

Abstract
Mitochondrial transcription factor A (TFAM) is an essential component of mitochondrial nucleoids. TFAM plays an important role in mitochondrial transcription and replication. TFAM has been previously reported to inhibit nucleotide excision repair (NER) in vitro but NER has not yet been detected in mitochondria, whereas base excision repair (BER) has been comprehensively characterized in these organelles. The BER proteins are associated with the inner membrane in mitochondria and thus with the mitochondrial nucleoid, where TFAM is also situated. However, a function for TFAM in BER has not yet been investigated. This study examines the role of TFAM in BER. In vitro studies with purified recombinant TFAM indicate that it preferentially binds to DNA containing 8-oxoguanines, but not to abasic sites, uracils, or a gap in the sequence. TFAM inhibited the in vitro incision activity of 8-oxoguanine DNA glycosylase (OGG1), uracil-DNA glycosylase (UDG), apurinic endonuclease 1 (APE1), and nucleotide incorporation by DNA polymerase γ (pol γ). On the other hand, a DNA binding-defective TFAM mutant, L58A, showed less inhibition of BER in vitro. Characterization of TFAM knockdown (KD) cells revealed that these lysates had higher 8oxoG incision activity without changes in αOGG1 protein levels, TFAM KD cells had mild resistance to menadione and increased damage accumulation in the mtDNA when compared to the control cells. In addition, we found that the tumor suppressor p53, which has been shown to interact with and alter the DNA binding activity of TFAM, alleviates TFAM-induced inhibition of BER proteins. Together, the results suggest that TFAM modulates BER in mitochondria by virtue of its DNA binding activity and protein interactions.
Keywords: TFAM, BER, tumor suppressor, ROS (reactive oxygen species), 8oxoG, OGG1
Introduction
Oxidative stress plays a major role in aging and several diseases of great public health impact, such as cancer, neurodegeneration and diabetes [1]. Under physiological conditions, reactive oxygen species (ROS), a major contributor to oxidative stress, are produced as by-products of aerobic metabolism in the mitochondria of eukaryotic cells. The mitochondrial DNA (mtDNA) sits on the inner side of the mitochondrial inner membrane, where most ROS are generated, rendering it highly susceptible to oxidative damage [2]. The first level of protection to the mtDNA is achieved by the formation of membrane-associated nucleoid-like structures, which are large complexes containing an average of 6 to 10 mtDNA molecules wrapped together by a complex of proteins [3].
A major and essential protein component of the mitochondrial nucleoid core complex is a 29 kDa nuclear-encoded high-mobility group (HMG) family protein called mitochondrial transcription factor A (TFAM) [4, 5]. HMG family proteins are recognized as essential constituents of mammalian chromosomes that play roles in chromatin structure and function [6]. TFAM plays significant roles in mtDNA replication, transcription, and the structure/organization of the mitochondrial nucleoid [7–10]. Furthermore, TFAM binds preferentially to DNA containing 8-oxoguanine (8oxoG) or cisplatin adducts [11, 12] and inhibits repair of a cisplatin adduct in vitro [13], but its role in mitochondrial DNA repair is not characterized.
Mitochondria have a robust level of base excision repair (BER) activity but little or no nucleotide excision repair (NER) activity [14, 15]. Our recent studies suggest that mammalian mitochondria contain enzymes that catalyze mismatch repair (MMR), including YB-1, a protein that can recognize DNA mismatches [16]. Experimental evidence for the presence of other DNA repair pathways in mitochondria is still lacking. Oxidative damage and mutations in mitochondrial DNA are associated with a variety of mitochondrial diseases [17] and aging [18–20], suggesting that loss of DNA repair capacity could contribute to disease onset and/or pathology. Therefore, understanding how mitochondrial proteins modulate mtDNA repair will extend our understanding of the molecular biology behind these pathologies.
Previously, our laboratory reported that BER proteins in mitochondria are localized to the inner-membrane and thus to the nucleoid stuctures [21] where TFAM is also found. Due to the physical co-existence and similarity in substrate (oxidatively damaged DNA) preference, it is possible that they might influence each other. The goal of this study was to explore the possible influence of TFAM on DNA repair in mitochondria. Focusing on BER, the ability of TFAM to bind to DNA lesions repaired by BER and its effect on repair of BER lesions was examined in vitro and in cell based assays. We found that TFAM reduces the activity of all three BER steps investigated, involving DNA glycosylase, AP-endonuclease and DNA polymerase γ in vitro. We further characterized the effects of TFAM KD on mtDNA damage accumulation, cell survival and 8-oxoG incision. We found that TFAM KD cells can accumulate more mtDNA damage, have moderate resistance to high concentrations of menadione and show higher 8OxoG incision than the control cells. To test the hypothesis that DNA binding by TFAM inhibited BER protein access, we generated a mutation in TFAM where a leucine within the HMG box A was replaced by alanine (L58A), to lower its affinity for DNA. L58A TFAM showed a reduced inhibition of 8OxoG incision. Moreover, we found that the tumor suppressor protein p53, a TFAM interacting partner which has been shown to alter TFAM binding to DNA [22], also relieved TFAM inhibition of the incision reaction. Taken together, our results suggest that TFAM binds normal and damaged DNA, limiting access to BER proteins, and that BER activity in mitochondria is affected by the modulation of the TFAM/DNA affinity by interacting proteins. To our knowledge, this is the first demonstration of an integral mechanism that directly modulates DNA repair efficiency in mitochondria.
Material and Methods
Cells
HeLa cells were obtained from ATCC and cultured in complete DMEM medium[Dulbecco's Modified Eagle Medium (Gibco BRL) supplemented with 10% fetal bovine serum, 50 µg/ml streptomycin and 50 U/ml penicillin (where necessary)] at 37 °C with 5% CO2.
siRNA transfection
TFAM protein expression in HeLa cells was transiently reduced with duplex siRNAs obtained from Dharmacon [ON-TARGETplus SMARTpool TFAM (M-019734-00); non-targeting negative control #1 (D-001210-01)]. The cells were seeded in 6-well dishes or 100 mm dishes and grown in DMEM supplemented with 10% FCS (no antibiotic) for 24 h to reach approximately 70% confluence; 50 nM of each siRNA were transfected into the cells using DharmaFECT 1 (Dharmacon), according to the manufacturer’s directions. After 24 h the media was replaced with complete DMEM. The cells were harvested for further use after 48 h of incubation.
