

Abstract
Modular proteins contain individual domains that are often connected by flexible, unstructured linkers. Using a model system based on the GB1 domain, we constructed tandem repeat proteins and investigated the rotational diffusion and long-range angular ordering behavior of individual domains by measuring NMR relaxation parameters and residual dipolar couplings. Although they display almost identical protein-solvent interfaces, each domain exhibits distinct rotational diffusion and alignment properties. The diffusion tensor anisotropy of the N-terminal domain (NTD) is D‖/D⊥ = 1.5–1.6, similar to that of single-GB1 domains (D‖/D⊥ = 1.6–1.7), whereas the value for the C-terminal domain (CTD) is D‖/D⊥ = 2.0–2.2. In addition, the two domains have different rotational correlation times. These effects are observed for linkers of three to 24 residues, irrespective of linker length. The NTD and CTD also differ in their degree of magnetic alignment, even with a flexible linker of 18 residues, exhibiting Da values of 7.7 Hz and 9.7 Hz, respectively. Our results suggest that diffusion differences and long-range influences may persist in modular protein systems, even for systems that have highly flexible linkers and exhibit no domain-domain or domain-linker interactions.
Introduction
A large number of proteins encode multiple functionalities that are frequently located on individual domains along the same polypeptide chain. The resulting modularity bestows distinct advantages on such systems because it offers a mechanism for controlling the spatial localization of binding elements (1) as well as chemical processes through modulation of the relative domain orientations (2). In this manner it is possible to influence on- and off-states in signaling, enzymatic reactions (3), and transport processes within a cell.
In nature, each modular protein system has evolved to encompass unique features that are exquisitely selected for the individual case. These may include specific transient domain-domain interactions and interdomain linkers of defined lengths, composition, and structure (or lack thereof), with dynamic properties tailored to the specific functions at hand. These features can be highly optimized, and, considering linker length and composition alone, small changes can result in a significant loss of activity (4).
Over the last three decades, NMR relaxation-based methodologies have provided a particularly fertile avenue for investigating protein dynamics and diffusion. In particular, the Lipari-Szabo (5) model-free (MF) formalism emerged as the most widely applied and successful approach (6,7). It has enabled the interpretation of NMR relaxation parameters in terms of global domain diffusion and internal molecular-dynamics parameters. However, because the MF formalism requires statistical independence of global and internal motions, it was primarily used to analyze the internal dynamics of relatively rigid systems (6).
Therefore, progress in experimentally characterizing and interpreting interdomain dynamics was initially modest. Work on systems with two or more domains has generally focused on proteins with a well-defined overall structure in which the individual domain diffusion tensors are very similar to the one measured for the overall system (8) or are assumed to be identical (9). In these types of systems, NMR relaxation and residual dipolar couplings (RDCs) often permit determination of the relative domain orientations. The relatively small degree of motion due to interdomain dynamics for these cases does not alter the overall protein-solvent interaction, and therefore the protein as a whole may still be described by a single diffusion tensor (10). However, work on interdomain dynamics has also been carried out on proteins with more interdomain flexibility using either individual diffusion tensors, fit to each domain (11), or internal slow-timescale motion parameters to describe the interdomain motion as a wobble-in-a-cone (12).
More recently, new models have been developed and refined that are designed to extract more detailed domain dynamic and structural information from NMR as well as small-angle scattering data (13–19). Paramagnetic approaches that consider the probabilities of allowed orientations can be used to impose both angular and translational constraints on flexibly linked domains (20). Such overall conformational constraints can also be obtained by approaches that use NMR relaxation and small-angle x-ray scattering (17), as well as NMR diffusion tensors (21). For example, using a model for interconversion between two distinct conformational states (ITS model), Ryabov and Fushman (14) performed an analysis of rather large conformational changes in the Lys48-linked di-ubiquitin system. With this approach it is possible to describe slow effective motions (30 ns) through large angles (up to ∼90°) for the two linked ubiquitin domains, as they interconvert between two well-defined conformational states. However, like the MF formalism, the ITS model requires the interdomain and overall protein motions to be independent, a condition that was met fortuitously in the di-ubiquitin case, since the two distinct states were shown to possess almost identical overall diffusion tensors. A very different dynamic model was considered for Xenopus Ca2+-ligated calmodulin. Here, the crystal structure was taken as the dynamically averaged reference frame for overall diffusion, with slow interdomain motions interpreted as fluctuations with respect to this frame (13). Therefore, the overall protein rotational correlation time and diffusion tensor anisotropy were assumed to be identical for both the N-terminal domain (NTD) and C-terminal domain (CTD), leaving the individual domain orientations and the slow interdomain motions to be optimized. Relaxation parameters were determined at multiple magnetic field strengths, and an extended MF approach (22) was applied. This approach allows for large-amplitude, continuous domain rotations, and, for the calmodulin case, the estimated interdomain motion for the NTD and CTD resulted in a good fit for a wobble-in-a-cone of semi-angle ∼30°. A continuum of orientations for the two structured domains was assumed based on the highly dynamic nature of several residues in the middle of the linker (23).
In this work, we did not develop a new model for interpreting domain dynamics data; rather, we constructed a model protein system that allows us to investigate a large range of interdomain motions using a very simple approximation of domain diffusion. We devised a generic double-domain system consisting of two non-interacting GB1-type domains with identical surface properties. We minimized the possibility of residual linker structure by using a flexible gly-gly-ser-based linker. Several double-GB1 (dGB1) proteins were constructed with linker lengths ranging from three to 24 residues. NMR T1, T2, and 15N−{1H} nuclear Overhauser enhancement (NOE) relaxation parameters were measured and used to fit two diffusion tensors per protein (one each for the NTD and CTD). In this way the effects of coupled motions between domains were included in the diffusion tensor of each individual domain, implicitly testing the limits of the hypothesis that the effects of the second domain may be treated as a perturbation on the diffusion tensor. Four models of domain diffusion were considered: isotropic, oblate, prolate, and fully anisotropic. The prolate model was found to fit the data best, using the least number of fitting parameters. The goodness of fit was best for the longer linker length proteins.
