

Abstract
Cationic cell-penetrating peptides (CPPs) are a promising vehicle for the delivery of macromolecular drugs. Although many studies have indicated that CPPs enter cells by endocytosis, the mechanisms by which they cross endosomal membranes remain elusive. On the basis of experiments with liposomes, we propose that CPP escape into the cytosol is based on leaky fusion (i.e., fusion associated with the permeabilization of membranes) of the bis(monoacylglycero)phosphate (BMP)-enriched membranes of late endosomes. In our experiments, prototypic CPP HIV-1 TAT peptide did not interact with liposomes mimicking the outer leaflet of the plasma membrane, but it did induce lipid mixing and membrane leakage as it translocated into liposomes mimicking the lipid composition of late endosome. Both membrane leakage and lipid mixing depended on the BMP content and were promoted at acidic pH, which is characteristic of late endosomes. Substitution of BMP with its structural isomer, phosphatidylglycerol (PG), significantly reduced both leakage of the aqueous probe from liposomes and lipid mixing between liposomes. Although affinity of binding to TAT was similar for BMP and PG, BMP exhibited a higher tendency to support the inverted hexagonal phase than PG. Finally, membrane leakage and peptide translocation were both inhibited by inhibitors of lipid mixing, further substantiating the hypothesis that cationic peptides cross BMP-enriched membranes by inducing leaky fusion between them.
Introduction
In recent years, considerable progress has been made in the design of macromolecular drugs based on peptides, oligonucleotides, and their mimics (1). The intracellular targets for many of these drugs are located in the cell nucleus and cytoplasm, necessitating the development of efficient and safe approaches for delivering macromolecules into specific cellular compartments. A major barrier for macromolecules en route to the cytoplasm and nucleus is the lipid bilayer of the cell plasma membrane, the same barrier that protects cells from viruses and bacterial toxins. One of the actively studied approaches for macromolecular delivery is based on cationic cell-penetrating peptides (CPPs) (2–4). In this work we explored the membrane interactions of HIV-1 TAT48-60 (TAT) peptide, a prototypic CPP. The delivery of functionally active macromolecular cargo to the cell cytoplasm and nucleus is facilitated by cationic CPP binding to ubiquitous cell-surface heparan sulfate proteoglicans (5–7). Inhibitors of clathrin-dependent endocytosis, caveolin/raft-dependent endocytosis, and macropinocytosis strongly impair cationic peptide-dependent delivery of functionally active proteins and oligonucleotides into the cell cytosol and nucleus (6,8–11). The relative role of different endocytic pathways as observed in different studies apparently depends on the specific CPP, cargo, and cell types used in each study (12,13). However, to reach targets in the cytosol and nucleus, CPP-cargo complexes internalized by any endocytic pathway would have to cross the lipid bilayer of the endosomal membrane and escape from the endosome. Apparently, endosomal escape is a limiting factor in endocytosis-dependent delivery of functionally active CPP-cargo conjugates into the cytosol and nucleus (14,15). Many viruses and toxins also utilize endocytosis to gain access to the cell cytosol and nucleus. Thus, understanding the mechanism of endosomal escape is of great practical importance.
In this work we propose a new model for the delivery of cationic CPPs and conjugated cargo into the cytosol. This model is based on both the characteristic multivesicular morphology of late endosomes and the fact that intraluminal vesicles are highly enriched with bis(monoacylglycero)phosphate (BMP), an anionic lipid specific to late endosomes. We hypothesize that cationic peptides induce leaky fusion (i.e., fusion associated with membrane permeabilization) between intraluminal vesicles, resulting in delivery of the peptide and cargo molecules into intraluminal vesicles. Subsequent back-fusion of intraluminal vesicles with the limiting membrane of late endosomes releases the peptide and cargo into the cell cytosol. To test this model, we focused on interactions of TAT peptide with liposomes of various lipid compositions, including those mimicking lipid bilayers of plasma membrane and late endosomes. We report that TAT peptide induces leakage of encapsulated probes and translocates across BMP-enriched bilayers mimicking the lipid composition of intraluminal vesicles of late endosomes, but not across membranes mimicking lipid composition of plasma membranes. TAT-induced leakage and translocation are linked to membrane fusion and inhibited by fusion inhibitors. These results substantiate the proposed model of endosomal escape and suggest that membrane fusion, which is known to be an important stage of intracellular delivery for membrane-enclosed macromolecules such as viral nucleic acids, may also be involved in delivery of membrane-free macromolecules.
Materials and Methods
Materials
N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-phosphatidylethanolamine (NBD-PE), N-(lisamine Rhodamine B sulfonyl)-phosphatidylethanolamine (Rh-PE), cholesterol (Chol), sphingomyelin (SM) from porcine brain, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (PE), phosphatidylinositol (PI) from soybean, 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC), dioleoyl-BMP, and 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (PG) were purchased from Avanti Polar Lipids (Alabaster, AL). A recent study suggested that naturally occurring BMPs have acyl chains in the sn-2(2′) position (16); however, 2,2′-diacyl BMP is not readily commercially available and is unstable due to 2,3-acyl migration (17). Therefore, we used commercially available sn-1/sn1′ of BMP with acyl chains at the sn-3(3′) positions. We also confirmed in a few experiments that we observe similar levels of lipid mixing and leakage for liposomes with PC/BMP (1:1) lipid composition using (S,S)-2,2′-Bisoleoyl-LBPA (Echelon-Biosciences, Salt Lake City, UT). 8-Aminonaphthalene-1,3,6-trisulfonic acid, disodium salt (ANTS), 8-hydroxypyrene-1,3,6-trisulfonic acid, trisodium salt (HPTS), and p-xylene-bis-pyridinium bromide (DPX) were purchased from Molecular Probes (Invitrogen, Carlsbad, CA). HIV-1 TAT peptide with the sequence GRKKRRQRRRPPQC was custom synthesized by SynPep (Dublin, CA) and purified to 95% by high-performance liquid chromatography.
Liposome preparation
Large unilamellar vesicles (LUVs) were prepared by extrusion. In brief, lipids dissolved in benzene/methanol (95:5, vol/vol) were freeze-dried under high vacuum overnight and the dried lipid powder was hydrated in an appropriate buffer at room temperature and then vortexed. The resulting lipid suspension was submitted to 10 successive cycles of freezing and thawing, and then extruded 10 times through two stacked Nucleopore polycarbonate filters of 100 nm pore size (Whatman Inc., Piscataway, NJ) using a LIPEX extruder (Northern Lipids, Burnaby, Canada) to produce LUVs. Liposome sizes were measured using dynamic light scattering on an N4Plus submicron particle size analyzer (Beckman Coulter, Brea, CA).