Measuring mtDNA damage using PCR: The assay was performed as described by Santos et al [23] with minor changes. The DNA was extracted using the QIAamp DNA blood extraction kit (QIAGEN) and initially quantitated using a nanodrop spectrophotometer (thermo scientific) and then again using the PicoGreen dsDNA Quantitation kit (Molecular Probes). Primers were identical to those previously published [23]. Using the human mitochondrial primer set 14841 (5`-TTT CAT CAT GCG GAG ATG TTG GAT GG-3`) and 5999 (5`- TCT AAG CCT CCT TAT TCG AGC CGA- 3`) we amplified a 8.9 kb product, verified using standard DNA electrophoresis. QPCR was conducted using an Eppendorf Mastercycler. The amount of PCR product was quantitated using Picogreen. The relative amount of initial mtDNA added to the QPCR reaction was normalized using a real time PCR TAQMAN® probe for the mitochondrial gene MTCOX-1 (Applied Biosystems) run on a 7900T fast real time PCR system (Applied Biosystems). Lesion quantitation was calculated using Poisson’s distribution.
Cell viability assays
For the cytotoxicity assays, 30,000 cells were plated in triplicate in 96-well microtiter plates with complete growth medium. The cells were washed in PBS and exposed to menadione (dissolved in DMEM without FBS). A range of doses was used as indicated in the figure legends, and incubated for 4 h. At the end of the treatment, the cells were washed with PBS and incubated in complete media for 18 h. Viability was determined by the ability of cells to reduce the WST-1 dye (Roche); 10 µl of WST-1 was added to each well for 3 h, at which point the absorbance (450 nm – 595 nm) was read.
Plasmid construction, expression, purification and western blotting of recombinant TFAM protein
Bacterial cell line DH5α (genotype: F− Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK−, mK+) phoA supE44 λ− thi-1 gyrA96 relA1) and BL21 DE3 (genotype: F− ompT hsdSB(rB−, mB−) gal dcm (DE3)) (Invitrogen) were used for cloning and expression of recombinant TFAM, respectively. Gateway Technology was used to construct an N-terminal- six-His-tagged human recombinant TFAM expression vector (Invitrogen). The full length human TFAM coding sequence was transferred from the entry vector pENTR™221 to the expression vector pDEST™17 to obtain pDEST17-TFAM according to manufacturer’s protocol. pDEST17-TFAM was further modified by PCR based quick change site directed mutagenesis to construct a pDEST17-TFAMΔC25, a C-terminal truncation mutant and pDEST17-TFAM-L58A mutant, respectively. All the primers used in this procedure are listed in Table 1.
Table 1. Names and sequences of primers used to create TFAM mutants.
Forward and reverse primers used in construction of L58A (leucine to alanine at 58th amino acid in TFAM protein) and ΔC-25 (truncation mutant generated by the removal of 25 amino acids from C-terminal domain of full length TFAM)
| Primer name | Primer Sequence |
|---|---|
| L58A forward | 5′-CCTGTAAGTTCTTACGCTCGATTTTCTAAAGAACAACTACCC-3′ |
| L58A reverse | 5′-GGGTAGTTGTTCTTTAGAAAATCGAGCGTAAGAACTTACAGG-3′ |
| ΔC-25-forward | 5′-P-TAGAACCCAGCTTTCTTG-3′ |
| ΔC-25-reverse | 5′-P-TTGTTCTTCCCAAGACTT-3′ |
Plasmid pDEST™17-TFAM was transformed into competent BL21 (DE3) cells (Novagen) and TFAM expression was induced by the addition of 0.4 mM IPTG. Induced cultures were incubated for 12–14 h at 16 °C while shaking at 225 rpm. Protein was purified by affinity chromatography, using Ni-NTA agarose beads and His-Bind purification kit (Novagen). The resulting eluate was dialyzed against storage buffer (25 mM Tris pH 7.4, 20% glycerol, 1 mM DTT, 0.5 mM EDTA and protease inhibitor cocktail (Roche)), separated into smaller aliquots, and stored at −80 °C until use. Fractions of purified protein were analyzed by SDS-PAGE followed by coomassie blue (Sigma) staining. The protein was judged to be 95% pure by Coomassie staining. Protein purity was further analyzed by western blot analysis using goat anti-TFAM polyclonal antibody (catalog # sc-19050, Santa Cruz Biotechnology) and mouse anti-his6 monoclonal antibody (cat #11922416001, Roche Applied Science) according to manufacturer’s instructions.
Cellular lysate preparation
HeLa cell pellet was washed in PBS and resuspended in a mixture of 100 µl of buffer A (10 mM Tris-HCl, pH 7.8 and 200 mM KCl) and 200 µl of buffer B (10 mM Tris-HCl, 600 mM KCl, 2 mM DTT, 2 mM EDTA, 40% glycerol, 0.2% NP-40, 0.5 mM PMSF) containing protease inhibitors (Roche). Microson Ultrasonic Cell Disruptor was used to sonicate this mixture. Sonication was done three times at power level 3 with a 10 second pulse time separated by one-minute intervals on ice. The sonicated mixture was incubated at 4 °C for 1.5 h on a rocker. The lysed cell mixture was centrifuged at 130000 ×g for 1 hour at 4°C to remove cell debris. The supernatant was dialyzed against buffer C (25 mM HEPES-KOH pH 8.0, 100 mM KCl, 1 mM DTT, 1 mM EDTA, 17% glycerol and 12 mM MgCl2) followed by another round of centrifugation for 10 minutes at 16,000 ×g. Protein concentrations were estimated by Bradford assay and the protein was stored at −80 °C until use.
DNA repair assays
A series of 91-mer DNA oligonucleotides (Midland Inc.) with various site specific single DNA damage adducts were used for in vitro incision assays and are listed in Table 2.