Our data show that even for flexible linkers of 24 residues in length, a significant mutual perturbation in the diffusion is observed. Rotational correlation times for both domains were > 5 ns, compared to 3.8 ns for the equivalent single-domain protein. Furthermore, for all linker lengths the NTD and CTD differed in their diffusion anisotropy and degree of magnetic alignment. This is a surprising finding, since both domains differ by only one internal amino acid and possess the same overall structure and identical protein surface residues. Our results raise interesting questions regarding the influence of modules on the dynamics and long-range ordering of multidomain proteins, even in cases of identical repeating domains.
Materials and Methods
Design of model protein constructs
Twelve specific GB domain constructs were created for this study, and a schematic illustration of these variants is provided in Fig. 1, together with their amino acid sequences. We use the following nomenclature for all proteins: the designation GB1 refers to the original GB1-T2Q mutant that has been used in a myriad of biophysical studies. The introduction of a glutamine at position 2, instead of the wild-type (WT) threonine, prevents N-terminal heterogeneity (24). GB1L7I represents a GB1 domain that differs from GB1 by a single core mutation, which is the sole core amino acid difference between the natural GB1WT and GB3WT domains. The third domain type used in this study is designated GB3β1 = GB1 M1T-Q2T-I6V-L7I because it contains the WT sequence of GB3 in its first β-strand, but otherwise is identical to GB1. The prefix “s”, as in sGB1, refers to a single-domain protein with seven and six residue tails before and after the GB1 domain, respectively. The prefix “d”, as in dGB1L7I-(n)-GB1, refers to double-domain proteins. These proteins possess the same tails as the single-domain proteins, as well as an interdomain linker comprising n residues. The full list of protein constructs that were investigated in this study is as follows: GB1, sGB1, sGB1L7I, sGB3β1, dGB1L7I-(3)-GB1, dGB1L7I-(6)-GB1, dGB1L7I-(12)-GB1, dGB1L7I-(18)-GB1, dGB1L7I-(24)-GB1, and dGB3β1-(6)-GB1. Other constructs employed in control experiments were dGB1-(6)-GB1 and dGB1-(6)-GB1L7I. Protein expression and purification were performed as described in the Supporting Material.
Figure 1.
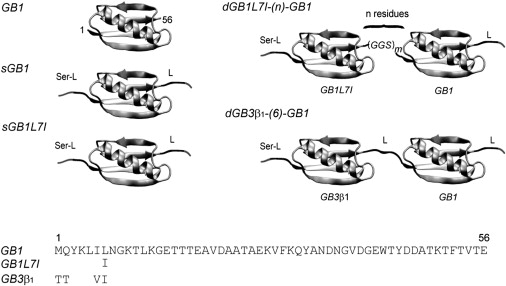
Model system employed in this work involves variants of the GB domain. The sGB construct consist of the GB1 domain with a tail, L = GGSGGS, after residue 56, and a prepended sequence of Ser-GGSGGS before residue 1. The two sequence variants are sGB1 and sGB1L7I. Two types of dGB constructs were investigated: 1) dGB1L7I-(n)-GB1, which consists of sGB1L7I linked to sGB1 with an n-residue interdomain linker (n = 3, 6, 12, 18, 24); and 2) dGB3β1-(6)-GB1.
Aligned protein samples
Proteins were aligned in alkyl poly(ethylene glycol)/n-hexanol liquid crystalline phase (25) (C12E5) in 25 mM sodium phosphate buffer, 0.02% NaN3, pH 6.5. The C12E5 surfactant/water ratio was 2.5% (w/v), with a molar ratio of surfactant to alcohol of 0.96. To ensure uniform alignment conditions for the different proteins, a stock solution of 10% C12E5 was diluted into the protein solutions. The samples were allowed to equilibrate in the spectrometer for several hours until a stable value of the deuterium quadrupolar splitting was reached before data collection. The measured quadrupolar splittings were 11.51 Hz, 11.02 Hz, 10.88 Hz, and 11.60 Hz for C12E5/protein solutions of sGB1, dGB1L7I-(18)-GB1, dGB1L7I-(3)-GB1, and GB1, respectively.
NMR spectroscopy
NMR spectra were recorded at 25°C on NMR samples containing 1.0 mM 15N-labeled or 13C/15N-labeled proteins in 25 mM sodium phosphate buffer, pH 6.5, containing 0.02% azide, using Bruker AVANCE600 and 800 spectrometers (Bruker BioSpin, Billerica, MA) equipped with 5 mm triple-resonance, three-axis gradient probes or z-axis gradient cryogenic probes. Backbone resonance assignments were carried out via HNCACB experiments (26) on samples of sGB1, sGB1L7I, and sGB3β1. The data were processed with nmrPipe (27), and assignments were made using the program CARA (28). Residual HN dipolar couplings were measured using in-phase/antiphase two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) interleaved experiments (29) at 800 MHz. For each experiment we used acquisition times of 242.7 ms (15N) and 60.0 ms (1H) with a data matrix of 1024 × 768 complex points for the proteins sGB1, dGB1L7I-(18)-GB1, and dGB1L7I-(3)-GB1. For GB1, acquisition times of 85.5 ms (15N) and 60.0 ms (1H) with a data matrix of 512 × 768 complex points were used. For the double-domain proteins, both time domains were linear-predicted, and for all proteins sine-bell and squared sine-bell window functions were applied to direct and indirect dimensions, respectively. In both dimensions the sine bells were shifted by 63° and truncated at 176°. After zero-filling, the digital resolution in the 15N-dimension was 0.52 Hz/point for sGB1, dGB1L7I-(3)-GB1, and dGB1L7I-(18)-GB1, and 0.73 Hz/point for GB1. The alignment tensors were determined by singular value decomposition (SVD) fitting of the RDCs using PALES (30) with the RDC-refined GB3 crystal structure as the input model (31).