Lipid mixing
TAT peptide-induced lipid mixing was measured by the dequenching of rhodamine PE (Rh-PE) fluorescence. Unlabeled liposomes of different lipid composition and liposomes labeled with a self-quenching concentration (5 mol %) of Rh-PE were prepared in buffers with pH 5.5 (150 mM NaCl, 10 mM MES) or pH 7.4 (150 mM NaCl, 10 mM Tris). Liposomes were added to a cuvette in a ratio of 1:10 of labeled to unlabeled liposomes for a total lipid concentration of 25 μM. Different concentrations of TAT peptide were added to induce lipid mixing between liposomes. All experiments were performed at room temperature. Lipid mixing between liposomes results in a dilution of Rh-PE and an increase in dye fluorescence due to a relief of self-quenching. Fluorescence changes induced by TAT were recorded under constant stirring using a Bowman-2 luminescence spectrometer (Aminco, Rochester, NY) with λex = 560 nm and λem = 585 nm. At the end of each recording, complete dequenching of the dye was induced by addition of Triton X-100 (0.1% v/v final concentration). The degree of dye quenching was calculated as Q(t) = 100 × (F(t) − F0)/(Ftriton − F0), where F(t), F0, and Ftriton are fluorescence at time t, before addition of peptide and after addition of Triton X-100, respectively. The initial rate of lipid mixing was estimated from the initial slope of the Q(t) curve. These experiments were carried out in disposable methacrylate 10 mm pathlength cuvettes in 2 mL of buffer under constant stirring with a magnetic stirrer bar.
Release of small water-soluble dye
To measure content release, liposomes were prepared in a buffer containing small water-soluble fluorescent ANTS together with a quencher DPX (12.5 mM ANTS, 45 mM DPX, 65 mM NaCl, and 10 mM HEPES for pH 7.4 or 10 mM MES for pH 5.5) or HPTS with DPX (1 mM HPTS, 5 mM DPX, 130 mM NaCl, and 10 mM HEPES for pH 7.4 or 10 mM MES for pH 5.5). After extrusion, unencapsulated dye and a quencher were removed by size exclusion chromatography using a PD-10 desalting column (Amersham Biosciences, Piscataway, NJ). Fluorescence changes induced by different concentrations of TAT were recorded under constant stirring using a Bowman-2 luminescence spectrometer (Aminco, Rochester, NY) with λex = 353 nm and λem = 520 nm for ANTS and λex = 416.4 nm and λem = 520 nm for HPTS. The extent of content leakage was calculated according to the following equation: % Leakage = 100 × (F(t) − F0) / (Ftriton − F0), where F(t), F0, and Ftriton are fluorescence intensities at time t, before addition of peptide and after the complete release of encapsulated dye by addition of 0.1% v/v of Triton X-100. These experiments were carried out in disposable methacrylate 10 mm pathlength cuvettes in 2 mL of buffer under constant stirring with a magnetic stirrer bar.
TAT peptide translocation into liposomes
Our assay for TAT translocation is based on the observation that binding of TAT-fluorescein to vesicles containing anionic lipids results in significant quenching of the fluorescein signal that then gradually recovers as we add more liposomes. Similar behavior was observed upon binding of TAT-fluorescein to the anionic polymer dextran sulfate. This could be explained by peptide crowding on the liposome surface or on the anionic polymer that leads to fluorescein self-quenching. Dye self-quenching was then released as we provided additional binding sites by adding more liposomes or dextran sulfate, and the peptide was diluted on the surface of the liposomes or polymer. Because binding of TAT to dextran sulfate is significantly stronger than binding to anionic liposomes (18,19), the addition of dextran sulfate to a mixture of BMP-containing liposomes and TAT peptide resulted in a fast inhibition of lipid mixing, content leakage, and reversal of liposome aggregation (see Fig. S1 in the Supporting Material). To measure TAT peptide translocation, we first added TAT-fluorescein (2 μM final concentration) to the buffer, followed by addition of liposomes up to a 25 μM final concentration. Binding of the peptide to the liposome surface resulted in a rapid partial quenching of peptide fluorescence. Slow additional quenching after this rapid initial fluorescence quenching indicated peptide translocation into the liposomes and binding to the inner monolayer of the liposome membrane. Then, at different times after addition of the liposomes, we added a high concentration of 500 kDa dextran sulfate (20 μM final concentration) to the cuvette. Competitive release of the TAT peptide from the outer surface of the liposomes and its binding to the dextran sulfate resulted in a partial recovery of fluorescence. Finally, we added Triton X-100 to solubilize liposomes and release translocated peptide from the inner monolayer of liposomes. This resulted in an additional increase in fluorescence that was used to measure peptide translocation. These experiments were carried out in disposable methacrylate 10 mm pathlength cuvettes in 2 mL of buffer under constant stirring with a magnetic stirrer bar.
Liposome aggregation
TAT-induced aggregation of liposomes was measured as an increase in the absorbance at 440 nm with the use of a DU-8 spectrophotometer (Beckman, Palo Alto, CA).
TAT-membrane binding
Fluorescein-tagged TAT (F-TAT) was incubated with multilamellar vesicles (MLVs) at different lipid/peptide molar ratios in Tris or MES buffer for 30 min at room temperature. The mixtures were then centrifuged at 14,000 rpm for 30 min to pellet MLVs with bound peptide. The supernatants were incubated with trypsin in Tris buffer for 30 min at 37°C to minimize a loss of fluorescence due to the binding of the peptide to polystyrene. The fluorescence intensity of the trypsin-treated supernatants was recorded at 520 nm (excitation at 480 nm) to determine the concentration of the free F-TAT. The concentration of the bound peptide was calculated from the difference between total amount of the peptide in the mixtures and free peptide found in the supernatants. We determined the apparent dissociation constant Kd by measuring the fraction of bound peptide, [P]b/[P]tot as a function of lipid concentration [L]. [P]b is the amount of bound peptide and [P]tot is the total peptide concentration. The data were fitted by the equation [Pb]/[Ptot] = [L]/(Kd + [L]). To estimate the fraction of the lipid in MLVs available for binding, we compared the fluorescence signal of the membrane-impermeable lipid dye FM4-64 in the presence of MLVs and in the presence of LUVs. Fluorescence of FM4-64 dramatically increased upon its membrane insertion, and thus the changes in FM4-64 fluorescence for the same amount of the probe give a measure of the total bilayer area accessible for the probe insertion. For the same total lipid amount, the signal in the presence of MLVs was ∼10 times smaller than for LUVs, indicating that only 5% of the total lipid in MLVs was available for FM4-64 and hence TAT binding (assuming that only half of the total lipids are accessible for binding in LUVs).
