

Abstract
The interaction of cytochrome c with ubiquinol-cytochrome c oxidoreductase (bc1 complex) has been studied for >30 years, yet many aspects remain unclear or controversial. We report the first molecular dynamic simulations of the cyt c-bc1 complex interaction. Contrary to the results of crystallographic studies, our results show that there are multiple dynamic hydrogen bonds and salt bridges in the cyt c-c1 interface. These include most of the basic cyt c residues previously implicated in chemical modification studies. We suggest that the static nature of x-ray structures can obscure the quantitative significance of electrostatic interactions between highly mobile residues. This provides a clear resolution of the discrepancy between the structural data and functional studies. It also suggests a general need to consider dynamic interactions of charged residues in protein-protein interfaces. In addition, a novel structural change in cyt c is reported, involving residues 21–25, which may be responsible for cyt c destabilization upon binding. We also propose a mechanism of interaction between cyt c1 monomers responsible for limiting the binding of cyt c to only one molecule per bc1 dimer by altering the affinity of the cytochrome c binding site on the second cyt c1 monomer.
Introduction
Cytochrome (cyt) c is an ancient and ubiquitous protein present across all kingdoms of life (1). Its well-known activity as an electron carrier in energy transduction has been widely studied and the role of cyt c in shuttling electrons between the bc1 complex and cytochrome c oxidase or photosynthetic reaction centers is well understood. In contrast, cyt c1, a component of the bc1 complex, is much less studied.
Unlike the majority of Type I cyts c, cyt c1 has a transmembrane helix and is roughly twice as big. Cyt c1 is a part of the high-potential chain of the bc1 complex catalytic mechanism (2,3): an iron sulfur protein (ISP) accepts one electron from quinol in the Qo site of cyt b, unbinds from cyt b, undergoes a conformational change and docks to cyt c1. The electron carried by the ISP transfers to the heme of cyt c1 and subsequently to the heme of cyt c. One turnover of cyt c is completed after undocking of the reduced species from the bc1 complex and reoxidation by cytochrome c oxidase or the photosynthetic reaction center.
The interaction between cyt c and its reaction partners, notably cytochrome oxidase and the bc1 complex, has long been studied as a model of molecular recognition. It is well established that the association of cyt c depends strongly on the ionic strength of the solution (4–8). This was taken to indicate the involvement of charged residues in the formation of the cyt c–cyt c1 or cyt c–oxidase complex. Because mitochondrial cyts c have an unusually large number of lysine residues (16–19 lysines out of 103–108 amino acids), these were the major focus of attempts to map the cyt c docking surface. Extensive studies, primarily using chemical modification of mammalian cyt c, have implicated a subset of lysines in the apparent affinity and steady-state turnover of cyt c. For binding to the bc1 complex, these experiments firmly established the importance of Lys-8, Lys-13, Lys-27, Lys-72, Lys-86, and Lys-87 residues in formation of the cyt c–c1 complex, with less-certain contributions from Lys-73 and Lys-79 (6,7,9–11). (Residue numbering for cyt c follows the vertebrate sequence; see Fig. S1 in the Supporting Material.) However, much less was known about the role of other residues involved in cyt c-c1 binding.
The crystal structure of the bc1 complex from yeast with bound iso-1-cyt c (one of two yeast cyts c) provided new opportunities to study the cyt c-c1 interaction. The structure, at 2.97 Å resolution, has cyt c1 fully oxidized and cyt c 75% oxidized (1KYO.pdb) (12). Surprisingly, it revealed a number of nonpolar interactions and a cation-π bond but no apparent salt bridges or hydrogen bonds in the cyt c-c1 interface. It was suggested that the substantial mismatch with the earlier reports identifying several lysine residues of cyt c might be due to differences in the amino-acid compositions of mammalian cyt c used in the previous works and iso-1-cyt c used in the structural studies (12). However, these changes affect only three residues and two are conservative: Lys-8 is replaced by threonine in yeast, Lys-13 is substituted by arginine, and Lys-72 is naturally trimethylated in yeast. Five other Lys residues previously implicated in binding and/or activity are common to both yeast and mammalian cyt c. However, according to the x-ray structure, Lys-27, Lys-73, and Lys-87, which are located near the cyt c-c1 binding interface, did not form any bonds with cyt c1, nor any other short-range electrostatic bonds. The positions of Lys-79 and Lys-86 were reportedly not well defined by the electron density maps (12).
Lange and Hunte (12) suggested that the cyt c-c1 complex is stabilized primarily through nonpolar interactions and that the conserved lysine residues contribute only through long-range electrostatic interactions and might be important for transient states or binding/unbinding events. A more recent crystal structure at higher resolution has cyt c and cyt c1 both reduced (3CX5.pdb, 1.9 Å) (13). This showed a similar binding pattern to that observed for the complex with mostly oxidized cytochromes, but also potentially two hydrogen bonds between Gln-170:NE2..Gln-16:O and Glu-201:OE2..Lys-11:NZ. (Cyt c1 residue numbers include the leader sequence, according to the yeast bc1 structure.) Lys-11, however, is not conserved between yeast and mammalian cyt c. As in the previous crystal structure, lysine residues at the interface were poorly defined and it was concluded that these residues cannot form hydrogen bonds or salt bridges with cyt c. However, the x-ray structure reveals no steric constraints preventing side-chain reorganization and formation of hydrogen bonds or salt bridges with cyt c1.
To address the discrepancy between the x-ray structure and the functional dependence on salt concentration, we explored the possible role of dynamic electrostatic interactions in the cyt c-cyt c1 binding interface. We describe the results of 10 independent molecular dynamics simulations of the bc1 complex in explicit membrane, or of just the water-soluble part of cyt c1 with cyt c bound. We show that most of the lysine residues previously identified in labeling experiments are highly dynamic and reorient from the positions modeled in the crystal structures to form hydrogen bonds or salt bridges with cyt c1, often with multiple partners. The protein-protein spacing does not change significantly, but the increased number of residues involved in the simulated cyt c-c1 interactions results in a significantly larger protein-protein contact area than reported for the complex (12). We conclude that the static nature of x-ray structures obscures the quantitative significance of nonbonded interactions between highly mobile residues, and that short-range electrostatic interactions are substantially involved in cyt c binding to cyt c1. In addition, local changes in the structures of cyt c and cyt c1 suggest mechanisms for differences in the thermodynamic properties of cyt c in its free and cyt c1 complexed forms. Our results also suggest how cyt c binding to one cyt c1 monomer affects the structure to lower the affinity of the cyt c binding site on the second cyt c1 monomer of the dimeric bc1 complex.