NMR experiments to determine 15N longitudinal relaxation times (T1), relaxation-compensated 15N transverse relaxation time (T2), and 15N−{1H} NOEs employed 1H-15N HSQC-based pulse sequences at 600 MHz (32–34). The concentrations of all 15N-labeled proteins were 1.0 mM, except for the sGB1L7I sample, which was 0.9 mM. The delays in the T1 experiment were 0, 100, 200, 300, 500, and 800 ms, and delays in the T2 experiment were 0, 19.2, 38.4, 76.8, 96.0, and 115.2 ms. The 15N-{1H} NOE experiments employed a 2.8 s repetition delay. The T1 and T2 relaxation data were fit using single exponential functions. The heteronuclear 15N-{1H} NOE values were obtained by taking a ratio of two experiments recorded with and without 1H saturation.
Analysis of diffusion tensors
In addition to the requirement of chemical shift resolution, the set of resonances used for analysis were selected based on the amino acid locations in the more rigid parts of the structure, as evidenced by suitably corrected (35) heteronuclear 15N-{1H} NOE values > 0.65, as well as the absence of conformational exchange (12). Selection of the most appropriate diffusion tensor model was performed using the program Tensor2 (36), assuming isotropic, axially symmetric (prolate and oblate) and fully anisotropic molecular diffusion models. All four models were applied to the individual domains of single- and double-domain proteins, sGB1, and dGB1L7I-(24)-GB1, using both the full available data set of 44 T1/T2 values for sGB1 and a subset of data comprising only 10 T1/T2-values. The latter values are also available for the dGB1L7I-(n)-GB1 system, which has a high degree of resonance overlap. All pertinent parameters for these models, together with the goodness of fit and F-statistics, are summarized in Table S1. Further analysis of the angular sampling and effect of reduced data set size on the fits is provided in the Supporting Material. Analysis of the diffusion of all protein domains was also performed using the Palmer group's r1r2_diffusion software approach for fitting the prolate diffusion tensor (37). This allowed a straightforward implementation of cross-validation of data.
Results and Discussion
Design of the model system
Our dual-domain model system is based on GB1, and we engineered it such, that the hydrodynamic, chemical, and magnetic properties of the two domains would be as close to identical as possible (Fig. 1). We chose an interdomain linker based on a repeating gly-gy-ser motif because of this sequence's flexibility (38), experimentally demonstrated random coil behavior (39), and lack of any possible complicating ionic interactions.
Because the linker between the domains influences the protein chemical shifts and possibly the dynamics of residues at the end of the first domain and the beginning of the second domain, we also added short peptide tails to the N- and C-termini to create a more symmetric environment. A repeating gly-gly-ser sequence was used for this purpose, replicating the composition of the linker region. This created a dGB1 protein for which the amide chemical shifts of the two domains superimpose perfectly at 600 MHz. Having achieved a model system with exceptional similarity in terms of the physical environments and solvent interactions, we then introduced a single internal mutation at position 7 of the NTD (L7I), which allowed us to spectroscopically distinguish the two domains. In addition, a protein was created that contained GB3β1 as the NTD. This second construct allowed us to measure the relaxation of a larger set of residues for each individual domain, albeit at a cost of introducing a difference of three external residues between the domains, which resulted in a somewhat different protein-solvent interface for the two domains (Fig. 1). Two representative 600 MHz 1H-15N HSQC spectra for these double-domain proteins, recorded for samples of dGB1L7I-(6)-GB1 and dGB3β1-(6)-GB1, are provided in Fig. 2. The excellent spectral quality allows many equivalent residues from the NTD and CTD to be clearly distinguished. In the dGB1L7I-(6)-GB1 and dGB3β1-(6)-GB1 spectra, 17 and 30 pairs of resonances, respectively, can be resolved and are marked in Fig. 2. Of these, a subset of 10 and 22 resonances, respectively, were selected from the rigid portions of the domains for diffusion tensor analysis (see Materials and Methods, and the Supporting Material).
Figure 2.
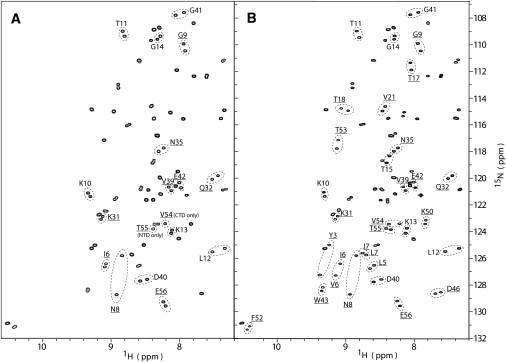
1H-15N HSQC spectra of dGB1 domain proteins. (A) 1H-15N HSQC spectrum of dGB1L7I-(6)-GB1. Pairs of resonances that are distinguishable between the GB1L7I (NTD) and the GB1 (CTD) are labeled with the residue name and number and circled. Ten pairs with 15N-{1H} NOE values > 0.65 were used for diffusion tensor determination (underlined). These pairs are from identical residues residing on the NTD and CTD, respectively, with the exception of T55 (NTD) and V54 (CTD), which possess no resolved counterpart in the other domain. The spectrum of the dGBL7I-(6)-GB1 protein is almost indistinguishable from those of the single-domain proteins sGB1 and sGB1L7I. (B) 1H-15N HSQC spectrum of dGB3β1-(6)-GB1. Thirty labeled pairs of resolved resonances are present, and 22 of these pairs exhibit 15N-{1H} NOE values > 0.65.
