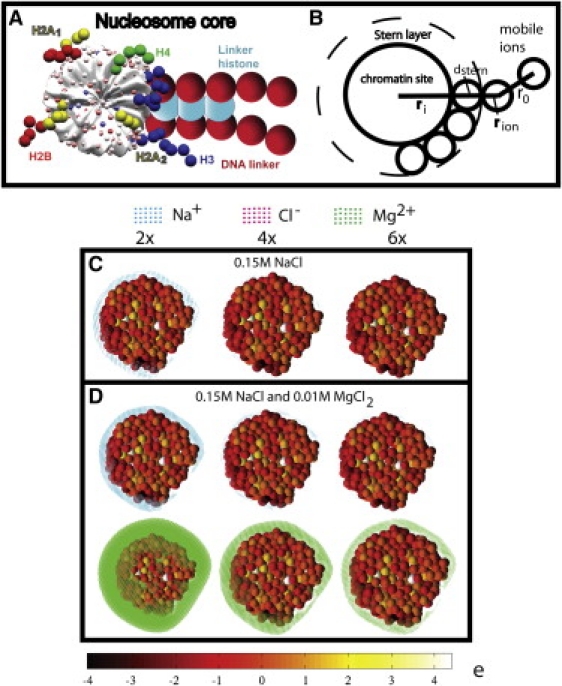
(A) The mesoscale model of the chromatin building block with four components: the nucleosome core (gray); the DNA linker (red); the histone tails H2A1 and H2A2 (yellow), H2B (red), H3 (blue), and H4 (green); and the linker histone (turquoise). The nucleosome core's irregular surface is represented by 300 sites (small colored spheres) whose charges are defined using the DiSCO program. (B) Ion exclusion zone around a chromatin interaction site used for calculating ionic densities. The zone has a thickness of dStern + rion from the surface of the chromatin site, where the Stern layer dStern = 4 Å and ion radius rion = 2 Å for Na+, Cl−, and Mg2+. Shielding charges are calculated within a shell of thickness r0 = 5 Å beyond the exclusion zone. (C) Distributions of Na+ (cyan dots) and Cl– (magenta dots) counterions around the mesoscale nucleosome core particle in 0.15 M NaCl salt at two (left), four (middle), and six (right) times the bulk concentration. The nucleosome core's 300 charges are colored-coded. Ionic charge density regions are shown as colored dots. (D) Distributions of counterions in mixed 0.15 M NaCl and 0.01 M MgCl2 salts at two, four, and six times the bulk concentration of the respective salt: Na+ (cyan dots) and Cl– (magenta) from monovalent salt (upper row); Mg2+ (green), and Cl– (magenta) from divalent salt (lower row).
Figure 2.

Counterion distributions around two 12-core chromatin conformations at 5 (upper row) and 7.5 (lower row) million Monte Carlo (MC) steps sampled at 0.15M NaCl salt condition starting with a zigzag conformation. The chromatin folds (left column), whose cores are labeled 1–12, are shown without their ionic clouds. DNA is shown as a tube (burgundy) wrapped around the histone core (white); other components are color-coded as follows: histone tails (H2A1 and H2A2, yellow; H2B, red; H3, blue; H4, green), linker DNA (gold), and linker histone (LH, turquoise). Ionic clouds at 0.15 M NaCl salt (middle column) and 0.15 M MgCl2 salt (right column) are shown at two times the bulk concentration. For the structures displaying ionic densities (middle and right columns), the nucleosome cores are colored burgundy, and Na+, Cl–, and Mg2+ are shown as cyan, magenta, and green dots, respectively.
Figure 3.
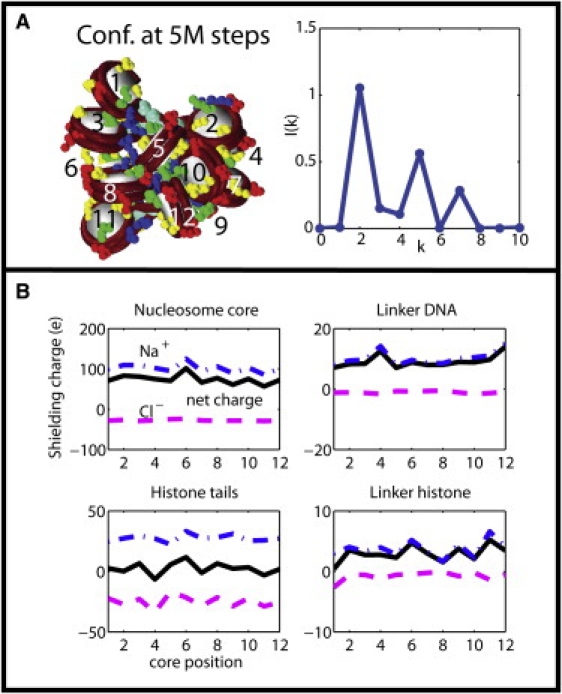
(A) Twelve-core chromatin conformation at 5,000,000 MC steps at 0.15 M NaCl salt with its internucleosome interaction intensity I(k) showing a dominant zigzag feature (peak at k = 2). (B) Shielding charges around the four chromatin components of chromatin structure in panel A. The net (solid line), Na+ (dash-dotted line), and Cl– (dashed line) shielding charges are plotted versus nucleosome core position. (Color figure reproduced in Fig. S5.)
Figure 4.