Effective molecular shape of DOBMP and DOPG lipids
To estimate the effective molecular shape of DOBMP and DOPG, we evaluated the effects of adding of small amounts of these lipids to egg PE on the temperature of the Lα–HII phase transition. We detected the Lα–HII phase transition using changes in NBD-PE fluorescence as described previously (20). In short, egg PE liposomes containing 0.1 mol % NBD-PE and 0, 5, or 10 mol % of either DOBMP or DOPG were prepared in basic buffer with pH 10 (5 mM sodium borate, 150 mM NaCl). The liposome suspensions incubated at different temperatures were acidified to pH 5.5 to induce liposome aggregation and, at elevated temperatures, the Lα–HII phase transition. The phase transition results in an increase in the NBD fluorescence. The dependence of the initial rate of change in NBD-PE fluorescence on the temperature was used to measure the temperature of the Lα–HII phase transition.
Escape of fluorescein-tagged dextran
PC or ILM liposomes labeled with 1 mol % Rh-PE were prepared in a buffer containing fluorescein-tagged dextran (5 mg/mL FD-70, 130 mM NaCl, 10 mM HEPES). We removed dextran that was not encapsulated into extruded liposomes by centrifugation on a Ficoll step gradient (0.75 ml 20% Ficoll, 1.5 mL 10% Ficoll, 0.5 mL buffer) for 1 h at 32,500 rpm using an SW-55 Ti rotor. Purified liposomes were collected from the interface between 10% Ficoll and buffer. Liposomes (500 μM) were incubated with TAT (40 μM) for 30 min and the released dextran was again removed on a Ficoll step gradient. The amount of dextran retained in liposomes was measured using fluorescein fluorescence (λex = 495 nm, λem = 520 nm) and normalized to a liposome concentration that was measured using rhodamine fluorescence (λex = 560 nm, λem = 585 nm).
Results
TAT peptide induces leakage of water-soluble probes from BMP-enriched liposomes
Because endosomal escape involves an increase in the permeability of the endosomal membrane for the peptide and conjugated cargo, we first tested whether TAT peptide induces leakage of a liposome-encapsulated, water-soluble probe comparable in size to a free peptide (∼500 Da for ANTS versus ∼1700 Da for TAT). We compared the effects of TAT addition to liposomes of different lipid compositions as follows: 1), a BMP/PC/PE (77:19:4) (ILM) mixture mimicking the phospholipid composition of intraluminal vesicles of late endosomes (16); 2), a PC/PE/PI/BMP (5:2:1:2) (LEM) mixture mimicking the overall phospholipid composition of late endosomes (16); and 3) a PC/PE/SM/Chol (1:1:1:1.5) (PM) mixture mimicking the lipid composition of the outer leaflet of the plasma membrane. The addition of TAT peptide to liposomes of ILM composition resulted in an efficient escape of liposome-encapsulated fluorescent probe ANTS both at acidic pH 5.5 characteristic of late endosomes (ILM; Fig. 1 a,) and, somewhat less efficiently, in neutral pH buffer (pH 7.4, ILM; Fig. 1 b). When TAT peptide was added to liposomes of LEM composition, we observed much less efficient dye leakage at pH 5.5 and almost no leakage in neutral pH buffer (pH 7.4; Fig. 1). No measurable leakage was observed when TAT peptide was added to PM liposomes at both neutral and acidic pH (PM; Fig. 1). The final extent of TAT-induced dye leakage increased with the peptide concentration at both pH 5.5 and pH 7.4 (Fig. 1, c and d). We observed similar results when ANTS was replaced with HPTS, another small water-soluble fluorescent probe (Fig. S2 A). To test whether TAT can induce release of large macromolecules from liposomes, we prepared ILM and PM liposomes encapsulating fluorescein-labeled 70 kDa dextran. Although TAT induced release of dextran from ILM liposomes, we did not observe release of dextran from PM liposomes (Fig. S2 B).
Figure 1.
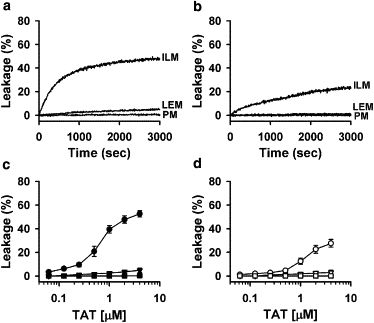
TAT peptide releases the encapsulated probe from ILM and LEM liposomes, but not from CPM liposomes. (a and b) Kinetics of dye dequenching due to the release of ANTS/DPX from 25 μM liposomes of ILM, LEM, and PM lipid composition upon addition of 2 μM TAT peptide at pH 5.5 (a) and pH 7.4 (b). (c and d) Dependence of dye dequenching, measured 50 min after the addition of TAT, on peptide concentration. Peptide was added to 25 μM of ILM (circles), LEM (triangles), and PM (squares) liposomes at pH 5.5 (c) and at pH 7.4 (d). Each data point in panels c and d represents the mean of four independent experiments, and error bars indicate the standard deviation (SD).
In brief, TAT permeabilizes liposomes mimicking the composition of late endosomal membranes, but not those mimicking the composition of the outer leaflet of the plasma membrane. The TAT-induced pores are large enough to allow the release of large macromolecules.