sGB1 domain diffusion tensors
Effect of the tail on diffusion
We explored the contribution of appended, unstructured residues on domain diffusion by comparing the prolate diffusion tensor of tailless GB1 with those of the six- to seven-residue tail-containing sGB1 and sGB1L7I. The results (presented in Fig. S1 A) show that an experimentally significant increase of the rotational correlation time of 0.86 ns, from τc = 2.95 ns to τc = 3.81 ns, is observed due to the tails. Furthermore, the anisotropy parameter, D‖/D⊥, increases by 0.33. This can be explained by the fact that the N- and C-terminal tails are located approximately along the long axis of diffusion of the tailless GB1 domain. The presence of the unstructured tails also appears to alter the orientation of the principal diffusion axis by ∼10°. The change in rotational correlation time indicates that the tails add hydrodynamic drag to the domain, and due to their constantly changing structure, this manifests itself as a change in the orientation of the principal diffusion axis of the fit time-independent, rigid-body diffusion tensor.
Effects of a modular environment on domain diffusion
Rotational correlation times
It is not clear, a priori, to what extent diffusion of a protein domain is altered when it exists in a modular arrangement of flexibly linked domains, compared to diffusion of the isolated domain. We set out to quantify the effect of the beads-on-a-string environment by comparing diffusion tensor fits obtained for the sGB1 isolated domain with those obtained for the individual domains in the dGB1 proteins dGB3β1-(6)-GB1 and dGB1L7I-(n)-GB1 (n = 3, 6, 12, 24 residues) using the prolate model (see Figs. 4 and 5). The results show that even for the flexible interdomain linker of 24 residues, domain diffusion is perturbed significantly, with the individual domain rotational correlation time increased to 5.2 ns and 5.0 ns for the NTD and CTD of dGB1L7I-(24)-GB1, respectively, compared to 3.8 ns for the sGB1 domain. This increase by 1 to 1.5 ns may be caused in part by the hydrodynamic drag of the long interdomain linker. However, the effect of the linker cannot be the dominant contributing factor, since domains separated by a linker of 12 residues also exhibited very similar rotational correlation times of 5.3 ns and 5.2 ns for the NTD and CTD, respectively. This small change of only 0.2 ns is incompatible with the >1 ns difference in correlation time between the GB1 domains in the single- versus double-domain contexts. Indeed, a more pronounced effect was seen for the shortest linker length: the domain rotational correlation times for the protein with a three-residue linker were 6.5 ns and 6.3 ns (NTD and CTD, respectively), representing a 65% and 70% increase compared to the sGB1 domain correlation time. The overall trend of larger domain rotational correlation times for smaller proteins with shorter interdomain linkers suggests a model of domain dynamics wherein the individual domain's semi-independent diffusion is increasingly perturbed and restricted by the neighboring domain as the linker is made shorter.
Figure 4.
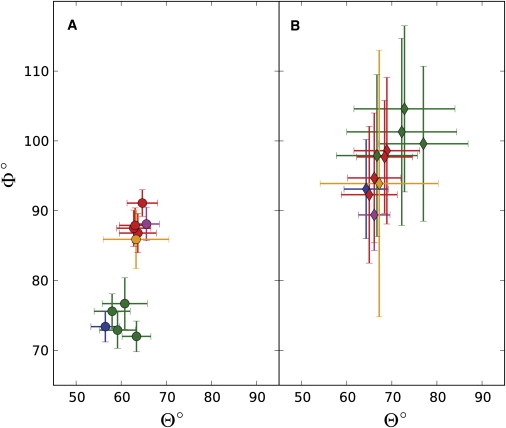
Orientations of the diffusion tensor principal axes in spherical coordinates. Orientations for the NTD and CTD of dGB1L7I-(n)-GB1 are shown in green and red, respectively, and those for dGB3β1-(6)-GB1 are shown in blue and purple, respectively. The principal diffusion axis orientation for sGB1 is shown in orange. (A) Fitting using all available data points for dGB1L7I-(n)-GB1 (10 points) and dGB3β1-(6)-GB1 (22 points). (B) Fitting using the full data sets with the data for V39 left out. The fit results for the NTD shift systematically when V39 is omitted, making the NTD and CTD diffusion axes indistinguishable. For sGB1, the data set corresponding to that available for dGB1L7I-(n)-GB1 was used. All error bars indicate a range of 2 RMSD.
Figure 5.
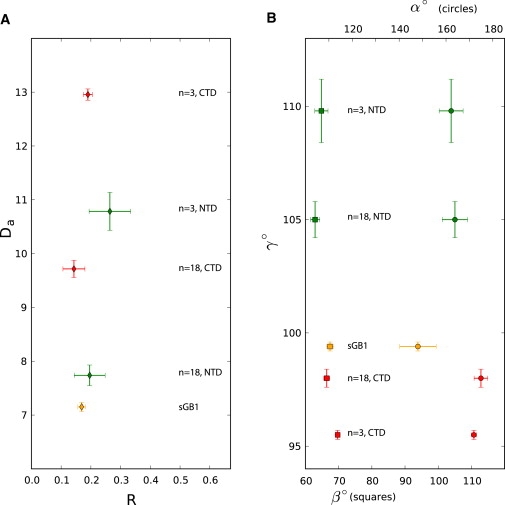
Alignment tensor comparison between sGB1 (orange) and dGB1L7I-(n)-GB1, NTD (green), and CTD (red) for n = 3 and n = 18 residue linkers. (A) Magnitude (Da) and rhombicity (R) of the alignment tensors. (B) Euler angles of the alignment tensors. The angles α (top scale, circles) and β (bottom scale, squares) are indicated by separate symbols for each value of γ. All error bars indicate a range of 2 RMSD.