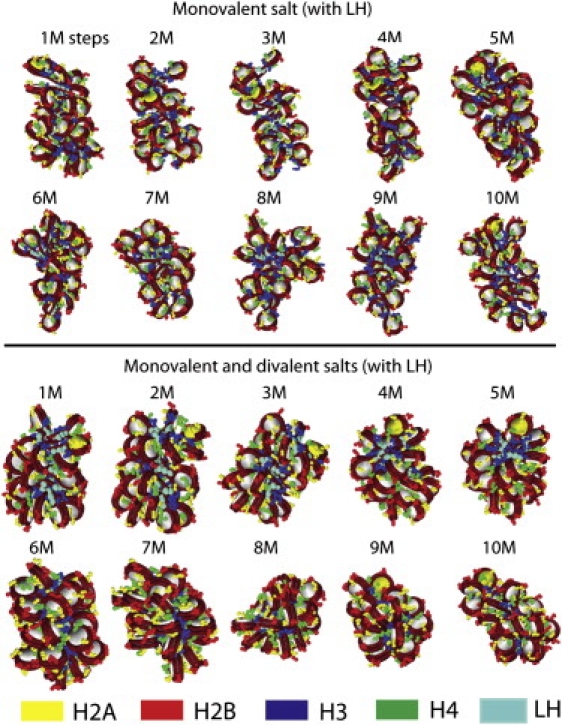
Ten snapshots of chromatin structures from two MC simulations of a 24-core nucleosome array at 0.15 M NaCl salt condition (upper set of structures) and mixed 0.15 M NaCl and MgCl2 salt condition (lower set). The starting configuration is a zigzag structure. (Color figure reproduced in Fig. S6.)
Figure 6.
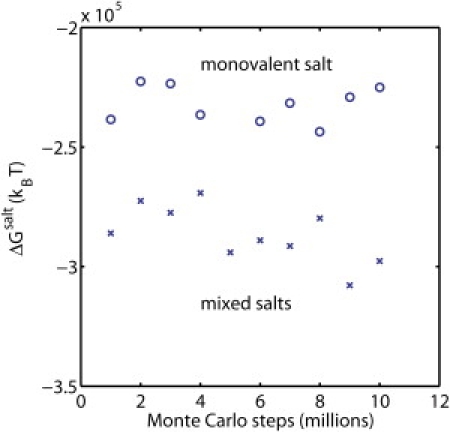
Electrostatic energies due to ionic effects for chromatin structures from two MC trajectories (see Fig. 4) at 0.15 M NaCl salt (○) and mixed 0.15 M NaCl and 0.01 M MgCl2 salts (×).
Materials and Methods
Below we present the mesoscale chromatin model and methods for analyzing ionic densities. In the Supporting Material, we detail methods for sampling chromatin conformations, analysis of charge distribution and fluctuations, and evaluation of internucleosome interaction patterns.
Mesoscale chromatin model
Our mesoscale chromatin model (18,23,32) incorporates the basic chromatin components: irregular nucleosome core, linker DNA, flexible histone tails, and linker histone. As shown in Fig. 1 A, each component is represented using beads or surface beads to coarse-grain protein (histone tails, linker histone) and DNA subunits and the protein/DNA complex (nucleosome core). The chromatin interaction sites are represented as spheres with given radii and charges, which interact via Lennard-Jones and electrostatic interactions; the linker DNA is also modeled with elastic bending and twisting energies. Below, we summarize the mesoscale model described in prior studies (18,32).
The nucleosome core model, based on the nucleosome crystal structure (PDB code 1KX5), is represented by 300 beads of radius 6 Å whose charges are evenly spaced over its nonuniform surface. The salt-dependent surface site charges are calculated using the DiSCO algorithm (16); see Section S1 in the Supporting Material.
The other chromatin components are also modeled as beads. The double-stranded DNA linker connecting two adjacent nucleosome cores is modeled as an elastic wormlike chain of discrete beads (33,34). Each DNA bead has a diameter of 30 Å and a charge of −17.65e based on the number of basepairs spanned by the bead. There are 10 histone tails per core: tails belonging to N- and C-termini of H2A (denoted as H2A1 and H2A2, respectively), H2B, H3, and H4 histones. The H2A1, H2A2, H2B, H3, and H4 histone tails are represented using 4, 3, 5, 8, and 5 beads, respectively, for a total of 50 tail beads per nucleosome to model the 250 or so histone tail residues. All tail beads have a radius of 9 Å but variable charges assigned by the modeling procedures described previously (see Table S4) (18). The linker histone is modeled using three beads to represent the C-terminal (two beads) and globular (one bead) domains of rat H1d linker histone (23). The globular domain bead has a radius of 17 Å and a charge of 8.32e, and the C-terminal beads have a radius of 18 Å and charge of 16.81e.
The model captures the essential structural characteristics and experimental data for macroscopic properties of chromatin, as we showed previously (15,18,23,31,32). In this work, the nucleosome core is represented by 300 sites, linker DNA (62 bp) by six sites, linker histone by three sites, and the 10 histone tails by a total of 50 sites. In the Supporting Material, we summarize the bare charges (i.e., at zero ion concentration) and radii for all chromatin beads in Table S4 and Table S5, respectively; we also provide in Section S5 in the Supporting Material justifications for the model resolution of each chromatin component. Thus, there are 4308 and 8616 interaction sites, respectively, for 12- and 24-core chromatin structures.
We use the mesoscale model to sample chromatin conformations in monovalent and divalent salts using a MC approach (see Section S1 in the Supporting Material). The monovalent salt effect on chromatin interactions is treated via effective salt-dependent charges derived from our DiSCO program (15,16). The divalent salt effect is treated phenomenologically based on experimental studies on DNA bending (24,25), as described in our previous studies (23).