TAT peptide induces fusion between liposomes with high content of BMP
Because the addition of TAT peptide to ILM and LEM liposomes induced their aggregation (Fig. S2 C), we tested whether TAT induces lipid mixing between liposomes. The efficiency of lipid mixing was quantified as the initial rate of dequenching of the lipid dye Rh-PE upon addition of different concentrations of TAT peptide to labeled (5 mol % of Rh-PE) and unlabeled liposomes mixed in a 1:10 ratio (Fig. 2). The addition of TAT peptide to ILM liposomes led to efficient lipid mixing at both acidic pH 5.5 (Fig. 2, a and c, ILM and solid circles) and neutral pH 7.4 (Fig. 2, b and d, ILM and open circles). When TAT peptide was added to LEM liposomes, lipid mixing was observed in both acidic and neutral pH buffers, although it was less efficient than lipid mixing between ILM liposomes (Fig. 2, LEM and triangles). In contrast, no lipid mixing was observed upon addition of TAT peptide to PM liposomes (Fig. 2, PM and squares). The efficiency of lipid mixing was dependent on peptide concentration for both ILM and LEM lipid compositions.
Figure 2.
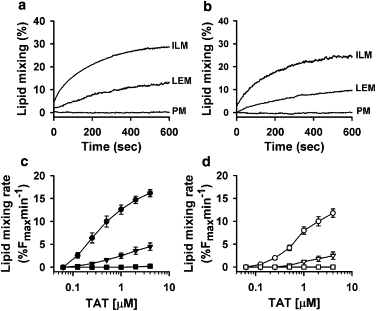
TAT induces lipid mixing for ILM and LEM liposomes, but not for CPM liposomes. (a and b) Kinetics of Rh-PE dequenching due to lipid mixing between ILM, LEM, and PM liposomes induced by 1 μM TAT at pH 5.5 (a) and pH 7.4 (b). (c and d) Dependence of the initial rate of lipid mixing induced by TAT on peptide concentration. TAT was added to 25 μM of a 1:9 mixture of labeled and unlabelled liposomes with ILM (circles), LEM (triangles), and PM (squares) lipid composition at pH 5.5 (c) and at pH 7.4 (d).Each data point in panels c and d represents the mean of four independent experiments, and error bars indicate the SD.
Leakage of liposome content in the presence of TAT precluded us from using a liposome content mixing assay to further confirm that TAT merges liposomal bilayers. Furthermore, significant aggregation of ILM and LEM liposomes in the presence of TAT would prevent detection of liposome fusion by liposome size measurements. However, we were able to reverse liposome aggregation by addition of dextran sulfate (Fig. S1 C), which binds TAT peptide significantly more strongly than anionic liposomes (18,19). Addition of dextran sulfate to a mixture of ILM liposomes and TAT peptide also resulted in a rapid inhibition of content leakage and lipid mixing, and reversal of liposome aggregation (Fig. S1, A and B). Thus, the addition of dextran sulfate at the end of the lipid mixing experiment allowed us to compare liposome sizes before and after incubation of liposomes with TAT peptide. Using dynamic light scattering, we measured ILM liposome sizes before and after 30 min incubation with TAT peptide to be 111 nm and 142 nm, respectively, with the polydispersity index of liposome distribution increasing from 0.092 to 0.148. These results, along with the lipid mixing data, indicate that TAT fuses ILM liposomes.
Leakage and lipid mixing depend on BMP content
To explore the role of BMP in TAT-induced liposome leakage and lipid mixing, we prepared liposomes from a set of PC/BMP mixtures with mole fractions of BMP varied from 0 to 60%. As we increased the mole fraction of BMP in the mixture, the efficiency of both dye leakage (Fig. 3, a and b) and lipid mixing (Fig. 3, c and d) increased. For all BMP-containing liposomes, leakage and lipid mixing at pH 5.5 were more efficient than at pH 7.4 (Fig. 3, b and d).
Figure 3.
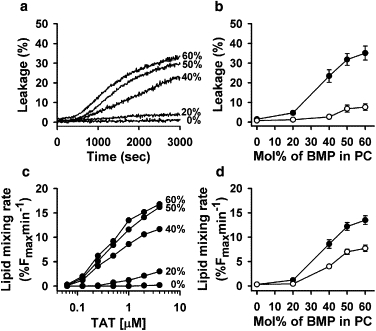
Efficiencies of TAT-induced aqueous probe leakage (a and b) and lipid mixing (c and d) rise with an increase in the mole fraction of BMP in liposomes. (a) The time course of encapsulated ANTS/DPX release from liposomes composed of different BMP/PC mixtures (25 μM total lipid concentration) induced by 2 μM TAT at pH 5.5. Mol % of BMP in each lipid composition is indicated on the figure. (b) Dependence of dye dequenching measured 50 min after addition of 2 μM TAT to 25 μM liposomes on the mole fraction of BMP in binary BMP/PC mixtures at pH 5.5 (solid symbols) or at pH 7.4 (open symbols). (c) Dependence of the initial rate of lipid mixing induced by TAT on the mole fraction of BMP in binary BMP/PC mixtures at pH 5.5 (the mol % of BMP is indicated in the figure). TAT was added to 25 μM of 1:9 mixtures of labeled and unlabeled liposomes. (d) Dependence of the initial rate of lipid mixing on mol% of BMP in BMP: PC binary mixtures at pH 5.5 (solid symbols) or at pH 7.4 (open symbols). TAT and total lipid concentration was 1 μM and 25 μM, respectively. Each data point on panels (b-d) represents the mean of three independent experiments and error bars indicate the standard deviation.
Although BMP is a predominant anionic lipid in late endosomes, we were interested in determining whether it can be replaced with another anionic lipid in vitro. The replacement of BMP in a PC/BMP (1:1) mixture with its structural isomer, PG, resulted in a significant inhibition of TAT-induced lipid mixing and aqueous probe leakage (Fig. 4). Similarly, replacement of BMP in the ILM mixture with PG resulted in a strong inhibition of dye leakage (Fig. S3). This inhibition cannot be explained by differences in TAT peptide binding because TAT peptide bound similarly to BMP- and PG-containing liposomes (Fig. S4), and induced a similar degree of liposome aggregation for BMP- and PG-containing liposomes (Fig. S5).
Figure 4.
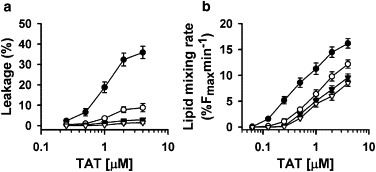
Replacement of BMP with structural isomer PG lowers the efficiencies of aqueous dye leakage and lipid mixing. (a) Dependence of ANTS/DPX release from liposomes composed of BMP/PC (1:1) (circles) or PG/PC (1:1) mixtures (triangles) at pH 5.5 (solid symbols) and pH 7.4 (open symbols) on TAT concentration. (b) Dependence of the initial rate of lipid mixing of BMP/PC (1:1) (circles) or PG/PC (1:1) (triangles) liposomes at pH 5.5 (solid symbols) and pH 7.4 (open symbols) on TAT concentration. The total lipid concentration was 25 μM in all experiments. Each data point represents the mean of three independent experiments, and error bars indicate the SD.