Anisotropy
The effect of the modular domain environment on the anisotropy parameters D‖/D⊥ consistently differs for the NTD and CTD by 0.35 ± 0.04. This difference in anisotropy for the NTD versus CTD is not an artifact of the small number (10) of T1/T2 data used for the dGB1L7I-(n)-GB1 diffusion tensor fits. A very similar result was obtained for the dGB3β1-(6)-GB1 protein (Fig. 3 A; NTD, blue, and CTD, purple), for which 22 T1/T2 data points were available for fitting.
Figure 3.
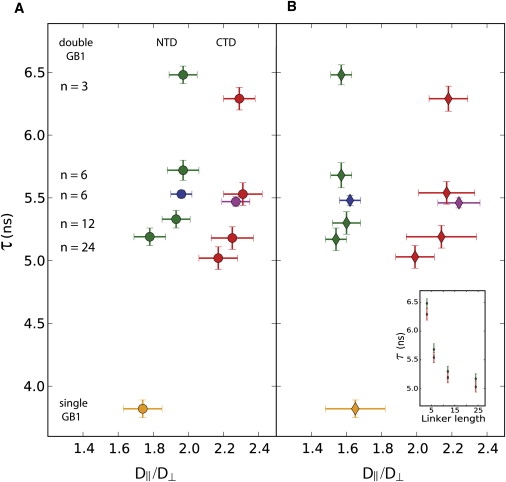
Effect of the modular protein environment on the individual domain diffusion parameters (correlation time, τ, and anisotropy factor, D‖/D⊥). Values for the NTD and CTD of dGB1L7I-(n)-GB1 are displayed in green and red, respectively, and those for dGB3β1-(6)-GB1 are colored in blue and purple, respectively. sGB1 is shown in orange. (A) Fitting using all available data points for dGB1L7I-(n)-GB1 (10 data points) and dGB3β1-(6)-GB1 (22 data points). (B) Fitting with a data set in which the data for V39 was omitted. All error bars indicate a range of 2 RMSD. (Inset) The correlation time, τ, is plotted versus the linker length for the NTD (green) and CTD (red) using the data set with the V39 data omitted. Equivalent results were obtained when the V39 data were included.
Essentially the same values for NTD and CTD anisotropy factors were obtained when fitting was carried out for subsets of data in which one T1/T2 value was omitted (Supporting Material), with one exception. If the T1/T2-value for V39 was left out, very large differences between NTD and CTD anisotropy factors were noted. As shown in Fig. 3 B and Table 1, the V39 T1/T2 data seem to represent a pivotal data point in defining the anisotropy of the NTD. A noticeable shift to smaller anisotropy values of D‖/D⊥ ∼ 1.6 compared to D‖/D⊥ ∼ 2.0 is seen after omission of the V39 data. In contrast, for the CTD, omission of the V39 T1/T2-value causes only a small shift in D‖/D⊥ and an increased uncertainty for all linker lengths, with no significant effect on the reduced χ2. Omission of the V39 T1/T2 data for both domains increases the anisotropy differences between NTD and CTD to 0.45−0.62. Again, similar results were obtained for dGB3β1-(6)-GB1, showing that the diffusion tensor fits with 22 data points are qualitatively the same as those with 10 data points (Fig. 3, A and B).
Table 1.
Diffusion tensor parameters for the prolate model fit to the 15N relaxation data for all proteins in this study
| n∗ | N†∗∗ | Θ‡ | Φ‡ | D‖/D⊥§ | τ (ns)¶ | χ2‖ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| GB1 | ||||||||||
| — | 44 | 59 | (6) | 97 | (8) | 1.40 | (0.05) | 2.95 | (0.03) | 0.67 |
| — | 10 | 59 | (14) | 102 | (12) | 1.39 | (0.11) | 2.95 | (0.07) | 0.77 |
| - | 9 | 59 | (16) | 102 | (26) | 1.40 | (0.12) | 2.95 | (0.07) | 0.92 |
| sGB1 | ||||||||||
| — | 44 | 58 | (4) | 84 | (2) | 1.73 | (0.06) | 3.81 | (0.03) | 2.27 |
| — | 10 | 63 | (7) | 86 | (4) | 1.74 | (0.11) | 3.82 | (0.07) | 2.77 |
| — | 9 | 67 | (13) | 94 | (19) | 1.65 | (0.17) | 3.82 | (0.07) | 3.14 |
| sGB1L7I | ||||||||||
| — | 44 | 59 | (4) | 85 | (3) | 1.74 | (0.06) | 3.81 | (0.03) | 2.29 |
| — | 10 | 63 | (8) | 88 | (5) | 1.73 | (0.11) | 3.82 | (0.09) | 3.59 |
| — | 9 | 69 | (13) | 99 | (16) | 1.63 | (0.11) | 3.82 | (0.10) | 3.75 |
| dGB1L7I-(n)-GB1 | ||||||||||
| NTD | ||||||||||
| 3 | 10 | 63 | (3) | 72 | (2) | 1.97 | (0.08) | 6.48 | (0.07) | 9.41 |
| 6 | 10 | 59 | (4) | 73 | (3) | 1.97 | (0.09) | 5.72 | (0.08) | 9.28 |
| 12 | 10 | 58 | (4) | 76 | (3) | 1.93 | (0.08) | 5.33 | (0.07) | 6.43 |
| 24 | 10 | 61 | (5) | 77 | (4) | 1.78 | (0.09) | 5.19 | (0.07) | 6.65 |
| 3 | 9 | 77 | (10) | 100 | (11) | 1.57 | (0.06) | 6.48 | (0.08) | 3.21 |
| 6 | 9 | 73 | (11) | 105 | (12) | 1.57 | (0.06) | 5.68 | (0.10) | 3.00 |
| 12 | 9 | 67 | (9) | 98 | (12) | 1.60 | (0.08) | 5.30 | (0.09) | 3.12 |
| 24 | 9 | 72 | (12) | 101 | (13) | 1.54 | (0.06) | 5.17 | (0.09) | 3.55 |
| CTD | ||||||||||
| 3 | 10 | 65 | (3) | 91 | (2) | 2.29 | (0.09) | 6.29 | (0.09) | 4.40 |
| 6 | 10 | 63 | (4) | 88 | (3) | 2.31 | (0.11) | 5.53 | (0.09) | 2.23 |
| 12 | 10 | 63 | (4) | 88 | (3) | 2.25 | (0.12) | 5.18 | (0.09) | 1.96 |
| 24 | 10 | 64 | (4) | 87 | (3) | 2.17 | (0.11) | 5.02 | (0.09) | 2.38 |
| 3 | 9 | 68 | (6) | 98 | (8) | 2.18 | (0.11) | 6.29 | (0.10) | 4.74 |
| 6 | 9 | 66 | (6) | 95 | (9) | 2.17 | (0.16) | 5.54 | (0.09) | 2.22 |
| 12 | 9 | 65 | (6) | 92 | (10) | 2.14 | (0.20) | 5.19 | (0.09) | 2.14 |
| 24 | 9 | 69 | (7) | 99 | (11) | 1.99 | (0.11) | 5.03 | (0.09) | 1.84 |
| dGB3β1-(6)-GB1 | ||||||||||
| NTD | ||||||||||
| 6 | 22 | 57 | (3) | 73 | (2) | 1.96 | (0.06) | 5.53 | −0.03 | 5.59 |
| 6 | 21 | 64 | (5) | 93 | (7) | 1.62 | (0.06) | 5.48 | −0.04 | 3.79 |