Methods
All atom coordinates were taken from the 1KYO.pdb (12). Fragments of antibody, used in crystallization, and Core I and II subunits of the bc1 complex were removed. The latter are not necessary for the bc1 complex catalytic activity (14), and are located on the opposite side of the membrane, >60 Å from the cyt c-c1 complex interface. This decreases the total number of atoms by ∼100,000. Both quinone binding sites of the bc1 complex, located ∼35 Å and ∼60 Å away, were left unoccupied. The 2Fe2S cluster of ISP was retained in five simulations performed on the entire bc1 complex and removed in two simulations. There was no noticeable difference in the cyt c-c1 interaction, as expected for short 10–20-ns simulations and a cofactor located >32 Å from the interface. When present, FeS cluster parameters were adopted from Izrailev et al. (15). Topology and parameters for trimethylated lysine of cyt c (M3Lys) were derived from lysine and tetramethylammonium (both present in CHARMM 27). C-heme topology and parameters were from Autenrieth et al. (16). B-hemes and all common residues were simulated with the standard CHARMM 27 force field (17). Structures for MD were generated with PSFGEN, MEMBRANE BUILDER, SOLVATE, and IONIZE plugins of VMD (18). The total system size with POPC membrane and all water molecules was 275,000 atoms. MD simulations were performed with NAMD2 (19).
Initially, all atoms except those located in the lipid tails were fixed and the system was brought to 310 K and equilibrated for 100 ps. Then, only nonhydrogen atoms of the protein were restrained and the system was packed for 150 ps at T = 310 K, P = 1 atm. Five simulations (freeA-E), were performed on this packed system with the same NPT settings and without any atom constraints. Two simulations (fixA, fixB) were set with frozen positions for atoms located >15 Å from either cyt c or c1. First, all atoms were released from constraints after the membrane packing step and the system was equilibrated for 1 ns. Then, all atoms farther than 15 Å from cyt c or from the water-soluble part of either of the two monomers of cyt c1 (residue numbers 62–262) were fixed, leaving ∼60,000 moveable atoms (Fig. 1). However, the forces from all atoms acting on the moveable parts were calculated.
Figure 1.
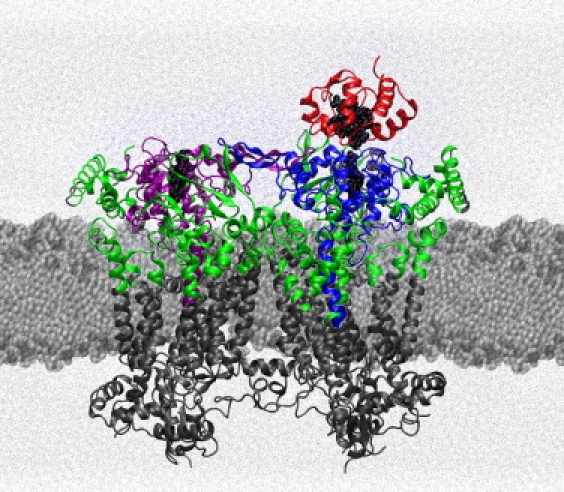
Structure of the yeast bc1 complex with cyt c bound to it. The yeast bc1 complex minus core subunits (coordinates from 1KYO.pdb (12)) is shown embedded in lipid bilayer with solvent. Colored areas show atoms of the complex not fixed in fixA and fixB simulations. Cyts c1, one in each monomer of the bc1 complex, are shown in blue and purple, cyt c in red, and all other polypeptide chains in green.
Three simulations of water-soluble cyt c1 with cyt c (cc1A-C) were performed in a water box, with the minimum distance between protein images resulting from the periodic boundary conditions always >15 Å. Pressure and temperature settings were as for the free bc1 dimer simulations.
All MD simulations were performed with the following parameters: cutoff distance 12 Å, time step 1 fs, and nonbonded forces calculated every two steps. Electrostatic forces were calculated with PME (20) every four steps. A Langevin bath and piston maintained temperature and pressure (21,22). The trajectories were evaluated from structures recorded at 10-ps intervals. Nonhydrogen atoms of the cyt c or c1 backbones were aligned against the 1KYO structure by least-squares fitting. Root-mean-square deviations (RMSDs) were then calculated for the backbone nonhydrogen atoms of each residue by comparison with the reference structure at each time point.
Contact areas between cyt c and cyt c1 were calculated as the difference between the sum of the solvent-accessible surface areas (SAS) of isolated cyts c and c1 and the SAS of the cyt c-c1 complex (23). SAS was calculated in VMD (18) using a probe radius of 1.4 Å.
Z-scores, a measure of the quality of the electron density map, were calculated for the 3CX5 structure using the Uppsala Electron Density Server (eds.bmc.uu.se (24)). This is defined as Z = (RSR–RSRavge)/σ, where RSR is the real space R-value for a particular residue, RSRavge the average real space R-value for all residues of the same type (e.g., lysine) in all structures with similar resolution in the PDB databank, and σ the standard deviation for RSRavge. Positive Z-scores indicate poorer electron density than expected from the overall resolution (in SD units); negative Z-scores imply better density.
Results
Structural changes in the backbones of cytochromes c and c1
We ran 10 independent MD simulations (Table S1): five 10–20 ns simulations of the entire bc1 dimer (minus core subunits) embedded in a POPC membrane in a water box (freeA-E); two 10-ns simulations of the whole complex but with only cyt c, the water-soluble parts of both cyt c1 subunits, and all atoms located closer than 15 Å allowed to move (fixA,B); three 85–95-ns simulations of a single water-soluble domain of cyt c1 with cyt c in a water box (cc1A-C). We define potentially significant, weak bonding interactions between pairs of atoms, following Lange and Hunte (12): C to C, O, or N distances ≤4 Å identify apolar bonds; two electronegative atoms (O or N) within 4 Å and with at least one H identify a potential hydrogen bond (H-bond). Electrostatic/polar interactions or bonds include salt bridges and potential H-bonds, as defined.
Cytochrome c
In all 10 simulations, the cyt c backbone RMSD from the starting crystal structure was <2 Å for most residues. However, there were two notable exceptions: a few N-terminal residues and a loop comprising residues 21–25 (Fig. 2). In both, the structural changes developed very fast and achieved the full magnitude in 0.5–5 ns suggesting a strong driving force behind them. While the flexibility of the N-terminus is not surprising, the sustained conformational change (≥5 Å) for the loop of residues 21–25 merits attention (see Discussion).
Figure 2.
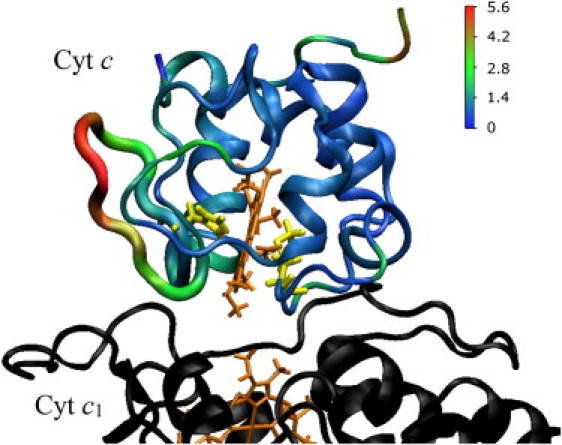
Backbone changes in cyt c bound to bc1 complex relative to the initial structure over the course of an MD simulation. The average backbone displacement (Å) was color-coded and mapped onto the cyt c structure in 1KYO. Hemes are drawn in tan, His and Met ligands in yellow.