An algorithm for computing ionic densities around mesoscale structures
Our algorithm for computing electrostatics with coarse-grained chromatin structures consists of three steps.
First, we assign the bare charges and radii at all interaction sites of each isolated chromatin component, as described above. The nucleosome's bare charges are assigned using our DiSCO software (15,16) at zero ionic concentration. At zero ionic strength, the DH potential in DiSCO reduces to the Coulomb potential and the assigned nucleosome bare charges simply reflect the mapping of all-atom potential with no screening to the Coulomb potential of the mesoscale model. Thus, the nucleosome's bare charges do not depend on the salt concentration; they are analogous to force-field partial charges of macromolecules which serve as input to a PB solver to compute potentials and ionic densities at given ion concentrations.
Second, we use the mesoscale chromatin model, radii of interaction sites, and bare charges from the first step to compute the electrostatic potential from the nonlinear PBE for a given chromatin conformation and ionic concentration.
Third, the ionic density is computed using a modified DH approximation relating ionic density to the electrostatic potential, as described below. Our methodology for computing electrostatic potentials and ionic densities can be generalized to other large, coarse-grained macromolecular structures whose atomic coordinates are not available.
In the second step, we use the DelPhi software v.4 (35) to obtain the mesoscale electrostatic potential; other PBE solvers, e.g., (36), can be used as well. To use the mesoscale model, we made some changes in DelPhi (i.e., assigning a unique atom type to each interaction site and recompiling DelPhi with larger parameter values for the number of atom and charge types) and its input charges (.crg) and radii (.siz) files. Thus, the input files have the same number of entries as the number of interaction sites. For nucleosome core and 12-core oligonucleosome arrays, we solve the PBE on a 201 × 201 × 201 mesh; the scale or grid/Å is varied (0.1–0.5) depending on the system size. For the larger 24-core arrays, we use a 301 × 301 × 301 mesh. We use standard DelPhi input parameters for temperature (24.85°C), interior dielectric (2), exterior dielectric (78.36), and ion radius of 2 Å (IONRAD parameter), which defines the Stern or ion exclusion layer in DelPhi; the convergence parameter maxc (for maximum change between iterations at any grid point) is set to 0.0001. Generally, chromatin in monovalent salt leads to faster convergence than in divalent salt; see Section S4 in the Supporting Material for a discussion on DelPhi convergence for 24-core arrays.
In the third step, we use the grid potential to calculate the distributions of ions or ionic clouds around a charged macromolecule using a modified DH approximation
| (1) |
where i designates ion type, qi is the ion charge (e.g., ±e, ±2e), ci is the bulk number concentration, and ϕ(r) is the electrostatic potential at position r. The function γ(r) is 1 for the original DH approximation (27), which is also known as the first DH approximation for ionic density (the second DH approximation linearizes the Boltzmann factor to yield the linear PBE). In this work, we compute all ionic densities at various bulk concentrations by using only the modified first DH approximation Eq. 1, with input bare charges that are not dependent on salt concentration. Because the finite size of ions is not accounted for by the nonlinear PBE, it significantly overestimates the ionic density near the charge sites on macromolecules (28,30,37). Based on previously derived ionic density functions from statistical mechanics (28,37), we use a simple step function to describe the ion exclusion zone so that γ(r) becomes
| (2) |
where ri is the radius of chromatin site i, the Stern diameter dStern = 4 Å, and the generic ion radius rion = 2 Å for both monovalent and divalent ions (see Fig. 1 B). Beyond the ion exclusion zone, the distribution of mobile ions is described by the PBE. A similar ion-accessibility function has been used to calculate ionic density for atomic models (30). Because we are using a mesoscale model with larger van der Waals radii (>6 Å), the ionic density is better behaved than for the atomic model where the molecular surfaces are only ∼2 Å from the charge centers.
Results and Discussion
Ionic atmospheres around the nucleosome core in monovalent and divalent salts
Because aggregation of isolated nucleosomes has been a subject of experimental (38) and computational (9) studies, characterizing the ionic atmosphere around the nucleosome core could help clarify the effects of ionic species and concentration on internucleosome association. Fig. 1 C shows the ionic distributions around the mesoscale nucleosome core model in the physiologically relevant 0.15 M NaCl salt. To reveal the charge shielding patterns, we show the local regions with high concentrations of Na+ (cyan) and Cl– (magenta) counterions greater than two, four, and six times the bulk concentration (cb). Due to charge neutralization, the net density is zero beyond the vicinity of the nucleosome core.
As expected, the sodium ions form a charge-shielding layer near the DNA that is wound around the periphery of the core histone proteins. The Cl ions form patches of negligible size near the mostly positively charged regions on the plane of the nucleosome core. The Na+ counterion layer around the nucleosomal DNA strongly depends on the cutoff density. At >2cb, Na+ charges form a thick, continuous layer around the nucleosomal DNA; at greater than four or six times cb, counterions only concentrate around high negatively charged (dark red) regions of the nucleosome core. The corresponding electrostatic potentials (Fig. S4) for these cases show that the most negative potential values are in the vicinity of the nucleosomal DNA, which give rise to increased cation density regions.