| CTD | ||||||||||
| 6 | 22 | 66 | (3) | 88 | (2) | 2.27 | (0.08) | 5.47 | −0.04 | 3.77 |
| 6 | 21 | 66 | (4) | 89 | (5) | 2.24 | (0.12) | 5.46 | −0.04 | 3.97 |
Errors (2 RMSD) in diffusion parameters are listed in parentheses.
Number of residues in the interdomain linker.
Number of T1/T2-values used to fit the diffusion model.
Polar (Θ) and azimuthal (Φ) angles of the principal axis of diffusion (D‖), based on the reference GB3 structure (PDB ID: 1p7e). The angles Θ and Φ are right-handed rotations about y and z, referenced to z and x, respectively.
Anisotropy, D‖/D⊥ ratio of the long over short diffusion axis in the prolate model.
Rotational correlation time, τ = 1/(2 Tr(D)).
Reduced Chi-squared, χ2red = χ2/(N − 4).
For the fits using nine and 21 data points, the T1/T2-value of residue V39 was omitted.
Orientation of the principal diffusion axis
For the fits obtained using the full 10 data points, the orientations of the principal diffusion axes for the NTD and CTD of dGB1L7I-(n)-GB1 become progressively more dissimilar as the linker length is decreased from 24 to three residues, as shown in Table 1 and Fig. 4 A. In particular, as the linker length is decreased, the Φ-value for the NTD becomes progressively smaller, whereas the Φ-value for the CTD becomes larger. This result can be rationalized as follows: In the shorter linker length proteins, the potential steric influences between domains increase, causing the principal diffusion axis of the domains to rotate with respect to each other. For reasons that are not clear at this time, for the longest linker length protein, the diffusion axis of the CTD is indistinguishable from that of the isolated single-domain sGB1 (Fig. 4 A, orange). In addition, the domain diffusion axes orientations for the dGB3β1-(6)-GB1 protein (Fig. 4 A; NTD, blue, and CTD, purple), obtained by fitting 22 data points per domain, are very similar to those of the dGB1L7I-(6)-GB1 NTD and CTD domains. However, as shown in Fig. 4 B, omission of data from residue V39 results in a shift of >20° in the orientation of the diffusion axis of the NTD of all proteins. Concomitantly, the uncertainty in the fit orientation dramatically increases for all domains, including sGB1. Thus, without the V39 T1/T2 data, it appears impossible to distinguish the orientation of the diffusion axes for the NTD and CTD.
Robustness of the dGB1 diffusion tensor fit
Because of the limited data set available, the stability of the fit parameters must be particularly scrutinized for effects of individual data. The analysis reveals two distinct effects. A small effect is observed in the tailed sGB1 domains and the CTDs of dGB3β1-(6)-GB1 and the dGB1L7I-(n)-GB1 proteins. For these domains, omission of the V39 data increases the uncertainty in the fit Φ, and a minor, insignificant increase in the value of Φ. This is most likely a sampling artifact, since in fits using larger data sets (44 points for sGB1 and 22 points for dGB3β1-(6)-GB1) this effect is essentially removed (Fig. S2 and Fig. S4). A second, more pronounced effect involves the fits for the NTD of the dGB1 proteins. These fits depend significantly on two residues: V39 and E42. Omission of either of these data caused a substantial decrease in the NTD's reduced χ2, bringing the goodness-of-fit for the NTD in line with the values obtained for the CTD, for all linker lengths (Table S1, Fig. S3, and Fig. S4). Whereas omission of the E42 data caused a small change in Θ of ∼10°, omission of the V39 data resulted in larger changes of Θ, Φ, and D‖/D⊥. These changes were also observed for the larger data set (22 points for dGB3β1-(6)-GB1), suggesting that these effects may not be due to sampling. Therefore, either limitations inherent to the independent domain diffusion model or specific local dynamics (chemical exchange) may play a role. Indeed, the difference between the V39 T2-values of the NTD and CTD decreases systematically with increasing linker length: T2CTD − T2NTD = 8, 6, 4, 2 ms for linker lengths of n = 3, 6, 12, 24 residues. In contrast, the difference between the E42 T2-values of the NTD and CTD does not vary with linker lengths. Irrespective of the changes in the NTD diffusion parameters caused by the peculiarities of the V39 and E42 data, the major findings of a significant difference between the D‖/D⊥ of NTD and CTD, and the observed domain rotational correlation time dependence on linker-length remain valid. Indeed, the fundamentally different diffusion of the two domains is supported by the observation that for the helical residues on the NTD and CTD a constant difference between the average T2 is seen for all linker lengths, i.e., <T2NTD − T2CTD> = 11 ± 1 ms (Fig. S5).