Table 2. Names and sequences of oligonucleotides used in the study.
Con= control oligonucleotide; OG = containing an 8-oxodG; U = containing a uracil; THF = containing a tetrahydrofuran abasic site analog; GAP =containing a single nucleotide gap.
| Name | Sequence |
|---|---|
| Con |
5′-TAATTAATGCTTGTAGGACATAATAATAACAATTGAATGTCT(G)CACA … 3′-ATTAATTACGAACATCCTGTATTATTATTGTTAACTTACAGA(C)GTGT … … GCCACTTTCCACACAGACATCATAACAAAAAATTTCCACCAAAC-3′ … CGGTGAAAGGTGTGTCTGTAGTATTGTTTTTTAAAGGTGGTTTG-5′ |
| OG |
5′-TAATTAATGCTTGTAGGACATAATAATAACAATTGAATGTCT(OG)CACA … 3′-ATTAATTACGAACATCCTGTATTATTATTGTTAACTTACAGA(C)GTGT … … GCCACTTTCCACACAGACATCATAACAAAAAATTTCCACCAAAC-3′ … CGGTGAAAGGTGTGTCTGTAGTATTGTTTTTTAAAGGTGGTTTG-5′ |
| U |
5′-TAATTAATGCTTGTAGGACATAATAATAACAATTGAATGTCT(U)CACA … 3′-ATTAATTACGAACATCCTGTATTATTATTGTTAACTTACAGA(C)GTGT … … GCCACTTTCCACACAGACATCATAACAAAAAATTTCCACCAAAC-3′ … CGGTGAAAGGTGTGTCTGTAGTATTGTTTTTTAAAGGTGGTTTG-5′ |
| THF |
5′-TAATTAATGCTTGTAGGACATAATAATAACAATTGAATGTCT(F)CACA … 3′-ATTAATTACGAACATCCTGTATTATTATTGTTAACTTACAGA(C)GTGT … … GCCACTTTCCACACAGACATCATAACAAAAAATTTCCACCAAAC-3′ … CGGTGAAAGGTGTGTCTGTAGTATTGTTTTTTAAAGGTGGTTTG-5′ |
GAP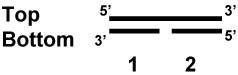
|
5′-TAATTAATGCTTGTAGGACATAATAATAACAATTGAATGTCTGCACA … 3′-ATTAATTACGAACATCCTGTATTATTATTGTTAACTTACAGA( )GTGT … … GCCACTTTCCACACAGACATCATAACAAAAAATTTCCACCAAAC-3′ … CGGTGAAAGGTGTGTCTGTAGTATTGTTTTTTAAAGGTGGTTTG-5′ |
Incision of an adduct present in the oligonucleotide substrate was performed under two different reaction conditions using either a whole cell lysate or purified recombinant proteins. In the former condition, 5 µg of whole cell lysate was incubated with 80 fmoles of 32P-labelled 91-mer, in the presence of 1X incision buffer (70 mM HEPES-KOH pH 7.6, 5 mM EDTA, 2 mM EGTA, 1 mM DTT, 75 mM NaCl, 10% glycerol) in a total reaction volume of 10 µl. Reactions were incubated for 5 h at 37 °C followed by treatment with 10 units of proteinase K (Invitrogen), for 10 minutes at 37 °C. The reactions were terminated by the addition of 10 µl of formamide gel loading dye (80% formamide, 10 mM EDTA, 1 mg/ml xylene cyanol FF, and 1 mg/ml bromophenol blue). The reactions were then heated at 95 °C for 6 minutes followed by electrophoresis on 15 % denaturing polyacrylamide and 7 M urea containing gels to resolve the substrate and product bands.
Purified recombinant proteins were obtained from the following sources: hOGG1 (α-isoform) and UDG were obtained from New England Biolabs, APE1 was obtained from Dr. David Wilson III (NIA). For in vitro incisions with purified components, increasing amounts of purified recombinant TFAM protein was incubated with 80 fmoles of 32P-labelled 91-mer in the presence of 1X EMSA buffer (10 mM Tris-Cl pH 7.4, 20 mM NaCl, 25 mM KCl, 0.5 mM EDTA, 0.5 mM DTT, 5% glycerol) for 15 minutes at 37 °C followed by the addition of the purified recombinant DNA repair protein as noted in figure. This was followed by incubation at 37 °C for 15 minutes. Reactions were stopped by the addition of 15 µl of formamide gel loading dye and heated at 95 °C for 6 minutes. Substrate and product bands were separated by electrophoresis as described above. Products on the gels were visualized by the Typhoon PhosphorImager (GE) and analyzed using ImageQuantTM (Molecular Dynamics). Incision activity was determined as the intensity of product bands relative to the combined intensities of substrate and product bands.
Pol γ was obtained from Dr. William Copeland (NIEHS) and used in a gap filling assay. Oligonucleotide bottom 1 and bottom 2 were duplexed to oligonucleotide top, thereby creating a substrate with a one nucleotide gap (sequences of the oligonucleotides are shown in Table 2). Upon incubation with 0.25 fmoles of Pol γ, in the presence or absence of increasing TFAM protein and 1X EMSA buffer, 32P-CTP incorporation was monitored by polyacrylamide gel electrophoresis followed by autoradiography. The Pol γ activity was quantitated by directly measuring the band intensity using ImageQuantTM (Molecular Dynamics).
Electrophoretic mobility shift assay (EMSA)
Various amounts of purified recombinant TFAM (wild type and mutant) was incubated with 80 fmoles of 32P-labelled control or 8oxoG containing 91-mer DNA substrates in the presence of 1X EMSA buffer (10 mM Tris-Cl pH 7.4, 20 mM NaCl, 25 mM KCl, 0.5 mM EDTA, 0.5 mM DTT, 5% glycerol). The reactions were incubated for 15 min at room temperature and the protein bound DNA substrate was separated from free substrate on a 5% non-denaturing polyacrylamide gel using 1X TBE (89 mM Tris, 89 mM borate, and 2 mM EDTA) by electrophoresis at 4 °C. Electrophoresis was conducted for 2 h at 150 volts and the products were visualized on a Typhoon PhosphorImager (GE) and analyzed using ImageQuantTM (Molecular Dynamics). The percent of DNA bound by the protein was determined by dividing the volume of the shifted band by the total intensity of the DNA in the lane.