Cytochrome c1
The differences between the crystal structure and the equilibrium structures of cyt c1 obtained in our MD simulations were substantially smaller than those for cyt c. Large changes were only observed for the tips (Glu-140, Gln-141, and Gly-142) of the extended β-strand fingers (residues 130–152), which connect the monomers of cyt c1. These fluctuations occurred to both cyt c1 monomers, and in some instances the RMSD exceeded 5 Å. This large-scale motion around the equilibrium position is consistent with the structural organization of the finger: an antiparallel β-strand with an overall length of ∼25 Å, and a flexible tip. In the crystal structure, the tip is attached to the other monomer of cyt c1 through a single hydrogen bond (Gly-142:O..Asn-161:ND), and there are no other polypeptide chains located closer than 5 Å to any part of the finger, nor are there any lipids closer than 5 Å in the MD simulations.
Other than the finger domain, there were three regions of the cyt c1 backbone with changes significantly greater than the average RMSD (∼1 Å) for residues of the entire globular domain: a cluster of acidic residues Asp-231, Asp-232, Glu-235, Glu-237, and Asp-238, and the neighboring Gly-239; part of a loop containing Gln-170, Gly-171, and Ala-172; and part of a loop with Ala-204, Gly-205, Val-206, and Ala-207. All these regions are located close to the cyt c, and the latter two loops also form the docking site for the ISP (see, e.g., 1BCC.pdb (25)).
Binding interface between cytochromes c and c1
The cyt c–cyt c1 contact area determined from the 1KYO structure was 880 Å2—the smallest contact area reported for any protein-protein complex. In seven out of 10 of our simulations, we observed a substantial increase in contact area over the first 3–5 ns, after which the contact area typically stabilized and fluctuated around average values of 1247–1555 Å2 (mean 1381 ± 168 Å2). The contact areas calculated from the bc1 dimer simulations were slightly smaller than for the simulations on the water-soluble cyt c1–cyt c complexes (mean 1499 ± 179 Å2). This may suggest that, despite the large distance from the cyt c-c1 binding interface, other bc1 subunits and the lipid membrane have an impact on cyt c docking, perhaps by decreasing the structural flexibility of cyt c1 or through long-range electrostatic interaction.
In two simulations, fixA and freeE, we initially observed partial unbinding of cyt c. During this event, the minimal contact area was ∼450 Å2 (Fig. 3 B, red trace). However, after 2–3 ns, cyt c re-bound to cyt c1, with an average contact area not statistically different from the values obtained in simulations where cyt c remained bound at all times. Finally, in one case (freeB), cyt c remained incompletely bound throughout the entire simulation (16.1 ns) and the contact area was 1090 ± 190 Å2.
Figure 3.
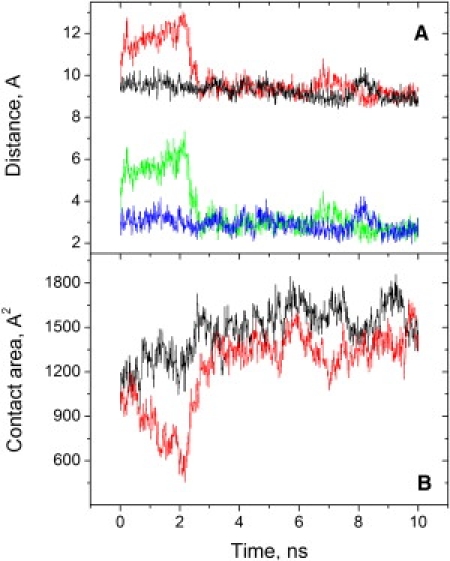
Two simulations of the cyt c-cyt c1 interaction. (A) Minimum distance between conjugated atoms of the hemes of cyts c and c1 (upper traces), and between any atoms (including hydrogens) of the hemes (lower traces). (B) Contact area between cyts c and c1. (Red and green, fixA; black and blue, fixB.)
In contrast with the substantial difference between the contact areas calculated from the crystal structure and from our MD simulations, the average minimum distance between conjugated atoms of the hemes of cyts c and c1 was 9.36 ± 0.49 Å in the simulations, compared to 9.4 Å in the crystal structure. The distances of closest approach between hydrogen atoms of the hemes of cyts c and c1 were typically 2.8–2.9 Å (Fig. 3 A, blue trace). This is too short to let water molecules in, yet presents a sizable vacuum gap on a tentative path for electron transfer between the hemes. During partial cyt c unbinding, the average distances were 2–3 Å greater than in the bound state, but water molecules entered between the hemes and the difference in electronic coupling could make electron transfer at least as fast in the more distant configuration (26).
Comparison of x-ray and MD structures
Reflecting the larger contact area between cyts c and c1, the MD simulations revealed many more residues forming interprotein bonds than seen in the crystal structure (Fig. 4). All five pairwise interactions reported by Lange and Hunte (12) to be involved in the cyt c–cyt c1 complex formation were present in all MD trajectories or segments thereof with bound cyt c (see also Table S2).
Figure 4.
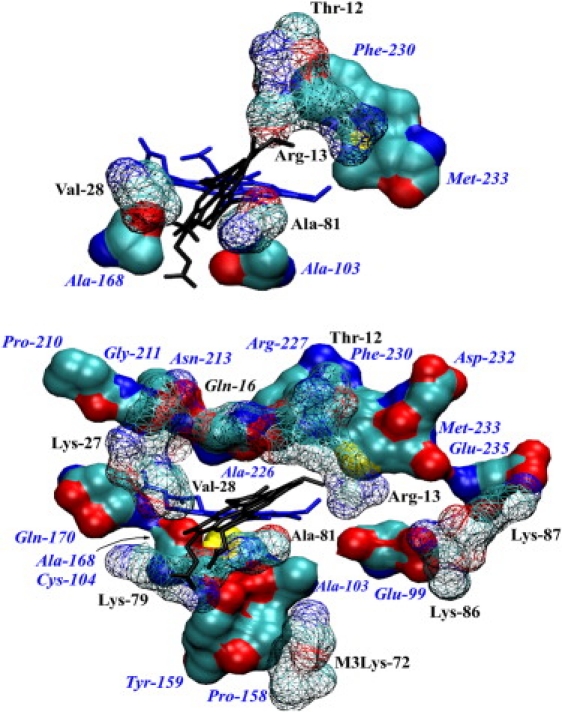
View of cyt c1 residues (solid surface beneath) interacting with cyt c (wire surface above) and their location relative to the hemes (blue, heme of cyt c1; black, heme of cyt c). (Top) Residues forming contact area in the 1KYO structure. (Bottom) Residues forming contact area in MD simulations (a snapshot from freeD). The views are similar but not identical. Shown are residues of cyt c (black labels) and c1 (blue labels) that have nonhydrogen atoms closer than 4 Å to nonhydrogen atoms from the other protein chain, with contact periods >70% for cyt c residues and >40% for cyt c1 residues (averaged over all trajectories for the bc1 complex, except freeB, which did not retain cyt c fully bound). Additional residues omitted for clarity are Ala-167 and Asn-169 of cyt c1 (see Table S2).