Fig. 1 D shows the Na+ (cyan) and Mg2+ (green) distributions around the nucleosome core in a typical physiological mixture of 0.15 M NaCl and 0.01 M MgCl2 salts. The distributions of Na+ at greater than two, four, and six times cb (0.15 M) are similar but not identical to the 0.15 M NaCl case (Fig. 1 C). In contrast, the Mg2+ atmosphere at >2cb (0.02 M) almost completely covers the nucleosome core and extends significantly beyond the nucleosome surface; see also the corresponding potential profile in Fig. S4. Even at greater than four and six times cb, a thick, continuous layer of dense divalent ions is evident around the nucleosomal DNA. Similar results are seen even for low concentrations of 0.001, 0.002, and 0.004 M MgCl2 (see Fig. S3). These results highlight the effectiveness of Mg2+ for shielding the highly charged DNA component of the nucleosome core. Although the qualitative features of the ionic atmosphere around DNA are observed in recent 200-ns all-atom molecular dynamic simulations of the nucleosome core with explicit NaCl salt at 0.15 M (26), our mesoscale computation requires only a fraction of the computational time compared to atomistic simulations. Typically, our computation requires <1 h on a single SGI Origin 3200 processor, whereas a 200-ns simulation of a nucleosome core with explicit water molecules and ions takes over 200,000 CPU hours. Another advantage of our mean-field calculations is that equilibration of ions (typically taking longer than 100 ns) is not an issue.
Experimental studies have shown that divalent ions like Mn2+ bind to the major groves of DNA near GG and GC basepairs (39). Our computed ionic clouds show screening of the phosphate backbone of the nucleosomal DNA and exhibit clusters of high densities (>2 cb) near highly negatively charged DNA regions. However, our mesoscale model does not have sufficient resolution to reveal basepair-specific binding of ions.
Analysis of ionic atmospheres and shielding charges around 12-core oligonucleosome conformations
Unlike the isolated nucleosome core, ionic atmospheres around chromatin components could be influenced by local fiber configurations. To examine this effect and salt concentration dependence, we consider two conformations from a 12-core array simulation for 10,000,000 MC steps starting from a compact zigzag conformation at 0.15 M NaCl salt condition (Fig. 2). Both conformations (at 5 and 7.5 million MC steps) have a compact fold, but the former, with wider distances between the nucleosome cores, shows more disorder than the latter conformation (see core labels 1–12 with no ionic densities). The different histone tails have distinct conformations: the H2A and H2B tails are on the exterior of folded structure, the H3 tails tend to fold into axis of the oligonucleosome structure and interact with the linker DNA, and the H4 tails exhibit properties of both H2A/B and H3 tails (18,19). The linker histones lie in the interior of the structure.
Fig. 2 also shows the clouds of Na+ (middle column) and Mg2+ (right column) around two 12-core conformations at >2 times cb of 0.15 M monovalent or divalent salt. At 0.15 M NaCl, both conformations show shielding of the negatively charged nucleosomal DNA (dark red) and linker DNA (gold) by Na+ counterions which appear as (cyan) bands around the nucleosome cores. Although the positively charged histone tails have irregular conformations, clusters of high density Cl– ions (magenta) can be seen in their vicinity. Charge shielding of the histone tails is less prominent than that for DNA because of the tails' smaller charge magnitudes. In the interior of the oligonucleosome fold, the charge distribution is more complex due to the tight packing of chromatin components with opposite charges (positively charged linker histones and negatively charged linker DNA); finer potential grid is also needed to resolve the distribution's details. Thus, analysis of two 12-core conformations illustrates that counterion screening patterns are determined by the chromatin shape and charge distribution.
For comparison with the monovalent salt case, Fig. 2 (right column) shows the ionic density distributions at 0.15 M MgCl2 salt; the nucleosome core has been observed to partially dissociate near 0.14 M MgCl2 salt (40). The nucleosome cores and unburied regions of histone tails on the surface of both chromatin conformations are almost completely covered by a thick layer of Mg2+ (green) and Cl– (magenta) counterions, respectively. The linker histones (turquoise) and DNA linkers (gold) are also neutralized by their counterion layers as seen near the top and bottom of the structures. The green regions effectively define the shape of the nucleosome core and the magenta regions the histone tail conformations; different histone tails seem to be similarly screened by the Cl– (magenta). This observation implies that the divalent salt provides a much more effective neutralization of the charged oligonucleosome surface regions compared with monovalent salt. Neutralization of oligonucleosome's charged surface reduces repulsive electrostatic interactions between nucleosome arrays, thereby facilitating array self-association in the presence of divalent ions (>0.002 M) as observed in experiments (7); array self-association is also dependent on H3 and H4 tail interactions or tail bridging effect, but not on linker histones (7).
The enhanced effect of divalent ions is characterized by the Boltzmann factor exp(−βqϕ) in the ion density function (Eq. 1), where q is ion charge, β = 1/KBT, and ϕ is the potential function. Thus, divalent ions alter the thermodynamic weight significantly relative to monovalent ions for the same potential and temperature. This ion density behavior can qualitatively account for the experimental observation that a 12-core oligonucleosome folding in 0.001–0.002 M MgCl2 salt can achieve the maximally compact fold of 55S (sedimentation coefficient value of the 30-nm fiber), whereas 0.1 M NaCl salt attains only a less compact ∼40S structure (5).