DNA strand destabilization assay
Different amounts of TFAM WT and mutant were incubated with 80 fmoles of 32P-labeled 91-mer containing 8oxoG adduct. The reaction was performed in 1X EMSA buffer (10 mM Tris-Cl pH 7.4, 20 mM NaCl, 25 mM KCl, 0.5 mM EDTA, 0.5 mM DTT, 5% glycerol) at 37 °C for 15 minutes. The product and substrate bands were separated on a 10% non-denaturing polyacrylamide gel with 1X TBE buffer at room temperature. Electrophoresis was conducted for 2 h at 150 volts. Products were visualized by autoradiography (as described in EMSA). The percent DNA strand destabilization was determined by the volume of the product band destabilized by the total intensity of the DNA in the lane.
Statistical Analysis
Statistical significance was calculated by conducting unpaired student t-test assuming null hypothesis with sigma plot software. The cutoff for the p-value was set to ≤0.05.
Results
Effect of TFAM on DNA binding in vitro
TFAM binds DNA cooperatively with relaxed sequence specificity [4]. TFAM binds to cisplatin GdpG crosslinks and 8oxoG containing DNA with greater specificity than normal, unmodified DNA [12, 22]. The affinity of TFAM for DNA substrates containing other common lesions such as uracil, abasic site or ssDNA gaps has not been studied. Thus, we tested TFAM binding affinity to a series of 91-nucleotides long DNA substrates with a mitochondrial DNA specific sequence (nucleotides 213 to 303 in the human mitochondrial genome) containing four modifications commonly generated spontaneously and by oxidative stresses 8-oxoG, uracil, AP-site and 1-nt gap (Table 2). In each case, increasing amounts of TFAM were incubated with control (no damage) or a single adduct (8oxoG, abasic-site, gap or uracil) containing substrate. TFAM showed modest specificity to 8oxoG containing DNA compared to normal DNA. TFAM bound 18% more 8oxoG DNA as compared to normal unmodified DNA at the highest concentration (110 fmoles) of the protein used (Fig. 1A and B). Although no significant preference for an abasic site, uracil or gap containing DNA substrates was observed (Fig. 1 C, D and E), TFAM bound efficiently to damage-containing DNA. These results suggested that DNA binding by TFAM could limit the access of DNA repair enzymes. Thus, we asked whether TFAM could modulate the activities of BER protein activities.
Fig. 1.

Differential binding of TFAM to 8oxoG lesion. A) Electrophoretic mobility shift assay showing a typical gel used for analysis of TFAM DNA binding to a 91-mer oligonucleotide containing either an 8oxoG (OG) or normal G (Con). The numbers on top of the panel indicate the amount of TFAM used in fmoles. DNA alone without any protein is represented by the (−) symbol. TFAM binding affinity to 8oxoG (B), Uracil (C), abasic site (D) and gap (E) containing DNA relative to control (con) DNA are shown. Data represents mean ± SD of three independent experiments.
Effect of TFAM on base excision repair proteins in vitro
The ability of TFAM to influence the activity of BER pathway enzymes was examined by conducting a series of in vitro assays in the presence and absence of TFAM. First, we tested the effect of TFAM on the incision activity of OGG1, which initiates BER of 8oxoG. There was a significant dose-dependent inhibition of OGG1 as the concentration of TFAM in the reaction increased. At the highest concentration used (110 fmoles), OGG1 activity was reduced by approximately 50% (Fig. 2A). To address the specificity of this inhibition, we tested the effect of TFAM on UDG, APE1, and pol γ, other proteins involved in the mitochondrial BER process. TFAM showed a similar ability to inhibit the incision activity of UDG, APE1, and incorporation activity of pol γ (Fig. 2B, C, D). At the highest concentration used, TFAM inhibited UDG, APE 1 and pol γ activities by 73, 87 and 45%, respectively. This global inhibition of BER enzymes could indicate that TFAM competitively inhibits access of these enzymes to the DNA substrate in all assays tested, due to its nonspecific DNA binding affinity. To test this possibility, experiments were repeated with a recombinant mutant TFAM that is defective in DNA binding, as described below.
Fig. 2.

Effect of TFAM on BER protein activities. Graphs represent percent of control activity (in absence of TFAM) of OGG1 (A), UDG (B), APE1 (C), and pol γ (D) activities, respectively, in the presence of increasing amounts of TFAM (13.75, 27.5, 55 and 110 fmoles). Inset in each graph show a typical gel image used in the analysis of effect of TFAM on the corresponding BER protein. The lane with (−) symbol represent the negative control with no repair protein and (+) symbol in each case represents the positive control corresponding to the DNA repair enzyme alone with no TFAM. The black triangle indicates the increasing amounts of TFAM. Data are mean ± SD of three independent experiments.