The nonpolar interaction between Thr-12 of cyt c and Phe-230 of cyt c1 was observed in at least 60% of trajectory points in all simulations, with a mean of 85%. Similarly, interactions of the side chain of Arg-13 of cyt c with Phe-230 and Met-233 of cyt c1 were present in all seven simulations performed with the bc1 complex dimer (mean 91%). Interestingly, in the three simulations performed with cyt c bound to a water-soluble domain of cyt c1, the Arg-13..Phe-230 cation-π interaction was replaced by an Arg-13..Glu-99 salt bridge while contact between Arg-13 and Met-233 was maintained.
The crystal structure apolar interaction between Val-28 of cyt c and Ala-168 of cyt c1 was present in >50% of frames in five out of seven simulations of the bc1 complex dimer. Moreover, in all 10 simulations Val-28 of cyt c was within 4 Å of 1–3 residues from the following list: Ala-167, Ala-168, Asn-169, Gln-170, and Gly-211. In the six simulations of the bc1 dimer where cyt c remained bound, nonhydrogen atoms of Val-28 of cyt c were within 4 Å of nonhydrogen atoms of cyt c1 in >85% of the frames. It was also more likely for Val-28 to form bonds with two or more different residues of cyt c1 than to not form any. This is reflected in the average number of bonding partners, which in four cases was in the range of 1.3–1.7, with standard deviations 0.6–0.8, suggesting highly dynamic interactions. Only in freeA was the average bond population <1 (0.76 ± 0.64), but still ≈70% of frames showed van der Waals contacts between Val-28 of cyt c and cyt c1.
The fifth bond between cyt c and bc1 unambiguously present in the 1KYO structure, Ala-81..Ala-103, was observed in all trajectories, or segments thereof, where cyt c was fully bound to the bc1 complex. Overall, the interaction of Ala-81 with cyt c1 was similar to that of Val-28 described above: in addition to Ala-103, at least one of the following residues was also within 4 Å of Ala-81 in >50% of the recorded frames: Cys-104, Tyr-159, and Asn-169, yielding an average contact time >90%.
Potential H-bonds and salt bridges
In the x-ray structure analysis, only two polar interactions, Lys-79:NZ..Ala-164:O and Lys-86:NZ..Glu-235:OE2, were considered, but both lysines were poorly resolved and Lange and Hunte (12) concluded that they did not contribute significantly to the cyt c-c1 interaction. In all MD simulations with fully bound cyt c, Lys-79:NZ interacted significantly with Ala-167:O, Ala-168:O, Asn-169:O, and Gln-170:OE1 of cyt c1, often with more than one simultaneously. It was also one of several partners to Gln-141:OE1 in the finger of the other cyt c1 monomer. On average, Lys-79 was involved in favorable electrostatic interactions with cyt c1 53 ± 8% of the time. However, none of our 10 simulations demonstrated direct involvement with Ala-164 of cyt c1 in cyt c docking. Thus, MD results confirm the mobility of Lys-79 and a substantial involvement in polar bonds, but reveal consistent interactions with cyt c1 via multiple partners (Table S2).
A salt bridge between Lys-86 of cyt c and Glu-235 of cyt c1 was observed in >30% of frames in only three MD simulations. However, Lys-86 and Lys-87 of cyt c compete for multiple partners in cyt c1, including several acidic side chains (Glu-99, Asp-232, and Glu-235) and the carbonyl oxygen of Met-233 (Fig. 5). There were two distinct interaction patterns for the NZ atoms of Lys-86 and Lys-87 of cyt c. In six simulations, among other electrostatic interactions, we observed a relatively stable salt bridge between Lys-86 and Glu-99 correlated with the presence of a salt bridge between Lys-87 and Glu-235. The other major binding mode was a salt bridge between Lys-87 and Asp-232 and a sequence of fast rearranging electrostatic interactions between Lys-86 and three or four of Glu-99, Asp-232, Met-233:O, and Glu-235. Notably, the sum of the fractions of time during which electrostatic bonds were formed by Lys-86 ranged from 1.78 in freeB to 2.37 in fixB. In other words, in this binding mode, Lys-86 formed salt bridges or hydrogen bonds with two or more residues of cyt c1 at the same time, on average. Lys-86 and -87 were involved in favorable electrostatic interactions (predominantly salt bridges) with cyt c1 86 ± 6% and 67 ± 8% of the time, respectively, and at least one was maintained by one or the other at all times (Table S2).
Figure 5.
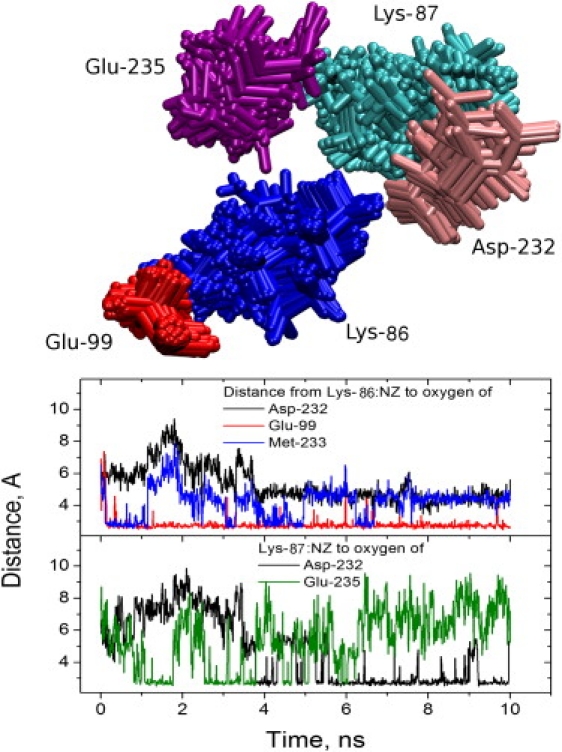
NZ atoms in Lys-86 and Lys-87 of cyt c form dynamic electrostatic bonds with carboxyl oxygens of Glu-99, Asp-232, and Glu-235 of cyt c1. (Top) Superimposed positions of the residues during the course of a simulation. (Bottom, upper panel) Minimum distance between NZ atom of Lys-86 from cyt c and the closest oxygen atom of Asp-232 (black), Glu-99 (red), and Met-233:O (blue trace) of cyt c1. (Bottom, lower panel) Minimum distance between the NZ atom of Lys-87 of cyt c and the closest oxygen of Asp-232 (black) and Glu-235 (green) of cyt c1.