Assessment of the validity of the rigid-body diffusion model
We validated our approach by comparing the quality of the fits obtained for the GB1 domain in various protein environments. As summarized in Table 1, the prolate diffusion tensor fit for the tailless GB1 domain has a reduced χ2-value of 0.7−0.9, depending on the data set. Therefore, no major systematic errors (such as possible conformational differences between the model structure that was used to fit the relaxation data and the actual solution structure) are unaccounted for. For comparison, the prolate diffusion tensor fits for both sGB1 and sGB1L7I yielded values of the reduced χ2 = 2.3 (full data set) to reduced χ2 = 3.6 (10 data points). This suggests that increased errors are introduced due to an indirect relaxation effect caused by the hydrodynamic drag of the flexible tails, or possibly direct relaxation effects imparted by the flexible tail on the several backbone amide nitrogens. It should be stressed, however, that the magnitude of the error is not unusually large. For instance, in the diffusion tensor fits for ubiquitin, a reduced χ2-value of 2.4 for the axially symmetric model was observed (37). The goodness-of-fit for the dGB1 proteins is also provided in Table 1 and shows that the CTD fits the model as well as the isolated tailed sGB1, with the exception of the protein with the shortest linker length (three residues). In contrast, the NTD exhibits a poorer fit, primarily due to the data of residues 39 and 42 (Table 1, Fig. S3, and Fig. S4).
Hydrodynamic coupling
The fundamental difficulty of studying interdomain dynamics of modular proteins stems from the fact that domain motion drastically alters the global, overall shape of the protein and therefore the overall protein-solvent interface. If the domain movements take place on a timescale similar to the overall protein rotational diffusion, the internal and global dynamics exhibit hydrodynamic coupling (10), and any approach that uses a time-independent protein diffusion model to describe such complex dynamics, by necessity, represents an approximation. In our model system, very long flexible linkers allow for a significant range of motion of the individual domains, with respect to each other. Therefore, we choose to fit the diffusion tensors separately to the individual domains instead of considering the protein as a flexible whole. This approach does not ignore the coupled diffusion of the domains, but rather results in the coupling effects becoming included implicitly in the parameters of the individual domain diffusion tensors.
Effects of a modular environment on domain alignment
Rhombicity
We assessed the effect of flexibly linking two domains by comparing the alignment tensor of the isolated sGB1 domain with those of the dGB1L7I-(n)-GB1 domains. The data for the proteins with three and 18 residue linkers are shown in Fig. 5. The alignment tensor for most tail-containing domains, whether in a single- or double-domain protein, exhibited very similar values for rhombicity, R = 0.14−0.19, with the exception of the NTD of dGB1L7I-(3)-GB1, which showed a rhombicity of R = 0.26 ± 0.07 (Fig. 5 A). Given the error range, however, this may not be a significant difference, especially by comparison with the tailless GB1 domain, for which a rhombicity of R = 0.37± 0.05 was determined (Table S2). Therefore, it appears that the modular domain environment does not have a substantial effect on the rhombicity of the domain alignment. This may be related to the axial location of the interdomain linkers in this particular system. Furthermore, the more significant effect comes not from the modular domain environment, but from adding six- to seven-residue-long flexible tails. This causes the alignment tensor to become almost axially symmetric, with any intrinsic domain-specific rhombic asymmetry becoming less pronounced. Therefore, it may be possible to exploit this surprisingly large effect for varying the alignment tensor of a protein by simply adding tails of specific length and composition.
Magnitude of alignment
In the double-domain context, we noted a variety of effects on the magnitude of the alignment tensor of the individual domains. For the protein with the 18-residue interdomain linker, the NTD exhibited very similar alignment as the isolated sGB1 with approximate values of Da = 7.7 Hz and Da = 7.2 Hz, respectively. Surprisingly, for the CTD, a Da-value almost 2 Hz larger than that for the NTD was obtained (Da = 9.7 Hz). This was especially surprising given the length of the linker (18 residues) and the protein domains' identical surface residues. Such a difference of ∼2 Hz is also observed when the interdomain linker is only three residues long. For this protein, the NTD and CTD exhibited values for Da of 10.8 Hz and 13.0 Hz, respectively. The fact that such differences in the magnitude of alignment are seen consistently parallels the differences found for the diffusion tensor D‖/D⊥ values for NTD and CTD.
Euler angles
The Euler angles of the alignment tensors for each domain are depicted in Fig. 5 B and listed in Table S2. No systematic dependence on linker length or domain identity was observed for either α (circles, top scale), or β (squares, bottom axis). For the angle γ (Fig. 5 B, vertical axis), however, the NTD and CTD differ by ∼7° for the protein with an interdomain linker of 18 residues, whereas for the protein with a three-residue linker γ differs by 14°. Furthermore, the γ Euler angle of the sGB1 domain is similar to that of the CTD for the long linker length protein. This effect is similar to that observed for the angle Φ of the principal axis of diffusion. In fact, the alignment tensor angle β exhibits a similar range, compared to the corresponding diffusion tensor angles β (Table S1, prolate and anisotropic models) and Θ (Table 1, prolate model).