L to A mutation creates a DNA binding and destabilization deficient TFAM mutant
Although previous studies suggested that deletion of the C-terminal 25 amino acids of TFAM reduced its DNA binding activity [24], EMSA experiments using a 32P-labeled 91-mer DNA substrate could not reproduce this result (Supplementary Fig. 1), and a similar observation was reported by Gangelhoff et al. [25]. It is possible that this discrepancy is due to use of different DNA binding assays in the studies; EMSA may not be sensitive enough to detect the effect of the C-terminal deletion on DNA binding. In addition, HMG box A but not HMG box B or C are required for DNA binding [25], and structural analysis of TFAM predicted that the conserved (58th amino acid) intercalating non-polar amino acid in the HMG box A is important for cooperative DNA binding and DNA strand destabilization activities of TFAM. Therefore, to construct a DNA binding-deficient TFAM mutant, a L58A (Leucine to Alanine) mutation was introduced into HMG box A domain of TFAM [25, 26]. The mutant protein was overexpressed and purified from an E.coli strain as described in Materials and Methods. EMSA assays were performed with wild type and the L58A TFAM mutant (Fig. 3A and B). As predicted, L58A TFAM demonstrated lower affinity for DNA than WT TFAM. In reactions with 60 or 120 fmoles TFAM, 12 and 33% less DNA was bound, respectively, than in reactions with WT TFAM (Fig. 3A). As predicted,the L to A mutation disables the intercalation capacity of TFAM, and therefore DNA strand destabilization of a 32P-labeled fork DNA substrate was studied. The fork substrate was incubated with a 60, 120 or 240 fmoles of either wild type (WT) or L to A mutant. At the highest concentration (240 fmoles; Supplementary Fig. 2) of protein used, no DNA destabilization activity was detected in the L to A mutant containing reactions whereas WT TFAM did destabilize the fork substrate. These data demonstrate that L58A TFAM has significantly lower affinity for DNA than WT TFAM.
Fig. 3.

EMSA of TFAM wild type and L58A mutant with 91-mer oligonucleotide. A) Typical EMSA gel used to quantitate the difference between binding affinity of wild type TFAM and L58A mutant. Protein bound fraction is shown by B and unbound free substrate is represented by UB. Lanes 1, 2, 3 and 4 are increasing concentrations of 30, 60, 120 and 240 fmoles of TFAM wild type (WT) and mutant (L58A). Lane 0 represents substrate alone without any protein. B) Data represents the percent mobility shift of protein bound DNA fraction with respect to the concentration of the protein used. Data are mean ± SD of three experiments. The * symbol indicates the p-value for the corresponding concentration of protein used (p-values 0.022 and 0.019, respectively at 30 and 60 fmoles).
The L58A TFAM mutant inhibits OGG1 to a lesser extent than WT TFAM
We predicted that L58A TFAM would inhibit BER enzymes in vitro less than wild type TFAM, due to its reduced affinity for DNA. To test this hypothesis, increasing quantities of WT or L58A TFAM (30, 60, 120, 240 fmoles) were incubated with 80 fmoles 32P labeled substrate containing a single 8oxoG lesion. L58A TFAM inhibited OGG1 incision activity less than WT TFAM (Fig. 4A), such that incision by OGG1 was 10% or 35% higher in reactions with 60 or 120 fmoles of the mutant protein (Fig. 4B) when compared to reactions containg the WT protein. This indicates that the partial reversal in the inhibition due to reduced DNA binding observed in the reactions containing L58A mutant is significant. This is consistent with the relative DNA binding affinities of the mutant and WT TFAM.
Fig. 4.

In vitro incision assay with TFAM wild type and L58A mutant. A) A typical gel used for determining the OGG1 activity in the presence of TFAM wild type (WT) or mutant (L58A) is shown. Lanes 1, 2, 3 and 4 are increasing concentrations of 30, 60, 120 and 240 fmoles of TFAM wild type (WT) and mutant (L58A). Lane (−) represents substrate alone without any protein and (+) represents positive control for the OGG1 protein alone with no TFAM. S and P represent substrate and product bands respectively. B) Graph represents OGG1 activity in the presence of increasing amounts of TFAM wild type (WT) and mutant (L58A). Data are mean ± SD of three independent experiments. The * symbol indicates the p-value for the corresponding concentration of protein used (p-values 0.008 and 0.02, respectively at 30 and 60 fmoles).
Effect of P53 on TFAM DNA binding and OGG1 incision activity
The data described above are consistent with our hypothesis that TFAM inhibits BER enzymes in vitro by competitive binding to the DNA substrate. Because the DNA binding activity is essential for the biological/cellular function of TFAM, we proposed that some cellular factors modulate its DNA affinity or otherwise limit its ability to inhibit DNA repair in vivo. Yoshida et al showed that p53 interacts with TFAM and reduces its DNA binding activity [22]. We therefore hypothesized that p53 may be one factor that modulates TFAM DNA binding affinity, in order to prevent TFAM from inhibiting DNA repair. To test this hypothesis, we first tested the effect of p53 on binding of TFAM to DNA Molar ratios of 1:0, 1:4, 1:8 and 1:16 of TFAM to p53 were used. Using our gel conditions, p53 alone caused the DNA to shift into the well (Fig. 5A, lane 6). When p53 and TFAM were combined with the DNA, we observed a dose dependent shift of the DNA into the well and a loss of the discrete TFAM:DNA bands (Fig. 5A, lanes 3–5). Due to the fact that the p53:DNA complex is shifted into the well, we cannot exclude the possibility that both TFAM and p53 are bound to these DNA molecules.
Fig. 5.

p53 alters TFAM DNA binding and reverses OGG1 inhibition by TFAM. A. A typical gel showing the effect of increasing concentrations of p53 (lanes 3–5 are 50, 100 and 200 ngs of p53, respectively) on TFAM DNA binding by EMSA. The ramp represents the increasing concentrations of p53. B. Upper panel shows a representative gel used to quantitate the OGG1 activity, in the presence of TFAM alone (1:0) and increasing amounts of p53 with respect to TFAM in the molar ratios of 1:4, 1:8 and 1:16. Graph represents the OGG1 activity in the presence of the two variables TFAM and p53. Data are mean ± SD of three independent experiments. The * symbol indicates the p-value of 0.0045 for 1:16 TFAM to p53 used.