Gln-16 is a universally conserved residue among various cytochromes c (see Fig. S1). It is located within the c-type cytochrome heme-binding motif CXXCH (CXQCH), close to the binding interface between cyt c and its binding partners, but, to the best of our knowledge, its role in cyt c binding has not been noted. The 1KYO crystal structure of the bc1 complex with bound cyt c (c-type hemes oxidized) does not reveal Gln-16 involvement in cyt c docking. A more recent structure with reduced c-type hemes (3CX5.pdb (13)) places the NE2 atom of Gln-170 of cyt c1 3.8 Å from the carbonyl oxygen of Gln-16. However, a sucrose molecule, an artifact of the crystallization conditions, was bound nearby and interacted directly with these and other residues in both proteins.
All our simulations showed extensive electrostatic interactions between Gln-16 and cyt c1. In the majority of frames, Gln-16 formed an H-bond with the backbone of Arg-227, i.e., Gln-16:NE2..Arg-227:O. Many other potential H-bonding pairs were simultaneously observed, including Gln-16:NE2 with Asn-213:OD1, Gly-211:O, and Ser-212:O, and Gln-16:OE1 with Arg-227:N and Asn-213:ND2. In effect, Gln-16 was in almost continuous polar interaction with cyt c1 (on average, in 88 ± 6% of frames).
Lys-27 is another highly conserved residue on the cyt c surface. As for Gln-16, the 1KYO structure shows all atoms of Lys-27 >4 Å away from cyt c1. In most MD simulations, however, the NZ atom of the side chain was bound through two hydrogen bonds to the carbonyl oxygens of Leu-15 and Gln-16 of cyt c. In addition, in all MD simulations there were H-bonds between the backbone atoms of Lys-27 of cyt c and Gln-170 and/or Pro-210 of cyt c1, either directly or through a shared water molecule.
Lys-72, located near the cyt c docking interface, is universally conserved but it is trimethylated in yeast, preventing H-bond formation but not charge-charge interactions. (This modification does not occur in higher organisms and its role is unclear.) In the 1KYO structure, trimethylated Lys-72 (designated M3Lys-72) does not show any bonds with cyt c1. In contrast, all MD simulations revealed that the trimethylamino group of M3Lys-72 was in almost constant contact (<4 A) with mostly electronegative atoms of cyt c1. In the six trajectories with cyt c consistently bound to bc1, the average contact time with one or more of Ala-103:O, Gly-157:O, Pro-158:O, and Tyr-159:OH of cyt c1 was 76 ± 11%, and 90 ± 3% with any nonhydrogen atom of cyt c1 (Table S2).
Both cyt c1 monomers interact with bound cyt c
One of the most surprising results from the work of Lange and Hunte (12) and Solmaz and Hunte (13) was that, even in the presence of excess cyt c, only one cyt c molecule was bound to the bc1 complex dimer. The origin of this asymmetry was not obvious from the x-ray structure, but in the MD simulations a significant difference was observed for the fingerlike domains of the two cyt c1 monomers. We observed rapid structural changes near the tip of the finger belonging to the cyt c1 monomer without cyt c bound, which resulted in effective bond contacts between the tip and cyt c bound to the other cyt c1 monomer (Fig. 6). On average, >80% of frames showed nonhydrogen atom contacts, and 75% of these were polar, especially the formation of dynamic hydrogen bonds between the side chain of Gln-141 of cyt c1 and one or more of the following atoms of cyt c: Ser-47:O/OG, Tyr-48:O, Asp-50:N, Asp-50:OD1/2, Ala-51:N, Gly-77:N, and Lys-79:NZ (Table S3). In the 3CX5 structure, the electron density corresponding to Asp-50 of cyt c is especially poorly defined for the nominal 1.9 Å resolution (Z-score: +5.54), while Gln-141 of cyt c1, Ser-47, Tyr-48, and Lys-79 of cyt c are all worse than average (Z-scores in the 1.5–1.8 range).
Figure 6.
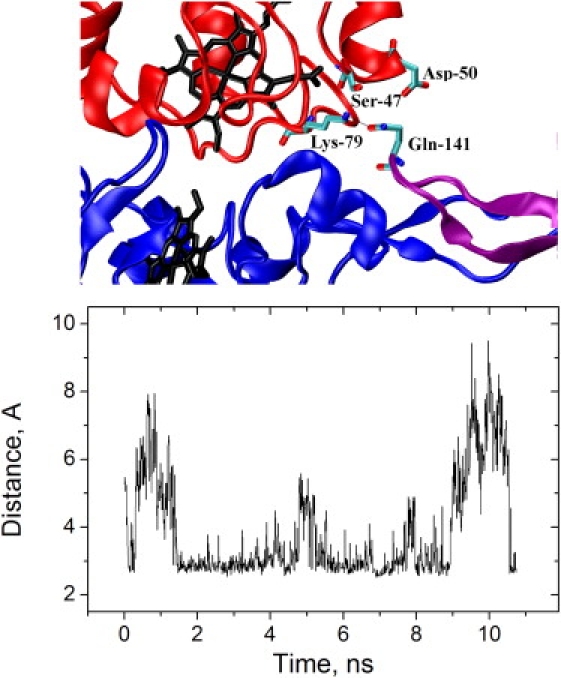
The second monomer of cyt c1 is also involved in cyt c binding. (Top) View of fingers of both cyts c1 (blue and purple) in the bc1 complex dimer and cyt c (red) bound to one of them (blue). In this snapshot, Gln-141:OE2 of c1 forms a H-bond with Lys-79:NZ of cyt c. Other cyt c residues shown (Ser-47 and Asp-50) are also significant partners with Gln-141 (see Table S3). (Bottom) Minimum distance (Å) between any electronegative atom of bound cyt c and any electronegative atom of the tip of the finger from the cyt c1 monomer without cyt c bound. Data from freeC.
Discussion
Conformational change in cyt c
Changes in the physico-chemical properties of cyt c upon binding to other proteins are a relatively well-established phenomenon. In addition to changes in redox potential upon binding of cyt c to cytochrome c oxidase (27) and photosynthetic reaction centers (28), Yu et al. (29) demonstrated a decrease in thermal stability of cyt c and an increase in stability of cyt c1 upon binding. The cyt c atom coordinates in both crystal structures of the bc1 complex with cyt c bound (1KYO (12) and 3CX5 (13)) are generally very similar to the structure of free cyt c. However, one region of the bound cyt c structure was poorly resolved, and the MD simulations showed a substantial and highly reproducible structural change in cyt c.
In all simulations we observed a fast (0.5–5 ns) conformational change in cyt c in the loop formed by residues 21–25, which flipped toward the cyt c1 surface. The magnitude of the displacement of the backbone atoms of Gly-23 and Gly-24 exceeded 5 Å. This is substantially more than the 2.97 Å global resolution of the 1KYO structure, but this region was poorly resolved. The conformational change is accompanied by the loss of several internal H-bonds involved in maintaining proper folding of cyt c near its histidine heme ligand, which would contribute to the thermal destabilization of cyt c in the cyt c-c1 complex.