TAT peptide translocates across membranes with high content of BMP
Although our membrane leakage experiments indicated that TAT-induced pores in BMP-enriched membranes are large enough to allow passage of aqueous probes comparable in size or even larger than TAT, we decided to directly verify that under these conditions TAT peptide itself crosses the liposomal membrane. To address this question, we used F-TAT peptide and took advantage of fluorescence quenching upon peptide binding to liposomes containing negatively charged lipids. After addition of negatively charged liposomes (25 μM final concentration) to F-TAT (2 μM final concentration) in a low pH buffer, we observed almost immediate fluorescence quenching, reflecting binding of the peptide to the outer monolayer of liposomes (Fig. 5 a, +liposomes). This fast initial quenching was followed by a more gradual decrease in fluorescence with a time constant of ∼100 s in the case of ILM liposomes, but not when BMP in liposome lipid composition was replaced with PG (Fig. 5 a, +liposomes). The addition of membrane-impermeable 500 kDa dextran sulfate (20 μM), which binds the peptide with a higher affinity than the liposomes (18,19), released the peptide from the outer surface of the liposomes and partially dequenched the fluorescence (Fig. 5 a, +DS). Because dextran sulfate rapidly stops liposome content leakage (Fig. S1 a), TAT peptide molecules that entered the inner volume of the liposomes were inaccessible for dextran sulfate, and thus remained bound to the inner monolayer of liposomes and quenched. These TAT peptide molecules were released from the internal volume of the liposomes upon addition of Triton X-100 at the end of the experiment. This release of the peptide resulted in a further fluorescence increase and gave us a measure of the amount of peptide that translocated into liposomes (Fig. 5 a, +Triton, ΔF). The amount of translocated peptide increased with the time between the addition of liposomes and the addition of dextran sulfate (Fig. 5 b). Of interest, the peptide inside liposomes remained entrapped (Fig. S6) even after extended incubation with dextran sulfate, suggesting that TAT-induced leakage of liposome content and peptide translocation require interliposome interactions.
Figure 5.
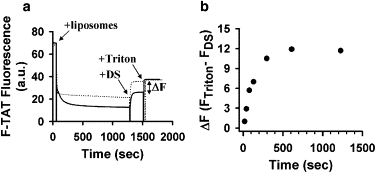
TAT peptide translocates into liposomes that mimic the phospholipid composition of intraluminal vesicles of late endosomes (ILM). (a) Change in the fluorescence of fluorescein-tagged TAT (2 μM) at pH 5.5 after the addition of 25 μM of ILM (solid line) and PG/PC/PE (77:19:4) (dotted line) liposomes (+liposomes), 20 μM dextran sulfate (+DS), and finally 0.1% Triton X-100 (+Triton). (b) Amount of translocated TAT-fluorescein as a function of incubation time with ILM liposomes. The amount of translocated peptide was assayed as an increase in fluorescence after the final addition of Triton X-100 (denoted as ΔF in panel a).
Liposome-liposome fusion is required for TAT-induced dye leakage and TAT translocation
To test whether TAT peptide-induced vesicle fusion and leakage are mechanistically coupled, we modified liposome composition in two different ways known to inhibit membrane fusion. In the first approach, we included 2 mol % of PEG-PE in our lipid composition. This lipid was previously shown to inhibit vesicle fusion by sterically hindering close opposition of fusing bilayers (21–23). In the second approach, we modified the lipid composition of our liposomes by including 20 mol % of LPC. LPC inhibits diverse fusion processes (24,25), presumably because its inverted-cone molecular shape does not match the curvature of the early-fusion intermediate stalk. In contrast, LPC fits into the edge of the lipid pore and therefore promotes formation of lipid pores in membranes (25), and thus is expected to promote dye leakage. In our experiments, both PEG-PE and LPC efficiently inhibited both lipid mixing (Fig. 6 b) and dye leakage (Fig. 6 a). This indicates that leakage and fusion induced by TAT peptide are mechanistically connected. TAT peptide translocation was also inhibited by incorporation of 2 mol % of PEG-PE into the ILM liposomes (Fig. S7).
Figure 6.
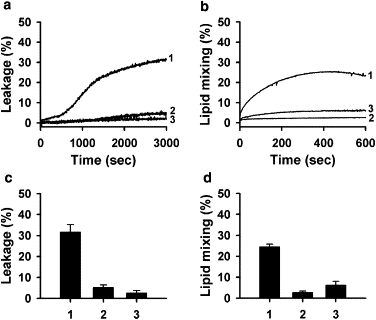
Incorporation of PEG-PE or LPC into the liposome lipid composition inhibits TAT-induced dye leakage and lipid mixing. The liposome lipid composition was modified by replacing corresponding amounts of PC in 1), basal BMP/PC (1:1); 2), a lipid mixture with 2 mol % of PEG-PE; or 3), 20 mol % of LPC. Effects of PEG-PE and LPC incorporation on the kinetics of dye dequenching due to release of ANTS/DPX from liposomes (a), the kinetics of Rh-PE dequenching due to lipid mixing between liposomes (b), the extent of dye dequenching measured 50 min after addition of the TAT peptide (c), and the initial rate of lipid mixing (d) are shown. The concentration of TAT was 2 μM and the total lipid concentration was 25 μM. These experiments were performed in pH 5.5 buffer. Each data point in panels c and d represents the mean of three independent experiments, and error bars indicate the SD.
Membrane fusion involves bending of lipid bilayers, and the propensity of membranes to fuse correlates with the effective shape of the lipids that constitute the bilayers (26,27). Similarly, the effective molecular shapes of lipids strongly affect the propensity of these lipids to form different phases. In particular, lipids that are cylindrical in shape (for example, PC) favor the formation of lamellar Lα phase, whereas cone-shaped lipids, such as PE, promote the formation of inverted hexagonal HII phase. Thus, one can evaluate the effective molecular shapes of lipids by assessing the effect of minor additions of these lipids on the temperature of the well-characterized Lα–phase transition of PE (20,28–30). We compared the effective molecular shapes of two lipids (BMP and PG) that have distinct effects on TAT-induced leakage and lipid mixing. We found that BMP increased the Lα–HII transition temperature of egg PE much less compared to PG (Fig. S8), indicating that the molecular shape of BMP is more cone-shaped than the almost cylindrical PG (31). Because cone-shaped lipids (lipids of negative spontaneous curvature) promote early fusion stages (25), inhibition of TAT-induced leakage upon substitution of BMP with PG further substantiates the key role of fusion in TAT-induced dye escape.