Distribution and fluctuations of shielding charges around chromatin components
To quantify the salt effects, we compute the shielding charges around nucleosome core, linker DNA, histone tails, and linker histone, which we label as α = 1, 2, 3, and 4, respectively. The shielding charge around a chromatin component α at core position j is computed using the formula
where ρ is the desired ion density (i.e., Na+, Mg2+, Cl–); Sαj is the set of grid points with nonzero density lying within a shell of distance r0 (typically, 5 Å) from the surface of the exclusion zone (see Fig. 1 B and Eq. 2) of a chromatin site; and ΔV denotes the constant local volume defined by the uniform grid (points ri) on which the PBE is solved. An ionic charge on a grid can be assigned to one or more chromatin components, depending on their proximity.
Fig. 3 B (or Fig. S5) displays the Na+ (blue line), Cl– (magenta), and net (black) charges near each chromatin component along the chain of the 12-core conformation at 5,000,000 steps in 0.15 M NaCl salt. Under this salt condition, the internucleosome interaction intensity I(k) for configurations from 1 to 10,000,000 MC steps (see Section S2 in the Supporting Material) has a strong peak at k = 2 and secondary peaks at k = 5 and 7, indicating the structure ensemble has a dominant zigzaglike structure with long-range internucleosome interactions (Fig. 3 A). Fig. 3 B shows that the shielding charge depends on chromatin component and core position. For the nucleosome core, the total Na+ charge varies between +86e and +125e and Cl– charge between −24e and –28e, with a net shielding charge in the range of 56–100e within a shell of = 5 Å wide outside of the exclusion zone of each interaction site. Fluctuations of the shielding Na+ charge around the nucleosome core reflect varying local packing environments in the chromatin fiber. For the negatively charged linker DNA, Na+ ions account for most of the total shielding charges and the amount of Cl– charge is near zero, as expected. On average, the histone tails appear to be surrounded by an equal number of Na+ and Cl– ions because of the interference of Na+ surrounding the parent nucleosome core. A visual inspection of Fig. 2 confirms that the histone tails are indeed surrounded by Cl– counterions. Similarly, the positively charged linker histones have apparent positive shielding charges because of their proximity to the linker DNA and its negative shielding charges.
Fig. S1 compares the concentration dependence of the total shielding charge around the chromatin components for the 12-core conformation at 5,000,000 MC steps in NaCl and MgCl2 salts (Fig. 2). At all concentrations computed (0.05–0.3 M), the total shielding charges are higher for divalent salt ions (Mg2+ and Cl–) than those for monovalent salt ions (Na+ and Cl–) for all chromatin components. Generally, the shielding charges for both monovalent and divalent ions increase/decrease linearly with the salt concentration.
In Section S3 in the Supporting Material, we analyze fractional distributions of shielding charges and their fluctuations to help identify the dominant effects of counterions and their variations due to interactions in the chromatin structure. Essentially, we find that counterions are condensed mainly around the nucleosome core (∼83%) and linker DNA (∼10%). This result depends on the amount of bare charges on these components but is not sensitive to ion concentration (see Table S1 and Table S2). Moreover, as discussed in Section S3 in the Supporting Material, screening charge fluctuations are correlated to the degree of conformational flexibility and interactions of chromatin components. Thus, the histone tails and nucleosome core have the largest and smallest charge fluctuations, respectively (see Table S3), and the ionic atmospheres of tails/parent nucleosome are correlated because of proximity and extensive contacts.
Analysis of chromatin folding trajectories with monovalent and divalent salts
To examine the effects of chromatin structure compaction and fluctuations on condensation of ions, we analyze and compare the folding trajectories of 24-core arrays with linker histone in monovalent and divalent salts. Fig. 4 (or Fig. S6) shows 10 snapshots of 24-core chromatin structures at 1–10,000,000 MC steps starting from a compact zigzag conformation at 0.15 M NaCl salt condition (upper set of structures) and mixed 0.15 M NaCl and MgCl2 salt condition (lower set). In monovalent salt, the nucleosome array displays significant structural fluctuations during MC evolution. In contrast, the oligonucleosome fold is quite stable under mixed monovalent and divalent salts, indicating the effectiveness of divalent salt in stabilizing the zigzag folding pattern over the entire simulation span.
Fig. 5 shows the ionic densities of two 24-core conformations at 10,000,000 MC steps under 0.15 M NaCl salt (top row) and mixed 0.15 M NaCl and MgCl2 salt (bottom row) conditions, accompanied by their chromatin folds (left column) and internucleosome interaction plots (right column). At the monovalent salt condition, the oligonucleosome exhibits a stretched fiber conformation with unpacked nucleosome cores and a prominent peak at k = 2 for nucleosome interactions indicating that the conformations maintain a zigzaglike structure (i.e., alternate nucleosomes are in closest contacts). At mixed salt conditions, a compact fiber forms. The internucleosome interaction plot has a strong peak at k = 2 with enhanced k = 1 contribution; this indicates that the structure ensemble favors a mostly zigzag structure with short-range (solenoidlike) interactions. The prevalence of this hybrid fiber with zigzag and solenoidal features was the subject of our prior work (23).
Figure 5.