At the same time, a number of residues of cyt c1 located near the cyt c-c1 binding interface showed lower fluctuation amplitudes in the monomer with cyt c bound compared to the monomer with unoccupied cyt c binding site. This is consistent with a higher denaturation temperature of cyt c1 in the presence of bound cyt c (29).
It is unlikely that the conformational change in cyt c is an artifact due to shortcomings of the force field, because no conformational changes were observed in simulations of free cyt c (coordinates for yeast iso-1-cyt c from 1CRG.pdb (30)). Furthermore, a number of computational studies on c-type cytochromes (free and bound to the photosynthetic reaction center), using the same force field parameters, did not report any conformational changes (16,31,32). However, we have observed a similar conformational change for cyt c bound to cytochrome c peroxidase, in three independent simulations (O. Kokhan, unpublished).
We do not exclude the possibility that there are indications of this conformational change in the raw data from which the 1KYO structure was derived. We have analyzed the electron density data file for the 3CX5 structure of the bc1 complex with reduced cyts c and c1, deposited with the Uppsala Electron Density Server (24), and it is clear that the accuracy of the electron density fitting corresponding to the residues of this cyt c loop was substantially lower than expected for a structure with 1.9 Å resolution (Z-scores: Glu-21 +2.17, Lys-22 +2.74, Gly-23 +5.02, Gly-24 +2.78, and Pro-25 +2.61; see also Fig. S2) This is despite the fact that reduced cyt c is significantly more compact and thermally stable than the oxidized form (4,33–35). In contrast, the same part of free yeast iso-1-cyt c is well defined in structures with a similar resolution. For example, in 1CRH (reduced) and 1CRG (oxidized) (30), this loop is defined better than average (Fig. S2). Such differences in the quality of the electron density data would be consistent with either increased loop flexibility when cyt c is bound to the bc1 complex, or an overlooked conformational change. The electron density data for 1KYO are not available for this analysis, but residues 23–25 of cyt c were manually positioned (12), strongly suggesting that this part was poorly resolved.
Electrostatic interactions between cytochromes c and c1
The widely reported, strong effect of ionic strength on cyt c-c1 binding may not be reliably inferred from kinetic assays but is convincingly established by equilibrium methods (8,11). The ability of moderate salt concentrations to weaken cyt c-c1 interaction is interpreted as an indicator of the involvement of electrostatic interactions in complex formation (8,36). Despite its modest size, cyt c has 16–19 lysine residues and selective chemical modification has implicated as many as eight of these in binding and for efficient turnover (e.g., (9,11)). Six of these are conserved in yeast iso-1-cyt c (Lys-27, -72, -73, -79, -86, -87). Thus, it was a surprise that the crystal structure of the bc1 complex with cyt c (1KYO) showed no H-bonds or salt bridges between cyts c and c1.
For the conserved lysine residues previously implicated in cyt c binding, but with no apparent short-range interactions seen in the crystal structures, two possibilities were suggested (12,13):
-
1.
They may be important for cyt c orientation on the cyt c1 surface or in the formation of transient complexes during binding and unbinding events.
-
2.
They may stabilize the cyt c-c1 complex through long-range electrostatic interactions.
However, involvement of the lysine residues only in transient intermediate states is not consistent with the equilibrium results (9,11). Furthermore, our MD simulations reveal that five of the six cyt c lysine residues conserved in yeast and previously implicated in the cyt c-c1 interaction (Lys-27, Lys-72, Lys-79, Lys-86, and Lys-87) reorient and are substantially involved in short-range (<4 Å) though rapidly fluctuating electrostatic bonds.
The large number of changes in position of side chains at the cyt c-c1 binding interface observed in the first 1–5 ns indicates that the starting structure had much higher energy. The relatively short lifetime of H-bonds and salt bridges and their fast rearrangements in MD simulations are consistent with the expected energies of only a few kcal/mol; for example, Kumar and Nussinov (37) calculated ΔΔG = −3.29 ± 3.11 kcal/mol for solvent-exposed salt bridges. With typical periods of protein normal mode oscillations of ∼0.1–10 ps, a bond lifetime can be shorter than 1 ns. An important feature observed in our simulations was the competition of various cyt c1 residues to form electrostatic bonds with residues Gln-16, Lys-27, Lys-79, Lys-86, and Lys-87 of cyt c. Each of these cyt c residues had at least two alternative binding sites on the cyt c1 surface with comparable occupancies, and was involved in short-range electrostatic interactions for 70–90% of the time (Table S2). These will be quantitatively much more significant than any long-range contributions. Based on our MD simulations and previous experimental reports on the cyt c-c1 interaction, we suggest that short-range electrostatic interactions, i.e., H-bonds and salt bridges, do indeed play an important role in cyt c binding and retention on the surface of cyt c1. However, multiple binding configurations and fast rearrangements result in delocalized electron density not amenable to refinement to a single set of atomic coordinates. The average positions of residues involved in electrostatic interactions over an entire MD trajectory are similar to those in the crystal structure, yet the interpretations and their implied roles in docking are very different: maintaining a significant bond with one of several possible partners in nearly all recorded structures versus positioned between several binding partners but too distant to form a H-bond or a salt bridge with any.
Cation-π interactions in cyt c docking
Hunte and co-workers have emphasized the importance of cation-π interactions in cyt c docking to its physiological binding partners, and especially to bc1. The contribution of the interaction between Arg-13 of cyt c and Phe-230 of cyt c1 was estimated, using CAPTURE (38), to be −7.07 kcal/mol for the 3CX5 structure (13). However, Paddock et al. (39) experimentally estimated the energy of a cation-π interaction in the interface between the photosynthetic reaction center and cyt c2 as only −0.6 kcal/mol, while values from −2.90 to −7.76 kcal/mol were calculated for three cocrystal structures. The calculated energies of the cation-π interaction for Arg-13..Phe-230 in three different cyt c-bc1 structures ranged from −3.65 to −7.34 kcal/mol.
Complexes of cytochrome c peroxidase with cyt c (2PCB and 2PCC (40), 2GB8 (41)) do not exhibit any energetically favorable cation-π interactions, undermining any general trend for cation-π interactions to be important for cyt c docking to its binding partners. Thus, at this time it seems advisable to defer discussion of the importance of this type of bond for cyt c docking to bc1 until some quantitative experimental data are available.
Similarities in cyt c-cyt c1 and cyt c–CCP interactions
A discrepancy between an expected domination of electrostatic forces and the results derived from a crystal structure is also seen for the cyt c–cytochrome c peroxidase (CCP) complex. In the crystal structure obtained with yeast iso-1-cyt c and yeast CCP (2PCC.pdb (40)), only one H-bond was observed, between Glu-290 of CCP and Asn-70 of yeast cyt c. Two other potential pairs (Asp-34..Lys-87 and Glu-290..Lys-73) were considered too distant (>4 Å) to form significant bonds. Indeed, analyses of static structures of this and other binary protein complexes have led to suggestions that a very limited involvement of ionic interactions may be a general property of redox complexes (23,42). However, this is often in conflict with ionic-strength-dependent binding similar to that described for cyt c.