To summarize our data, TAT-induced permeabilization of BMP-enriched bilayers detected as a release of aqueous probes from liposomes and entry of TAT peptide into liposomes is mechanistically linked to liposome fusion, indicating that the peptide crosses BMP-containing bilayers by leaky fusion.
Discussion
The mechanism of cationic CPP-mediated delivery into the cytosol and nucleus remains hotly debated. Although several groups have reported that at higher peptide concentrations (>10 μM) cationic CPPs can directly cross the plasma membrane (32–36), there is ample experimental evidence that endocytosis is a necessary step in delivery of CPP-conjugated cargo into cytosol (for review, see Edenhofer (37)). In both cases, CPPs have to cross the lipid bilayer; however, their interactions with endosomal (especially late endosomal) and plasma membrane may differ significantly because these membranes are exposed to environments with different pH values (pH 5.5–6.0 in endosomes versus pH 7.4 at plasma membrane) and have significantly different lipid compositions. Indeed, the transmembrane pH gradient has been suggested to play an important role in endosomal escape of pAntp and bPrPp peptides (38). Although extensive studies on the interactions of cationic CPPs with protein-free liposomes of different compositions have led to the proposal of several different mechanisms of translocation across the lipid bilayer (39–43), the possible role of differences between the lipid composition of endosomal and plasma membranes in the mechanism of CPP translocation has not previously been explored. Therefore, in this work we investigated whether the lipid composition of endosomal membranes plays an important role in the mechanism by which cationic peptides enter cytosol.
Given the cationic nature of most CPPs, we were especially intrigued by the fact that intraluminal vesicles of late endosomes are highly enriched with a specific anionic lipid, BMP (16,44). This is in stark contrast to the outer leaflet of the plasma membrane, which normally contains a very small amount of negatively charged lipids, although the cell surface is negatively charged due to the presence of various proteoglycans, including heparan sulfate, which is responsible for TAT peptide binding (5–7). It was previously shown that TAT peptide efficiently binds to negatively charged liposomes, whereas binding to uncharged liposomes is very small (18). Our experiments revealed that TAT peptide binds strongly to BMP-enriched liposomes, fuses them, releases liposome-encapsulated aqueous probes, and translocates into the inner volume of the liposomes. None of these effects were observed upon addition of TAT peptide to PM liposomes. The promotion of liposome content leakage, fusion, and peptide translocation at pH 5.5, which is characteristic of late endosomes, further supports the relevance of the observed effects for endosomal escape of peptide, although the mechanism of pH dependence remains unclear. Similar specificity toward endosomal membranes was recently reported for octa-arginine-modified liposomes that fused with endosomes but not with the plasma membrane (45).
Are TAT translocation, lipid mixing, and release of aqueous probes for lipid bilayers enriched in BMP mechanistically coupled? One may hypothesize that TAT translocates directly through the lipid bilayer, and liposome leakage and lipid mixing are both unrelated to translocation. The finding that several modifications of our experimental system inhibited all of these effects of TAT in concert rather than uncoupled them argues against this hypothesis. Replacement of BMP with PG inhibited lipid mixing, leakage, and TAT translocation. Furthermore, the incorporation into the lipid bilayer of fusion inhibitor PEG-PE inhibited not only lipid mixing but also leakage and TAT translocation. Finally, TAT peptide that entered liposomes remained entrapped after liposome leakage and lipid mixing were stopped by addition of dextran sulfate.
Furthermore, our data suggest a mechanistic link between leakage and lipid mixing induced by TAT. Both of the inhibitors of lipid mixing used in our work (PEG-PE and LPC) also inhibited the escape of encapsulated dye. This is especially instructive in the case of inverted cone-shaped LPC because although LPC is a fusion inhibitor, it promotes lipid pore formation (46) and would be expected to promote fusion-independent dye leakage. Moreover, inhibition of both lipid mixing and leakage by replacement of fusion-promoting, cone-shaped BMP with cylindrical PG also indicates that lipid mixing and leakage are mechanistically linked. Note that we do not suggest that leakage and fusion events induced by TAT occur simultaneously. This is unlikely, because lipid mixing appears to be more efficient than leakage, and for some lipid compositions we observed significant lipid mixing with no leakage. Rather, we suggest that leakage and fusion share a common intermediate—possibly the stalk-like intermediate that is involved in diverse fusion reactions. It is important to note that late endosomes are not only enriched with BMP and have acidic pH, they also have a multivesicular morphology that permits a fusion-dependent mechanism of leakage and translocation that is unlikely to occur on the plasma membrane or in early endosomes.
The release of encapsulated dye coincidentally with membrane fusion was previously observed for a number of fusion systems (47-49), but was often considered to be a side effect of biologically relevant, nonleaky fusion reactions (47). Although concomitant leakage is arguably undesirable in many biological fusion reactions (e.g., exocytosis), we propose that it is instrumental for drug delivery by cationic peptides. Given that the surface glycoproteins of some nonenveloped viruses have structural similarities to proteins that mediate fusion of enveloped viruses (50), it is tempting to suggest that these nonenveloped viruses employ similar leaky fusion-dependent mechanisms for endosomal escape and infection. What could be the mechanism of leaky fusion? Positive lateral tension generated along the rim of the expanding stalk facilitates nucleation of the pores in the vicinity of the rim (48,51). Expansion of the pores that develop within hemifusion structures results in fusion without leakage. We speculate that once TAT peptide brings membranes into close contact, the formation of multiple stalk intermediates within an extended contact zone brings about lipid mixing and promotes the formation of lipid pores outside hemifusion structures, allowing dye release and peptide translocation. Within this scenario, leakage and fusion are mechanistically connected through a common intermediate stalk, but do not necessary proceed simultaneously or with comparable efficiency. The replacement of BMP with the less fusogenic lipid PG increases the energy of the stalk intermediate and consequently inhibits both lipid mixing and leakage. TAT peptides may lower the energy of the stalk intermediate and/or just facilitate close opposition of interacting membranes.