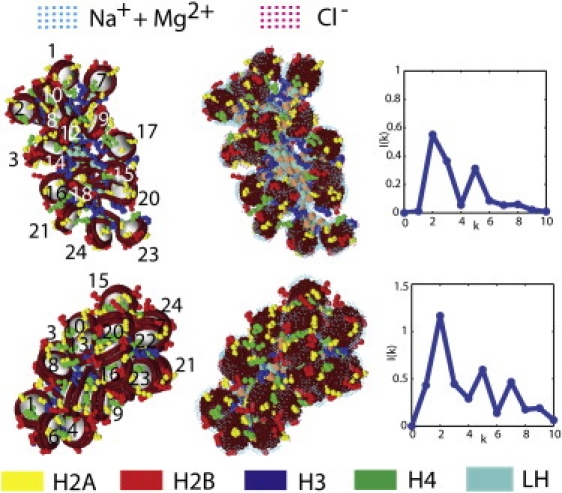
Distributions of counterions around an open (upper row) and compact (lower row) 24-core chromatin conformations from MC simulations at 10,000,000 steps sampled at 0.15 M NaCl salt and mixed 0.15 M NaCl and MgCl2 salts, respectively. (Left column) Chromatin folds with labeled cores (1–24) without their ionic clouds; (middle column) ionic distributions of conformations evaluated at 0.15 M NaCl salt (top) and mixture of 0.15 M NaCl and 0.01 M MgCl2 salts (bottom); (right column) internucleosome interaction intensity I(k) plots. Cation and anion density regions >1.5 times the bulk ionic concentration are displayed. (Color figure reproduced in Fig. S7.)
Shown in Fig. 5 (or Fig. S7) are the cation (cyan) and anion (magenta) densities >1.5cb; for the mixed salts case, the cation and anion densities include contributions from monovalent (0.15 M NaCl) and divalent (0.01 M MgCl2) salts. For the open conformation in NaCl salt, the Na+ ions form a layer around the nucleosomal DNA and linker DNA in the exposed chromatin stem and Cl– ions form clusters near the histone tails, as expected. For the compact conformation in the mixed salt environment, the cations form a layer around the exposed areas of the nucleosome cores to neutralize the negatively charged DNA but the cation condensation layer on the nucleosomal DNA in the interior of the chromatin fiber is much less significant due to tight chromatin compaction.
Next, we analyze the effects of salt on the Gibbs free energy of chromatin conformation as it evolves during MC simulations with monovalent and mixed salts. This energy is computed as the difference between grid energies with and without salt (35,41),
where c1 and c2 are concentrations of salts 1 and 2 (see computing details in Section S4 in the Supporting Material). Fig. 6 shows the energies of 10 structures at intervals of 1,000,000 MC steps for the two salt conditions (Fig. 5). The Gibbs free energy due to salt is negative because the counterions neutralize the charged chromatin structure. The average energies for the monovalent and mixed salt conditions are −232,160 and −286,480 kBT, respectively, showing that an addition of just 0.01 M of divalent salt lowers the free energy by ∼20%. Due to equilibrium conformational fluctuations, the energies for both salt conditions fluctuate by up to 9–13% about their mean values. Lower ion free energy values are associated with open structures where all chromatin components are accessible to ionic screening, whereas higher free energy values are associated with more compact structures. For example, for the monovalent salt case, the distorted, noncompact fiber structures at 4, 6, and 8,000,000 MC steps have lower energies than the fiberlike structures at 2, 3, and 10,000,000 steps.
We also analyzed the dependence of the average shielding charge for the four chromatin components on folding trajectory for the monovalent salt condition (see Fig. S2). Although the average shielding charges are nonuniform, the shielding patterns do not vary significantly during the folding trajectory because most structures remain fiberlike at 0.15 M NaCl salt condition. Shielding charges show greater fluctuations with core position within a structure as the orientations of individual cores can vary greatly (Fig. 3 B).
Our computation of ionic densities and charges is performed using fixed chromatin conformations. In principle, the ionic distribution should be computed self-consistently as the chromatin conformations are sampled during an MC trajectory. This work requires significantly higher computational efforts; conformational changes demand updating of ionic shielding effects, which involves solving the PBE and computing the salt-mediated effective charges (as described in our DiSCO program) as the trajectory evolves (15).
Conclusions
At the time of this writing, work on ionic shielding of the chromatin structure has been limited to analysis of the ionic atmosphere of the nucleosome core particle using atomistic simulations (26). Langevin dynamics has also been used to simulate aggregation behavior of isolated nucleosomes with explicit ions, but the modeling of oligonucleosome is coarse (9). In this article, we developed an approach for analyzing the ionic atmospheres of chromatin structures by using a combination of our previously-developed mesoscale chromatin model (18,23), Poisson-Boltzmann theory, and a modified Debye-Hückel (DH) approximation relating ionic density to electrostatic potential. We have also developed methods for computing shielding charges and their fluctuations in chromatin structure to help analyze the influence of conformational changes and divalent salt on charge screening patterns (see Section S3 in the Supporting Material). These tools for analyzing ionic atmosphere can be applied to any large, coarse-grained macromolecular complexes, including those whose atomic structures are not available.
Our analyses of the ionic atmospheres of the nucleosome core and oligonucleosome conformations show that divalent ions provide an enhanced screening compared with monovalent salt by forming a dense layer of counterions around nucleosomal and linker DNA, unburied regions of histone tails, and exposed chromatin surfaces. The neutralization of chromatin subunits and exposed surfaces promotes chromatin compaction. Charge screening is also sensitive to local and global chromatin conformations (Figs. 2 and 3); for example, the net shielding charge around the nucleosome core in a 12-core fiber spans a large range, from 56e to 100e. Interestingly, our screening charge correlation measure reflects the conformational flexibility and interactions among chromatin components (e.g., large charge correlations measured for histone tails and its interactions with the parent nucleosome core; see Table S3). Additionally, our analysis of chromatin folding trajectories shows that the free energy due to ions fluctuates by ∼10% about the mean value, and an addition of physiological levels (∼0.01 M) of divalent ions can lower the free energy by 20% (Fig. 6). These qualitative and quantitative insights provide a detailed characterization of ionic screening which is consistent with our general understanding of ionic screening in polyelectrolytes and chromatin folding studies. In particular, the dominance of counterion screening of nucleosomal DNA is similar to findings in atomistic simulations (26) and crystallographic experiments on binding of divalent ions (39). Our studies of 12- and 24-core arrays suggest that charge screening by divalent ions is especially effective on the exposed surfaces of chromatin structures. The neutralization of chromatin's surface is likely a prerequisite for array self-association in the presence of divalent ions as observed in experiments (7). Furthermore, the dependence of charge shielding on the local and global chromatin conformations implies that ionic screening should be dynamically computed during chromatin sampling simulations.