Next we evaluated the effect of p53 on OGG1 incision in the presence of TFAM. These assays were performed with 1:0, 1:4, 1:8 and 1:16 molar ratios of TFAM to p53. In the absence of TFAM or p53, OGG1 incised 62% of the substrate (Fig. 5b lane 1). In this experiment, TFAM inhibited OGG1 by approximately 39% (Fig. 5b, lanes 3 versus 2); however, p53 reversed this inhibition in a dose-dependent manner (Fig. 5b lanes 3 versus 4–6). This effect was not evident at the lowest p53 concentration (1:4 TFAM:p53), but became evident at a 1:8 ratio of TFAM:p53. At a 1:16 molar ratio of TFAM:p53, OGG1 activity increased approximately 2-fold (Fig. 5b). Control experiments show that p53 does not stimulate OGG1 activity directly (Supplementary Fig. 3) within the concentration range of protein used in our experiment. These data indicate that p53 stimulates DNA repair in the presence of TFAM, possibly by interacting with and altering its DNA binding activity, thereby making it accessible to OGG1
Effect of TFAM knockdown on damage accumulation in mtDNA
To determine if TFAM knockdown altered the mtDNA damage load, we used the Q-PCR approach. In this assay, a 8.5 kb fragment of mtDNA is amplified and its relative amplification reflects how much damage is present in mtDNA because any polymerase blocking lesion will lower the amount of DNA amplified. When we measured the relative amplification of the 8.5 kb PCR fragment from DNA prepared from scrambled and TFAM siRNA treated cells, TFAM knockdown cells consistently amplified less than scrabled DNA samples (Fig. 6). From the relative amplification values, the number of lesions per 10 Kb were calculated using the Poisons distribution and TFAM knockdown cells had an average of 0.21±0.11 lesions per 10 Kb more than scramble treated cells.
Fig 6.

Effect of TFAM knockdown on damage accumulation in the mitochondrial DNA. Table shows long (8.9kb) PCR amplification from mtDNA obtained from three biological replicates of TFAM knockdown in HELA cells. Ratio of average PCR amplifications (AVG) from Scr (scrambled) to TFAM KD was calculated. This was converted into lesions per 10kb by Poisson’s distribution. Panel A shows the relative amplification of an 8.5-Kb mtDNA fragment from DNA purified from control (Scr) and knockdown (TFAM) cells. Panel B shows the relative amplification of the 8.5 kb PCR product amplified without any contamination.
To investigate if this elevated lesion frequency in TFAM knockdown cells correlated with mtDNA mutations, we performed the chloramphenicol resistance-mitochondrial mutation assay [27]. Cells were subjected to chloramphenicol treatment, which selects for mtDNA mutations within the mitochondrial ribosomal genes. After seven days, the number of surviving colonies was counted. In this assay, the total number of colonies, and correspondingly mtDNA mutations, was slightly higher in TFAM knockdown cells than in the the control cells (data not shown). Together this data suggests that the presence of TFAM is beneficial for the cell and that it can decrease the mtDNA damage load and mutation frequency.
Effect of TFAM knockdown on cell survival in the presence of menadione
Treatment of cells with DNA damaging agents elicits a complex response that includes DNA repair activity. Sensitivity of cells to oxidative-damage inducing agents is thus an indirect indicator of the cell’s ability to repair the damage and survive. Three days after transfection, TFAM knockdown cells were treated with increasing concentrations of menadione and cell survival was measured. Western blotting was performed before menadione treatment, to confirm efficient knockdown of TFAM (Fig. 7A). Although, survival was significantly higher in TFAM knockdown cells than in control cells (Fig. 7B), it is only observed at a single point. At lower dose of menadione (20 and 40 µM), there was none to minimal effect, while at 60 µM menadione cell survival was ~5-fold higher. The mechanism by which this mild effect on cell survival occurs is not known. This could possibly be attributed to the fine balance between damage and repair levels in these cells.
Fig. 7.

Cell survival assay. A) Western blot of TFAM and GADPH (loading control) protein levels in TFAM control (Scr) and knockdown (TFAM) cellular lysates corresponding to the survival assay on day (three days after transfection). B) Graph represents relative cell survival for scrambled and TFAM knockdown siRNA treated cells after treatment with increasing concentrations of menadione. Data are mean ± SD of three experiments. The * symbol indicates p values of 0.0023 and 0.0076, respectively for 40 and 60 µM menadione treatments.
Effect of TFAM knockdown on overall 8oxoG incision activity
8oxoG incision activity was quantified in whole cell lysates from TFAM knockdown and control (scrambled siRNA) cells. In vitro 8oxoG incision activity in cellular lysates increased by ~2 fold (from 12.3% to 22.7%) in TFAM knockdown cells (Fig. 8A). The direct cause of the increase in 8oxoG incision activity was examined by measuring the level of αOGG1, the primary 8oxoG glycosylase in eukaryotic cells. Western blot analysis of αOGG1 showed that the level of this protein was not up-regulated in TFAM knockdown cells (Fig. 8B) indicating that the effect was dues to changes in TFAM levels. Together, this result is in agreement with our in vitro incision and survival data, supporting the idea that TFAM might inhibit DNA repair by competing for the substrate.
Fig. 8.

Effect of TFAM knockdown on overall 8oxoG incision activity A) Data are percent 8oxoG incision activity observed in control (Scr) and TFAM knockdown (TFAM) Hela cell lysates. Inset is a representative gel for the incision assay where s=substrate band and p=product band. Results are means ± SD of three experiments. B) Western blot analysis of OGG1 protein levels in either the control (Scr) or knockdown (TFAM) cellular lysates used in panel ‘A’ is shown.
Discussion
This study examines the possible role of TFAM in mitochondrial DNA repair. We show here that TFAM binds to DNA containing 8oxoG, uracil, an abasic site or a gapped DNA substrate. Although our results agree with a previous report, that TFAM preferentially binds 8oxoG containing DNA [12], we observed only a modest preference for the damage-containing DNA over intact DNA. This is probably due to the sequence and length of the substrate used here. On the other hand, TFAM did not show any preference for the uracil, abasic site or gap containing substrates tested, indicating that TFAM’s DNA binding activity is not lesion-dependent and is largely nonspecific. Next, TFAM was evaluated for its ability to modulate in vitro BER enzymatic activities. Our results revealed that TFAM inhibited all four BER core activities in vitro, including OGG1, UDG, APE1, pol γ and additionally OGG1 in our cell based assay. To independently confirm that it was TFAM’s DNA binding activity which interfered with the BER enzymatic activities, a DNA binding defective TFAM was created. We observed a loss of inhibition when using the DNA binding-defective TFAM mutant thus supporting our hypothesis that TFAM modulates DNA repair by competitively binding to the DNA substrate. However, we also propose that cellular factors, such as p53, may attenuate TFAM binding to mtDNA, thus increasing the rate of DNA repair in vivo. This could be one of the many characteristic features that define p53 as a stress response protein.