On the basis of our results, we propose the following model of intracellular entry for cationic CPP-mediated delivery of conjugated cargo: CPPs bound to heparan sulfates on the plasma membrane are internalized through diverse pathways of endocytosis. The low content of anionic phospholipids in the leaflets of the plasma membrane and early endosomes that are exposed to CPPs minimizes peptide interactions with the lipid bilayers of these membranes. However, once CPPs reach the lumen of a multivesicular late endosome, they bind to BMP-enriched intraluminal vesicles. CPP-mediated leaky fusion between these vesicles allows the peptide to enter into the lumen of intraluminal vesicles. CPP-induced leaky fusion between intraluminal vesicles and the limiting membrane is less likely to occur because of a relatively low BMP content in the limiting membrane and a strong dependence of leakage efficiency on BMP. Finally, the BMP-dependent back-fusion of intraluminal vesicles with limiting membrane that may proceed independently of CPP (52) releases CPP and CPP-associated cargo into the cytosol.
Although our results demonstrate a leaky fusion mechanism of CPP escape for protein-free membranes, it remains to be tested whether this mechanism underlies CPP entry into the cell cytosol. Given that only a small fraction of internalized CPP/CPP-cargo conjugate escapes into the cytosol, this may prove to be a challenging task.
Acknowledgments
We thank Sergei Pourmal for a critical reading of the manuscript.
This research was supported by the Intramural Research Program of the National Institute of Child Health and Human Development, National Institutes of Health (L.V.C.).
Supporting Material
References
- 1.Juliano R.L., Astriab-Fisher A., Falke D. Macromolecular therapeutics: emerging strategies for drug discovery in the postgenome era. Mol. Interv. 2001;1:40–53. [PubMed] [Google Scholar]
- 2.Morris M.C., Deshayes S., Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol. Cell. 2008;100:201–217. doi: 10.1042/BC20070116. [DOI] [PubMed] [Google Scholar]
- 3.Belting M., Wittrup A. Macromolecular drug delivery: basic principles and therapeutic applications. Mol. Biotechnol. 2009;43:89–94. doi: 10.1007/s12033-009-9185-5. [DOI] [PubMed] [Google Scholar]
- 4.Lebleu B., Moulton H.M., Gait M.J. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Deliv. Rev. 2008;60:517–529. doi: 10.1016/j.addr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Console S., Marty C., Ballmer-Hofer K. Antennapedia and HIV transactivator of transcription (TAT) “protein transduction domains” promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J. Biol. Chem. 2003;278:35109–35114. doi: 10.1074/jbc.M301726200. [DOI] [PubMed] [Google Scholar]
- 6.Richard J.P., Melikov K., Chernomordik L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005;280:15300–15306. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- 7.Wadia J.S., Stan R.V., Dowdy S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 8.Potocky T.B., Menon A.K., Gellman S.H. Cytoplasmic and nuclear delivery of a TAT-derived peptide and a beta-peptide after endocytic uptake into HeLa cells. J. Biol. Chem. 2003;278:50188–50194. doi: 10.1074/jbc.M308719200. [DOI] [PubMed] [Google Scholar]
- 9.Ferrari A., Pellegrini V., Beltram F. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol. Ther. 2003;8:284–294. doi: 10.1016/s1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan I.M., Wadia J.S., Dowdy S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release. 2005;102:247–253. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Nakase I., Niwa M., Futaki S. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004;10:1011–1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Jones A.T. Gateways and tools for drug delivery: endocytic pathways and the cellular dynamics of cell penetrating peptides. Int. J. Pharm. 2008;354:34–38. doi: 10.1016/j.ijpharm.2007.10.046. [DOI] [PubMed] [Google Scholar]
- 13.Melikov K., Chernomordik L.V. Arginine-rich cell penetrating peptides: from endosomal uptake to nuclear delivery. Cell. Mol. Life Sci. 2005;62:2739–2749. doi: 10.1007/s00018-005-5293-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abes R., Moulton H.M., Lebleu B. Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: structure-activity studies. Nucleic Acids Res. 2008;36:6343–6354. doi: 10.1093/nar/gkn541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Sayed A., Futaki S., Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi T., Beuchat M.H., Gruenberg J. Separation and characterization of late endosomal membrane domains. J. Biol. Chem. 2002;277:32157–32164. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 17.Hayakawa T., Hirano Y., Kobayashi T. Differential membrane packing of stereoisomers of bis(monoacylglycero)phosphate. Biochemistry. 2006;45:9198–9209. doi: 10.1021/bi060722o. [DOI] [PubMed] [Google Scholar]
- 18.Ziegler A., Blatter X.L., Seelig J. Protein transduction domains of HIV-1 and SIV TAT interact with charged lipid vesicles. Binding mechanism and thermodynamic analysis. Biochemistry. 2003;42:9185–9194. doi: 10.1021/bi0346805. [DOI] [PubMed] [Google Scholar]
- 19.Ziegler A., Seelig J. Interaction of the protein transduction domain of HIV-1 TAT with heparan sulfate: binding mechanism and thermodynamic parameters. Biophys. J. 2004;86:254–263. doi: 10.1016/S0006-3495(04)74101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong K.L., Baldwin P.A., Papahadjopoulos D. Fluorometric detection of the bilayer-to-hexagonal phase transition in liposomes. Biochemistry. 1988;27:3947–3955. doi: 10.1021/bi00411a009. [DOI] [PubMed] [Google Scholar]
- 21.Chams V., Bonnafous P., Stegmann T. Influenza hemagglutinin mediated fusion of membranes containing poly(ethylene-glycol) grafted lipids: new insights into the fusion mechanism. FEBS Lett. 1999;448:28–32. doi: 10.1016/s0014-5793(99)00333-6. [DOI] [PubMed] [Google Scholar]
- 22.Holland J.W., Hui C., Madden T.D. Poly(ethylene glycol)—lipid conjugates regulate the calcium-induced fusion of liposomes composed of phosphatidylethanolamine and phosphatidylserine. Biochemistry. 1996;35:2618–2624. doi: 10.1021/bi952000v. [DOI] [PubMed] [Google Scholar]