Many molecular theories for electrolyte modeling of biological molecules are currently under development (28,30). The major challenge is to account for the effects of finite ion size and ion-ion correlations, which are especially important for obtaining quantitative electrostatic potential fields near charged sites of macromolecules (28,29). These effects are of course included in simulations with explicit ions (9,26), which require much greater computing requirements. Standard Poisson-Boltzmann theory, however, does not account for all these effects, produces unphysical ionic densities near charged molecular sites (28,30), and incorporates only the leading term approximation of multivalent effects with no ion correlation terms, which become significant at elevated multivalent concentrations (42,43). Our simple compromise involves a correction of the DH approximation via an exclusion zone in the ionic density which accounts for finite ion size (Eqs. 1 and 2). In future studies, both the finite ion size and ion correlation effects could be incorporated based on statistical mechanics theory, such as the hypernetted chain equation (29), to better describe ionic atmospheres around macromolecules. Better algorithms for solving the PBE are also needed to obtain electrostatic potentials and energies for large and highly charged oligonucleosome arrays.
Acknowledgments
We thank Ogi Perišić and Rosana Collepardo for generating the chromatin trajectories and Qing Zhang for discussions on the DiSCO program.
This work was supported by National Institutes of Health grant No. R01GM055164.
Supporting Material
References
- 1.Shogren-Knaak M., Ishii H., Peterson C.L. Histone H4–K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Wang G.G., Allis C.D., Chi P. Chromatin remodeling and cancer. Part I: Covalent histone modifications. Trends Mol. Med. 2007;13:363–372. doi: 10.1016/j.molmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Widom J. Physicochemical studies of the folding of the 100 Å nucleosome filament into the 300 Å filament. Cation dependence. J. Mol. Biol. 1986;190:411–424. doi: 10.1016/0022-2836(86)90012-4. [DOI] [PubMed] [Google Scholar]
- 5.Schwarz P.M., Hansen J.C. Formation and stability of higher order chromatin structures. Contributions of the histone octamer. J. Biol. Chem. 1994;269:16284–16289. [PubMed] [Google Scholar]
- 6.Cano S., Caravaca J.M., Daban J.R. Highly compact folding of chromatin induced by cellular cation concentrations. Evidence from atomic force microscopy studies in aqueous solution. Eur. Biophys. J. 2006;35:495–501. doi: 10.1007/s00249-006-0057-7. [DOI] [PubMed] [Google Scholar]
- 7.Schwarz P.M., Felthauser A., Hansen J.C. Reversible oligonucleosome self-association: dependence on divalent cations and core histone tail domains. Biochemistry. 1996;35:4009–4015. doi: 10.1021/bi9525684. [DOI] [PubMed] [Google Scholar]
- 8.de Frutos M., Raspaud E., Livolant F. Aggregation of nucleosomes by divalent cations. Biophys. J. 2001;81:1127–1132. doi: 10.1016/S0006-3495(01)75769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korolev N., Lyubartsev A.P., Nordenskiold L. Computer modeling demonstrates that electrostatic attraction of nucleosomal DNA is mediated by histone tails. Biophys. J. 2006;90:4305–4316. doi: 10.1529/biophysj.105.080226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wocjan T., Klenin K., Langowski J. Brownian dynamics simulation of DNA unrolling from the nucleosome. J. Phys. Chem. B. 2009;113:2639–2646. doi: 10.1021/jp806137e. [DOI] [PubMed] [Google Scholar]
- 11.Stehr R., Kepper N., Wedemann G. The effect of internucleosomal interaction on folding of the chromatin fiber. Biophys. J. 2008;95:3677–3691. doi: 10.1529/biophysj.107.120543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aumann F., Lankas F., Langowski J. Monte Carlo simulation of chromatin stretching. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2006;73:041927. doi: 10.1103/PhysRevE.73.041927. [DOI] [PubMed] [Google Scholar]
- 13.Voltz K., Trylska J., Smith J. Coarse-grained force field for the nucleosome from self-consistent multiscaling. J. Comput. Chem. 2008;29:1429–1439. doi: 10.1002/jcc.20902. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y., Lyubartsev A.P., Nordenskiold L. Computer modeling reveals that modifications of the histone tail charges define salt-dependent interaction of the nucleosome core particles. Biophys. J. 2009;96:2082–2094. doi: 10.1016/j.bpj.2008.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beard D.A., Schlick T. Modeling salt-mediated electrostatics of macromolecules: the discrete surface charge optimization algorithm and its application to the nucleosome. Biopolymers. 2001;58:106–115. doi: 10.1002/1097-0282(200101)58:1<106::AID-BIP100>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q., Beard D.A., Schlick T. Constructing irregular surfaces to enclose macromolecular complexes for mesoscale modeling using the Discrete Surface Charge Optimization (DiSCO) algorithm. J. Comput. Chem. 2003;24:2063–2074. doi: 10.1002/jcc.10337. [DOI] [PubMed] [Google Scholar]