Although TFAM globally inhibits all four BER enzymatic activities tested in this study, the function of TFAM in vivo may not be to inhibit DNA repair or mtDNA metabolic processes. More likely the primary purpose of TFAM is to compact and protect the mtDNA, much the way histones do for nuclear DNA. This concept is supported by both the in vitro incision assays and the Q-PCR assay, in which TFAM knockdown cells were shown to have a higher level of endogenous DNA damage. However, such packaging and protection also implies that the presence of TFAM on mtDNA limits the access to DNA repair and other mtDNA metabolic enzymes.. If we use nuclear histones as an analogy, it is well known that histones (reviewed in [28]) and some high mobility group proteins bind to DNA and slow the DNA repair processes [6, 11]. Furthermore, it is well established that chromatin remodeling, which requires the transient dissociation of the histone from DNA, is a regulated process that is required for most DNA metabolic reactions, including replication, transcription and DNA repair [29, 30]. Thus, our study raises the question of how TFAM’s DNA binding is regulated in mitochondria. While there probably are many cellular factors involved in regulating the association and dissociation of TFAM from mtDNA, we show here that one such factor may be p53, a known TFAM interacting protein [22]. The mechanisms regulating this function of p53 are not yet known, but may involve C-terminal phosphorylation of p53, which may stimulate its interaction with TFAM [31]. Clearly, translocation of p53 to mitochondria, which has been characterized previously [2, 32], is a likely mechanism by which p53-mediated release of TFAM from mtDNA could be regulated. Further studies are necessary to examine whether other proteins modulate TFAM binding to mtDNA in vivo, thereby facilitating mtDNA metabolic transactions including DNA repair.
Ultimately, the cellular response to persistent and/or extensive DNA damage is apoptosis. Cells from TFAM knockout mouse embryos demonstrate increased apoptosis due to DNA fragmentation [33]. As stated above, the primary function of TFAM is as a structural component of the mtDNA nucleoid, and TFAM DNA binding activity provides a shielding effect that minimizes ROS attack which would otherwise cause deleterious effects [34]. In addition, our data support a model where association of TFAM with DNA can prevent BER proteins from freely accessing the DNA. This inhibition may also be important in elimination of futile DNA repair cycles. This spurious incision may be the reason for the increased incision activity observed in the TFAM knockdown whole cell lysates used in our experiment (Fig. 8A). In this context, it has been demonstrated earlier that futile, incomplete BER cycles may lead to significant mutagenesis [35]. In the case of abasic sites, which are common products of ROS attack to the DNA, mechanisms preventing free access of APE1 may help protect the mtDNA from accumulating single-strand breaks resulting from APE1 activity. Moreover, incomplete BER of replicating mtDNA could also result in double strand breaks, which could ultimately lead to mtDNA deletions. Recently, several groups have proposed a role for incomplete DNA repair as a mechanism leading to accumulation of deletions in the mtDNA [36, 37].
In conclusion, this study enhances our understanding of the biological function of TFAM in mitochondria, which may be more complex than previously thought. TFAM modulates the access of DNA repair proteins to mtDNA. Therefore the cell may have evolved mechanisms, which can modulate TFAM’s DNA binding activity in response to biological stimuli. We have demonstrated that TFAM’s interaction with the tumor suppressor p53 modifies TFAM DNA binding and has the capacity to promote BER in mtDNA. In other words, TFAM could be an important regulator of BER due to its interplay with p53 or other interacting partners as well as mtDNA.
Supplementary Material
Acknowledgments
We greatly appreciate the gift of purified pol gamma from Dr William C. Copeland, Laboratory of Molecular Genetics, National Institute of Environmental Health Sciences, National Institutes of Health, USA. We would like to thank Dr. Singh DK and Dr. Ramamoorthy M for critically reading this manuscript. This work was supported by the National Institutes of Health Intramural Program of the National Institute on Aging (Z01-AG000733-14).
References
- 1.Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol Regul. Integr. Comp Physiol. 2007;292:R18–R36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 2.de Souza-Pinto NC, Harris CC, Bohr VA. p53 functions in the incorporation step in DNA base excision repair in mouse liver mitochondria 19. Oncogene. 2004;23:6559–6568. doi: 10.1038/sj.onc.1207874. [DOI] [PubMed] [Google Scholar]
- 3.Iborra FJ, Kimura H, Cook PR. The functional organization of mitochondrial genomes in human cells. BMC. Biol. 2004;2:9. doi: 10.1186/1741-7007-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures 2. Mol. Biol. Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parisi MA, Clayton DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. 1991;252:965–969. doi: 10.1126/science.2035027. [DOI] [PubMed] [Google Scholar]