A very different picture of the electrostatic interactions at the yeast cyt c-CCP binding interface is given by the NMR structure of the complex (2GB8.pdb (41)). Upon analyzing this structure, which contains the 20 lowest energy states obtained under similar ionic strength conditions as 2PCC, it is clear that there are substantially more interprotein electrostatic interactions than was seen in the x-ray structure. In addition to the single H-bond in 2PCC, there are at least 13 more pairs of atoms potentially forming interprotein electrostatic bonds in some or all of the reported 20 lowest energy states in the 2GB8 structure (Table S4).
A detailed analysis of cyt c-CCP interactions falls outside the scope of this work. However, the universally conserved Lys-86 and Lys-87 residues of cyt c exhibit the same dynamic behavior in the 2GB8 NMR structure of the cyt c-CCP complex as we described for the cyt c-c1 interface (Fig. 5), based on our MD simulations (Fig. S3).
In contrast with the electrostatic bonds, the nonpolar interactions were identical in the NMR and x-ray structures. The contact area between cyt c and yeast CCP, calculated from all configurations of the 2GB8 structure, was 1440 ± 54 Å2. This is similar to the values calculated for the cyt c–c1 contact area from our simulations, and significantly larger than the 1170 Å2 calculated from the 2PCC x-ray structure (23), which was the lowest known protein-protein contact area at that time.
Our finding of dynamic short-range interactions in the cyt c-c1 complex, and this brief review of cyt c-CCP, indicate that any generalizations on the role of ionic residues in binding interfaces should be tested with methods providing a dynamic treatment of the interface, e.g., as offered by MD simulations.
Crosstalk between cyt c1 monomers
Allosteric and regulatory interactions between components of the bc1 complex have been widely proposed, but with little consensus (2,3,43–48). To the best of our knowledge, however, there are no previous suggestions of functional interaction between the two cyts c1 of the bc1 complex, nor has a role been discussed for the fingerlike β-strands of cyt c1, which physically connect the two monomers. This structural feature of cyt c1 is universally conserved in mitochondria and in many bacteria.
In our MD simulations we observed that the tip of the finger belonging to the monomer without cyt c bound to it was able to form dynamic H-bonds with cyt c. We suggest that the rigid β-strand could transfer the motion of the tip along its length toward the base and affect the position of a cluster of acidic residues (notably Glu-99) involved in cyt c docking, thereby affecting the affinity of one monomer after cyt c binding to the other. This structural mechanism, with mechanical motion spanning over 30 Å, may explain why in all three available crystal structures of the bc1 complex cocrystallized with excess cyt c (1KYO, 3CX5 with iso-1-cyt c, and 3CXH with iso-2-cyt c, which is quite different from iso-1) only one cyt c1 monomer had cyt c bound to it, while the other monomer had an empty cyt c binding site (12,13).
Acknowledgments
This work was supported by National Institutes of Health grants No. R01-GM053508 (to C.A.W) and Nos. R01-GM086749 and P41-RR05969 (to E.T.). The simulations were performed using TeraGrid resources (grant No. MCA06N060) and the Turing cluster of the Computational Science and Engineering program at the University of Illinois at Urbana-Champaign.
Supporting Material
References
- 1.Bertini I., Cavallaro G., Rosato A. Cytochrome c: occurrence and functions. Chem. Rev. 2006;106:90–115. doi: 10.1021/cr050241v. [DOI] [PubMed] [Google Scholar]
- 2.Crofts A.R., Holland J.T., Kuras M.G. The Q-cycle reviewed: how well does a monomeric mechanism of the bc1 complex account for the function of a dimeric complex? Biochim. Biophys. Acta. 2008;1777:1001–1019. doi: 10.1016/j.bbabio.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swierczek M., Cieluch E., Osyczka A. An electronic bus bar lies in the core of cytochrome bc1. Science. 2010;329:451–454. doi: 10.1126/science.1190899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Margoliash E., Schejter A. Cytochrome c. Adv. Protein Chem. 1966;21:113–286. doi: 10.1016/s0065-3233(08)60128-x. [DOI] [PubMed] [Google Scholar]
- 5.Yu C.-A., Yu L., King T.E. Kinetics of electron transfer between cardiac cytochrome c1 and c. J. Biol. Chem. 1973;248:528–533. [PubMed] [Google Scholar]
- 6.Speck S.H., Ferguson-Miller S., Margoliash E. Definition of cytochrome c binding domains by chemical modification: kinetics of reduction with beef mitochondrial reductase and functional organization of the respiratory chain. Proc. Natl. Acad. Sci. USA. 1979;76:155–159. doi: 10.1073/pnas.76.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith H.T., Ahmed A.J., Millett F. Electrostatic interaction of cytochrome c with cytochrome c1 and cytochrome oxidase. J. Biol. Chem. 1981;256:4984–4990. [PubMed] [Google Scholar]
- 8.Sarewicz M., Borek A., Osyczka A. Demonstration of short-lived complexes of cytochrome c with cytochrome bc1 by EPR spectroscopy: implications for the mechanism of interprotein electron transfer. J. Biol. Chem. 2008;283:24826–24836. doi: 10.1074/jbc.M802174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed A.J., Smith H.T., Millett F. Effect of specific lysine modification on the reduction of cytochrome c by succinate-cytochrome c reductase. Biochemistry. 1978;17:2479–2483. doi: 10.1021/bi00606a003. [DOI] [PubMed] [Google Scholar]
- 10.König B.W., Osheroff N., Margoliash E. Mapping of the interaction domain of purified cytochrome c1 on cytochrome c. FEBS Lett. 1980;111:395–398. doi: 10.1016/0014-5793(80)80835-0. [DOI] [PubMed] [Google Scholar]
- 11.Rieder R., Bosshard H.R. Comparison of the binding sites on cytochrome c for cytochrome c oxidase, cytochrome bc1 complex, and cytochrome c1. J. Biol. Chem. 1980;255:4732–4739. [PubMed] [Google Scholar]
- 12.Lange C., Hunte C. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proc. Natl. Acad. Sci. USA. 2002;99:2800–2805. doi: 10.1073/pnas.052704699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solmaz S.R.N., Hunte C. Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. J. Biol. Chem. 2008;283:17542–17549. doi: 10.1074/jbc.M710126200. [DOI] [PubMed] [Google Scholar]
- 14.Deng K., Shenoy S.K., Yu C.-A. Reconstruction of mitochondrial processing peptidase from the core proteins (subunits I and II) of bovine heart mitochondrial cytochrome bc1 complex. J. Biol. Chem. 2001;276:6499–6505. doi: 10.1074/jbc.M007128200. [DOI] [PubMed] [Google Scholar]