- 23.Mori A., Chonn A., Cullis P.R. Stabilization and regulated fusion of liposomes containing a cationic lipid using amphipathic polyethyleneglycol derivatives. J. Liposome Res. 1998;8:195–211. [Google Scholar]
- 24.Chernomordik L.V., Vogel S.S., Zimmerberg J. Lysolipids reversibly inhibit Ca(2+)-, GTP-, and pH-dependent fusion of biological membranes. FEBS Lett. 1993;318:71–76. doi: 10.1016/0014-5793(93)81330-3. [DOI] [PubMed] [Google Scholar]
- 25.Chernomordik L., Kozlov M.M., Zimmerberg J. Lipids in biological membrane fusion. J. Membr. Biol. 1995;146:1–14. doi: 10.1007/BF00232676. [DOI] [PubMed] [Google Scholar]
- 26.Chernomordik L.V., Kozlov M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008;15:675–683. doi: 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blumenthal R., Clague M.J., Epand R.M. Membrane fusion. Chem. Rev. 2003;103:53–69. doi: 10.1021/cr000036+. [DOI] [PubMed] [Google Scholar]
- 28.Janes N. Curvature stress and polymorphism in membranes. Chem. Phys. Lipids. 1996;81:133–150. [Google Scholar]
- 29.Lee Y.C., Taraschi T.F., Janes N. Support for the shape concept of lipid structure based on a headgroup volume approach. Biophys. J. 1993;65:1429–1432. doi: 10.1016/S0006-3495(93)81206-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tate M.W., Gruner S.M. Lipid polymorphism of mixtures of dioleoylphosphatidylethanolamine and saturated and monounsaturated phosphatidylcholines of various chain lengths. Biochemistry. 1987;26:231–236. doi: 10.1021/bi00375a031. [DOI] [PubMed] [Google Scholar]
- 31.Alley S.H., Ces O., Templer R.H. X-ray diffraction measurement of the monolayer spontaneous curvature of dioleoylphosphatidylglycerol. Chem. Phys. Lipids. 2008;154:64–67. doi: 10.1016/j.chemphyslip.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Takeuchi T., Kosuge M., Futaki S. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem. Biol. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- 33.Ziegler A., Nervi P., Seelig J. The cationic cell-penetrating peptide CPP(TAT) derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: optical, biophysical, and metabolic evidence. Biochemistry. 2005;44:138–148. doi: 10.1021/bi0491604. [DOI] [PubMed] [Google Scholar]
- 34.Tünnemann G., Ter-Avetisyan G., Cardoso M.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008;14:469–476. doi: 10.1002/psc.968. [DOI] [PubMed] [Google Scholar]
- 35.Ter-Avetisyan G., Tünnemann G., Cardoso M.C. Cell entry of arginine-rich peptides is independent of endocytosis. J. Biol. Chem. 2009;284:3370–3378. doi: 10.1074/jbc.M805550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothbard J.B., Jessop T.C., Wender P.A. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J. Am. Chem. Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 37.Edenhofer F. Protein transduction revisited: novel insights into the mechanism underlying intracellular delivery of proteins. Curr. Pharm. Des. 2008;14:3628–3636. doi: 10.2174/138161208786898833. [DOI] [PubMed] [Google Scholar]
- 38.Magzoub M., Pramanik A., Gräslund A. Modeling the endosomal escape of cell-penetrating peptides: transmembrane pH gradient driven translocation across phospholipid bilayers. Biochemistry. 2005;44:14890–14897. doi: 10.1021/bi051356w. [DOI] [PubMed] [Google Scholar]
- 39.Binder H., Lindblom G. Charge-dependent translocation of the Trojan peptide penetratin across lipid membranes. Biophys. J. 2003;85:982–995. doi: 10.1016/S0006-3495(03)74537-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herce H.D., Garcia A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA. 2007;104:20805–20810. doi: 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakai N., Takeuchi T., Matile S. Direct observation of anion-mediated translocation of fluorescent oligoarginine carriers into and across bulk liquid and anionic bilayer membranes. ChemBioChem. 2005;6:114–122. doi: 10.1002/cbic.200400256. [DOI] [PubMed] [Google Scholar]
- 42.Terrone D., Sang S.L., Silvius J.R. Penetratin and related cell-penetrating cationic peptides can translocate across lipid bilayers in the presence of a transbilayer potential. Biochemistry. 2003;42:13787–13799. doi: 10.1021/bi035293y. [DOI] [PubMed] [Google Scholar]
- 43.Thorén P.E., Persson D., Nordén B. Membrane destabilizing properties of cell-penetrating peptides. Biophys. Chem. 2005;114:169–179. doi: 10.1016/j.bpc.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi T., Startchev K., Gruenber J. Localization of lysobisphosphatidic acid-rich membrane domains in late endosomes. Biol. Chem. 2001;382:483–485. doi: 10.1515/BC.2001.059. [DOI] [PubMed] [Google Scholar]
- 45.Khalil I.A., Kogure K., Harashima H. Octaarginine-modified liposomes: enhanced cellular uptake and controlled intracellular trafficking. Int. J. Pharm. 2008;354:39–48. doi: 10.1016/j.ijpharm.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Chernomordik L.V., Kozlov M.M., Chizmadzhev Y.A. The shape of lipid molecules and monolayer membrane-fusion. Biochim. Biophys. Acta. 1985;812:643–655. [Google Scholar]
- 47.Engel A., Walter P. Membrane lysis during biological membrane fusion: collateral damage by misregulated fusion machines. J. Cell Biol. 2008;183:181–186. doi: 10.1083/jcb.200805182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katsov K., Müller M., Schick M. Field theoretic study of bilayer membrane fusion: II. Mechanism of a stalk-hole complex. Biophys. J. 2006;90:915–926. doi: 10.1529/biophysj.105.071092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frolov V.A., Dunina-Barkovskaya A.Y., Zimmerberg J. Membrane permeability changes at early stages of influenza hemagglutinin-mediated fusion. Biophys. J. 2003;85:1725–1733. doi: 10.1016/S0006-3495(03)74602-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dormitzer P.R., Nason E.B., Harrison S.C. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature. 2004;430:1053–1058. doi: 10.1038/nature02836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kozlovsky Y., Chernomordik L.V., Kozlov M.M. Lipid intermediates in membrane fusion: formation, structure, and decay of hemifusion diaphragm. Biophys. J. 2002;83:2634–2651. doi: 10.1016/S0006-3495(02)75274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le Blanc I., Luyet P.P., Gruenberg J. Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell Biol. 2005;7:653–664. doi: 10.1038/ncb1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.