- 6.Reeves R, Adair JE. Role of high mobility group (HMG) chromatin proteins in DNA repair 5. DNA Repair (Amst) 2005;4:926–938. doi: 10.1016/j.dnarep.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, Takio K, Hamasaki N, Kang D. Human mitochondrial DNA is packaged with TFAM 2. Nucleic Acids Res. 2003;31:1640–1645. doi: 10.1093/nar/gkg251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanki T, Ohgaki K, Gaspari M, Gustafsson CM, Fukuoh A, Sasaki N, Hamasaki N, Kang D. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA 16. Mol. Cell Biol. 2004;24:9823–9834. doi: 10.1128/MCB.24.22.9823-9834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maniura-Weber K, Goffart S, Garstka HL, Montoya J, Wiesner RJ. Transient overexpression of mitochondrial transcription factor A (TFAM) is sufficient to stimulate mitochondrial DNA transcription, but not sufficient to increase mtDNA copy number in cultured cells. Nucleic Acids Res. 2004;32:6015–6027. doi: 10.1093/nar/gkh921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pohjoismaki JL, Wanrooij S, Hyvarinen AK, Goffart S, Holt IJ, Spelbrink JN, Jacobs HT. Alterations to the expression level of mitochondrial transcription factor A, TFAM, modify the mode of mitochondrial DNA replication in cultured human cells 16. Nucleic Acids Res. 2006;34:5815–5828. doi: 10.1093/nar/gkl703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease 1. Proc. Natl. Acad. Sci. U. S. A. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida Y, Izumi H, Ise T, Uramoto H, Torigoe T, Ishiguchi H, Murakami T, Tanabe M, Nakayama Y, Itoh H, Kasai H, Kohno K. Human mitochondrial transcription factor A binds preferentially to oxidatively damaged DNA 12. Biochem. Biophys. Res. Commun. 2002;295:945–951. doi: 10.1016/s0006-291x(02)00757-x. [DOI] [PubMed] [Google Scholar]
- 13.Adair JE, Kwon Y, Dement GA, Smerdon MJ, Reeves R. Inhibition of nucleotide excision repair by high mobility group protein HMGA1 4. J. Biol. Chem. 2005;280:32184–32192. doi: 10.1074/jbc.M505600200. [DOI] [PubMed] [Google Scholar]
- 14.Cullinane C, Bohr VA. DNA interstrand cross-links induced by psoralen are not repaired in mammalian mitochondria. Cancer Res. 1998;58:1400–1404. [PubMed] [Google Scholar]
- 15.Wilson DM, III, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair (Amst) 2007;6:544–559. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 16.de Souza-Pinto NC, Mason PA, Hashiguchi K, Weissman L, Tian J, Guay D, Lebel M, Stevnsner TV, Rasmussen LJ, Bohr VA. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria 4. DNA Repair (Amst) 2009;8:704–719. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Souza-Pinto NC, Bohr VA. The mitochondrial theory of aging: involvement of mitochondrial DNA damage and repair. Int. Rev. Neurobiol. 2002;53:519–534. doi: 10.1016/s0074-7742(02)53018-x. [DOI] [PubMed] [Google Scholar]
- 19.Kujoth GC, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations and apoptosis in mammalian aging. Cancer Res. 2006;66:7386–7389. doi: 10.1158/0008-5472.CAN-05-4670. [DOI] [PubMed] [Google Scholar]
- 20.Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. PLoS. Genet. 2007;3:e24. doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stuart JA, Mayard S, Hashiguchi K, Souza-Pinto NC, Bohr VA. Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulate fraction. Nucleic Acids Res. 2005;33:3722–3732. doi: 10.1093/nar/gki683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Itoh H, Kang D, Kohno K. P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA 9. Cancer Res. 2003;63:3729–3734. [PubMed] [Google Scholar]
- 23.Santos JH, Mandavilli BS, Van HB. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol. Biol. 2002;197:159–176. doi: 10.1385/1-59259-284-8:159. [DOI] [PubMed] [Google Scholar]
- 24.Ohgaki K, Kanki T, Fukuoh A, Kurisaki H, Aoki Y, Ikeuchi M, Kim SH, Hamasaki N, Kang D. The C-terminal tail of mitochondrial transcription factor a markedly strengthens its general binding to DNA. J. Biochem. 2007;141:201–211. doi: 10.1093/jb/mvm020. [DOI] [PubMed] [Google Scholar]
- 25.Gangelhoff TA, Mungalachetty PS, Nix JC, Churchill ME. Structural analysis and DNA binding of the HMG domains of the human mitochondrial transcription factor A 4. Nucleic Acids Res. 2009;37:3153–3164. doi: 10.1093/nar/gkp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kienhofer J, Haussler DJ, Ruckelshausen F, Muessig E, Weber K, Pimentel D, Ullrich V, Burkle A, Bachschmid MM. Association of mitochondrial antioxidant enzymes with mitochondrial DNA as integral nucleoid constituents 1. FASEB J. 2009;23:2034–2044. doi: 10.1096/fj.08-113571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prigione A, Cortopassi G. Mitochondrial DNA deletions and chloramphenicol treatment stimulate the autophagic transcript ATG12 9. Autophagy. 2007;3:377–380. doi: 10.4161/auto.4239. [DOI] [PubMed] [Google Scholar]
- 28.Jagannathan I, Cole HA, Hayes JJ. Base excision repair in nucleosome substrates 14. Chromosome. Res. 2006;14:27–37. doi: 10.1007/s10577-005-1020-7. [DOI] [PubMed] [Google Scholar]
- 29.Hasan S, Hottiger MO. Histone acetyl transferases: a role in DNA repair and DNA replication 2. J. Mol. Med. 2002;80:463–474. doi: 10.1007/s00109-002-0341-7. [DOI] [PubMed] [Google Scholar]
- 30.Li B, Carey M, Workman JL. The role of chromatin during transcription 10. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 31.Wong TS, Rajagopalan S, Townsley FM, Freund SM, Petrovich M, Loakes D, Fersht AR. Physical and functional interactions between human mitochondrial single-stranded DNA-binding protein and tumour suppressor p53 2. Nucleic Acids Res. 2009;37:568–581. doi: 10.1093/nar/gkn974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sansome C, Zaika A, Marchenko ND, Moll UM. Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells 2. FEBS Lett. 2001;488:110–115. doi: 10.1016/s0014-5793(00)02368-1. [DOI] [PubMed] [Google Scholar]
- 33.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice 60. Nat. Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 34.Silva JP, Larsson NG. Manipulation of mitochondrial DNA gene expression in the mouse 3. Biochim. Biophys. Acta. 2002;1555:106–110. doi: 10.1016/s0005-2728(02)00263-3. [DOI] [PubMed] [Google Scholar]
- 35.Glassner BJ, Posnick LM, Samson LD. The influence of DNA glycosylases on spontaneous mutation 4. Mutat. Res. 1998;400:33–44. doi: 10.1016/s0027-5107(98)00045-1. [DOI] [PubMed] [Google Scholar]
- 36.Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons 2. Hum. Mol. Genet. 2009;18:1028–1036. doi: 10.1093/hmg/ddn437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM. What causes mitochondrial DNA deletions in human cells? 6. Nat. Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.