- 15.Izrailev S., Crofts A.R., Schulten K. Steered molecular dynamics simulation of the Rieske subunit motion in the cytochrome bc1 complex. Biophys. J. 1999;77:1753–1768. doi: 10.1016/S0006-3495(99)77022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Autenrieth F., Tajkhorshid E., Luthey-Schulten Z. Classical force field parameters for the heme prosthetic group of cytochrome c. J. Comput. Chem. 2004;25:1613–1622. doi: 10.1002/jcc.20079. [DOI] [PubMed] [Google Scholar]
- 17.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 18.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 19.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darden T., York D., Pedersen L. Particle mesh Ewald: an N∗log(N) method for Ewald sums in large systems. J. Chem. Phys. B. 1993;98:10089–10092. [Google Scholar]
- 21.Feller S.E., Zhang Y.H., Brooks B.R. Constant pressure molecular dynamics simulation—the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 22.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 23.Lo Conte L., Chothia C., Janin J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 24.Kleywegt G.J., Harris M.R., Jones T.A. The Uppsala Electron-Density Server. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z., Huang L., Kim S.H. Electron transfer by domain movement in cytochrome bc1. Nature. 1998;392:677–684. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 26.Page C.C., Moser C.C., Dutton P.L. Natural engineering principles of electron tunneling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 27.Schroedl N.A., Hartzell C.R. Oxidative titrations of reduced cytochrome aa3: influence of cytochrome c and carbon monoxide on the midpoint potential values. Biochemistry. 1977;16:4961–4971. doi: 10.1021/bi00642a004. [DOI] [PubMed] [Google Scholar]
- 28.Larson J.W., Wraight C.A. Preferential binding of equine ferricytochrome c to the bacterial photosynthetic reaction center from Rhodobacter sphaeroides. Biochemistry. 2000;39:14822–14830. doi: 10.1021/bi0012991. [DOI] [PubMed] [Google Scholar]
- 29.Yu C.A., Steidl J.R., Yu L. Microcalorimetric studies of the interactions between cytochromes c and c1 and of their interactions with phospholipids. Biochim. Biophys. Acta. 1983;736:226–234. doi: 10.1016/0005-2736(83)90288-2. [DOI] [PubMed] [Google Scholar]
- 30.Berghuis A.M., Guillemette J.G., Brayer G.D. The role of a conserved internal water molecule and its associated hydrogen bond network in cytochrome c. J. Mol. Biol. 1994;236:786–799. doi: 10.1006/jmbi.1994.1189. [DOI] [PubMed] [Google Scholar]
- 31.Autenrieth F., Tajkhorshid E., Luthey-Schulten Z. Role of water in transient cytochrome c2 docking. J. Chem. Phys. B. 2004;108:20376–20387. [Google Scholar]
- 32.Pogorelov T.V., Autenrieth F., Luthey-Schulten Z.A. Cytochrome c2 exit strategy: dissociation studies and evolutionary implications. J. Phys. Chem. B. 2007;111:618–634. doi: 10.1021/jp064973i. [DOI] [PubMed] [Google Scholar]
- 33.Cruanes M.T., Rodgers K.K., Sligar S.G. Protein electrochemistry at high pressure. J. Am. Chem. Soc. 1992;114:9660–9661. [Google Scholar]
- 34.Takano T., Dickerson R.E. Conformation change of cytochrome c. II. Ferricytochrome c refinement at l.8 Å and comparison with the ferrocytochrome structure. J. Mol. Biol. 1981;153:95–115. doi: 10.1016/0022-2836(81)90529-5. [DOI] [PubMed] [Google Scholar]
- 35.Trewhella J., Carlson V.A.P., Heidorn D.B. Differences in the solution structures of oxidized and reduced cytochrome c measured by small-angle x-ray scattering. Biochemistry. 1988;27:1121–1125. doi: 10.1021/bi00404a007. [DOI] [PubMed] [Google Scholar]
- 36.Engstrom G., Rajagukguk R., Millett F. Design of a ruthenium-labeled cytochrome c derivative to study electron transfer with the cytochrome bc1 complex. Biochemistry. 2003;42:2816–2824. doi: 10.1021/bi027213g. [DOI] [PubMed] [Google Scholar]
- 37.Kumar S., Nussinov R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999;293:1241–1255. doi: 10.1006/jmbi.1999.3218. [DOI] [PubMed] [Google Scholar]
- 38.Gallivan J.P., Dougherty D.A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. USA. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paddock M.L., Weber K.H., Okamura M.Y. Interactions between cytochrome c2 and the photosynthetic reaction center from Rhodobacter sphaeroides: the cation-π interaction. Biochemistry. 2005;44:9619–9625. doi: 10.1021/bi050651d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelletier H., Kraut J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 41.Volkov A.N., Worrall J.A.R., Ubbink M. Solution structure and dynamics of the complex between cytochrome c and cytochrome c peroxidase determined by paramagnetic NMR. Proc. Natl. Acad. Sci. USA. 2006;103:18945–18950. doi: 10.1073/pnas.0603551103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crowley P.B., Carrondo M.A. The architecture of the binding site in redox protein complexes: implications for fast dissociation. Prot. Struct. Funct. Bioinf. 2004;55:603–611. doi: 10.1002/prot.20043. [DOI] [PubMed] [Google Scholar]
- 43.Gopta O.A., Feniouk B.A., Mulkidjanian A.Y. The cytochrome bc1 complex of Rhodobacter capsulatus: ubiquinol oxidation in a dimeric Q-cycle? FEBS Lett. 1998;431:291–296. doi: 10.1016/s0014-5793(98)00768-6. [DOI] [PubMed] [Google Scholar]
- 44.Gutierrez-Cirlos E.B., Trumpower B.L. Inhibitory analogs of ubiquinol act anti-cooperatively on the yeast cytochrome bc1 complex: evidence for an alternating, half-of-the-sites mechanism of ubiquinol oxidation. J. Biol. Chem. 2002;277:1195–1202. doi: 10.1074/jbc.M109097200. [DOI] [PubMed] [Google Scholar]
- 45.Covian R., Trumpower B.L. Regulatory interactions in the dimeric cytochrome bc1 complex: the advantages of being a twin. Biochim. Biophys. Acta. 2008;1777:1079–1091. doi: 10.1016/j.bbabio.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu C.-A., Cen X., Xia D. Domain conformational switch of the iron-sulfur protein in cytochrome bc1 complex is induced by the electron transfer from cytochrome bL to bH. Biochim. Biophys. Acta. 2008;1777:1038–1043. doi: 10.1016/j.bbabio.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 47.Cooley J.W., Lee D.W., Daldal F. Across membrane communication between the Qo and Qi active sites of cytochrome bc1. Biochemistry. 2009;48:1888–1899. doi: 10.1021/bi802216h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarewicz M., Dutka M., Osyczka A. Magnetic interactions sense changes in distance between heme bL and the iron-sulfur cluster in cytochrome bc1. Biochemistry. 2009;48:5708–5720. doi: 10.1021/bi900511b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.