- 17.Sun J., Zhang Q., Schlick T. Electrostatic mechanism of nucleosomal array folding revealed by computer simulation. Proc. Natl. Acad. Sci. USA. 2005;102:8180–8185. doi: 10.1073/pnas.0408867102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arya G., Zhang Q., Schlick T. Flexible histone tails in a new mesoscopic oligonucleosome model. Biophys. J. 2006;91:133–150. doi: 10.1529/biophysj.106.083006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arya G., Schlick T. Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci. USA. 2006;103:16236–16241. doi: 10.1073/pnas.0604817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schlick T., Perisic O. Mesoscale simulations of two nucleosome-repeat length oligonucleosomes. Phys. Chem. Chem. Phys. 2009;11:10729–10737. doi: 10.1039/b918629h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perisic O., Collepardo R., Schlick T. Modeling studies of chromatin fiber structure as a function of DNA linker length. J. Mol. Biol. 2010 doi: 10.1016/j.jmb.2010.07.057. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wedemann G., Langowski J. Computer simulation of the 30-nanometer chromatin fiber. Biophys. J. 2002;82:2847–2859. doi: 10.1016/S0006-3495(02)75627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grigoryev S.A., Arya G., Schlick T. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl. Acad. Sci. USA. 2009;106:13317–13322. doi: 10.1073/pnas.0903280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumann C.G., Smith S.B., Bustamante C. Ionic effects on the elasticity of single DNA molecules. Proc. Natl. Acad. Sci. USA. 1997;94:6185–6190. doi: 10.1073/pnas.94.12.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rouzina I., Bloomfield V.A. DNA bending by small, mobile multivalent cations. Biophys. J. 1998;74:3152–3164. doi: 10.1016/S0006-3495(98)78021-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Materese C.K., Savelyev A., Papoian G.A. Counterion atmosphere and hydration patterns near a nucleosome core particle. J. Am. Chem. Soc. 2009;131:15005–15013. doi: 10.1021/ja905376q. [DOI] [PubMed] [Google Scholar]
- 27.McQuarrie D.A. Harper & Row; New York: 1976. Statistical Mechanics. [Google Scholar]
- 28.Chu V.B., Bai Y., Doniach S. Evaluation of ion binding to DNA duplexes using a size-modified Poisson-Boltzmann theory. Biophys. J. 2007;93:3202–3209. doi: 10.1529/biophysj.106.099168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carnie S.L., Torrie G.M. The statistical-mechanics of the electrical double-layer. Adv. Chem. Phys. 1984;56:141–253. [Google Scholar]
- 30.Gruziel M., Grochowski P., Trylska J. The Poisson-Boltzmann model for tRNA: assessment of the calculation set-up and ionic concentration cutoff. J. Comput. Chem. 2008;29:1970–1981. doi: 10.1002/jcc.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beard D.A., Schlick T. Computational modeling predicts the structure and dynamics of chromatin fiber. Structure. 2001;9:105–114. doi: 10.1016/s0969-2126(01)00572-x. [DOI] [PubMed] [Google Scholar]
- 32.Arya G., Schlick T. A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments. J. Phys. Chem. A. 2009;113:4045–4059. doi: 10.1021/jp810375d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allison S.A., Austin R., Hogan M. Bending and twisting dynamics of short linear DNAs: analysis of the triplet anisotropy decay of a 209 base pair fragment by Brownian simulation. J. Chem. Phys. 1989;90:3843–3854. [Google Scholar]
- 34.Heath P.J., Gebe J.A., Schurr J.M. Comparison of analytical theory with Brownian dynamics simulations for small linear and circular DNAs. Macromolecules. 1996;29:3583–3596. [Google Scholar]
- 35.Rocchia W., Alexov E., Honig B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. B. 2001;105:6507–6514. [Google Scholar]
- 36.Baker N.A. Poisson-Boltzmann methods for biomolecular electrostatics. Methods Enzymol. 2004;383:94–118. doi: 10.1016/S0076-6879(04)83005-2. [DOI] [PubMed] [Google Scholar]
- 37.Vlachy V. Ionic effects beyond Poisson-Boltzmann theory. Annu. Rev. Phys. Chem. 1999;50:145–165. doi: 10.1146/annurev.physchem.50.1.145. [DOI] [PubMed] [Google Scholar]
- 38.Mangenot S., Leforestier A., Livolant F. Phase diagram of nucleosome core particles. J. Mol. Biol. 2003;333:907–916. doi: 10.1016/j.jmb.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 39.Davey C.A., Richmond T.J. DNA-dependent divalent cation binding in the nucleosome core particle. Proc. Natl. Acad. Sci. USA. 2002;99:11169–11174. doi: 10.1073/pnas.172271399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorigo B., Schalch T., Richmond T.J. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 2003;327:85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 41.Sharp K.A., Honig B. Calculating total electrostatic energies with the nonlinear Poisson-Boltzmann equation. J. Phys. Chem. 1990;94:7684–7692. [Google Scholar]
- 42.Hansen J.P., Lowen H. Effective interactions between electric double layers. Annu. Rev. Phys. Chem. 2000;51:209–242. doi: 10.1146/annurev.physchem.51.1.209. [DOI] [PubMed] [Google Scholar]
- 43.Angelescu D.G., Linse P. Monte Carlo simulation of the mean force between two like-charged macroions with simple 1:3 salt added. Langmuir. 2003;19:9661–9668. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.