Influence of data set size on alignment tensor
Because several resonances experience small shifts in the IPAP spectrum recorded in the C12E5 alignment medium, and given the naturally increased overlap due to the RDC splittings, fewer data points are safely available for alignment tensor fitting for the dGB1L7I-(n)-GB1 proteins than for the relaxation analysis. We imposed stringent selection criteria for the RDC measurement to avoid any complications caused by resonance overlap. Therefore, we selected data for only eight residues to fit the tensor orientations in the different dGB1L7I-(n)-GB1 constructs. We tested the validity of using such a small data set for single-domain GB1 proteins by comparing alignment tensor parameters, using eight and 38 data points for sGB1, and seven and 30 data points for the tailless GB1 (Table S2). Gratifyingly, the Da values for the two data sets differ only by 0.30 Hz and 0.24 Hz for GB1 and sGB1, respectively, and thus are similar in magnitude to the combined uncertainties for these fits (0.25 Hz and 0.23 Hz). Likewise, the rhombicities are also essentially the same, lying within the 2 RMSD from Monte Carlo simulations. The largest angle variation is seen for α, with differences of ∼12° for GB1 and ∼25° for sGB1. For the angles β and γ, differences of <4° and <2° were obtained, respectively, which are substantially less than the systematic, linker-length-dependent differences in γ-values observed for the NTD and CTD in the double-domain proteins.
Modular environment: dynamics and long-range order
Because of the almost identical solvent-protein interfaces of the two domains, the dGB1 model system investigated here provides a unique opportunity to interpret the domain diffusion and alignment in terms of the modular domain environment. Our results clearly indicate that for a protein with a 12-residue-long flexible linker, the individual domain correlation times, anisotropy, and diffusion tensor orientations change very little with a further increase in linker length (Figs. 4 and 5). However, at these linker lengths the domains have not reached the limit of independent diffusion, as indicated by the substantial difference in rotational correlation times between the domains of the dGB1 and sGB1 (τ > 5 ns and τ = 3.8 ns, respectively). Furthermore, for linker lengths of 12−24 residues, the nearly identical domains experience distinctly different diffusion anisotropies, with the NTD possessing a value of D‖/D⊥ = 1.5–1.6, similar to that of single GB1 (D‖/D⊥ = 1.6–1.7), whereas the CTD anisotropy is substantially larger, with D‖/D⊥ = 2.0–2.2. (It should be emphasized again that these values for D‖/D⊥ of the NTD are dependent upon omission of the V39 T1/T2-value, and inclusion of this data point yields D‖/D⊥ = 1.8–1.9 for the NTD.) Furthermore, for the protein with domains separated by an 18-residue linker, the RDCs measured in C12E5 reveal that whereas the NTD alignment magnitude of Da = 7.7 Hz differs little from that of the single-domain protein (Da = 7.2 Hz), the alignment of the CTD is substantially larger, with Da = 9.7 Hz. Therefore, the NTD and CTD influence each other's dynamics and the orientational distribution differently, even for long linker lengths.
The same qualitative differences between NTD and CTD diffusion anisotropy and alignment magnitude are observed for a three-residue linker. Therefore, it is tempting to interpret these differences as resulting from the steric occlusion of one domain by the other during the course of interdomain diffusion. The different degrees of occlusion of the two domains is somewhat surprising, but may be the result of the fact that the GB1 domain is not a perfect prolate shape, due to differences between the N- and C-termini of the protein. Media that work predominantly by a mechanism of steric occlusion of a rigid domain, such as C12E5, result in an alignment that depends on the overall shape of the rigid domain (40). Similarly, when domains sterically occlude each other's motion in a two-domain protein with a flexible linker, the NTD and CTD alignments are expected to differ from those of the isolated domains in a way that reflects the restricted distribution of conformations of the multidomain protein in solution. This is indeed what the RDC Da-value indicates for the protein with a three-residue linker. However, for the protein with an 18-residue linker, the distribution of orientations of the NTD is not significantly restricted as compared to the isolated sGB1 domain, whereas the CTD is. This parallels the diffusion anisotropy results for proteins with long linkers.
Conclusions
Our data show that in multi-domain proteins the flexible linker and the individual domains play significant and different roles. The addition of flexible tails of six to seven residues in length to the protein domain caused an increase in diffusion anisotropy, as well as reduced rhombicity of the alignment tensor. For double-domain proteins, flexible linker residues in excess of 12 residues appeared to have little further effect on the individual domain diffusion. The observed significant differences between the diffusion anisotropy and magnitude of alignment for the NTD and CTD may reflect the intrinsic asymmetric arrangement of the protein domains in a modular protein, due to the polarity of the polypeptide chain and the shape of the domain.
The model system results presented here also provide useful reference data that can be used to assess interdomain influences in other, more complex multidomain proteins. For example, the contribution of a specific natural linker composition to the dynamics and alignment of a protein of interest may be derived to a first approximation by substituting it into the dGB1 system. In addition, further extension of this model system to include more linked domains, as well as non-symmetrically connected domains, is being developed in combination with computational and theoretical methods.
Acknowledgments
We thank Jinwoo Ahn for useful discussions regarding protein expression and purification, and Mike Delk for technical support in the NMR instrumentation.
This work was supported by a grant from the National Science Foundation (MCB 0814905 to R.I.) and startup funds from the University of Pittsburgh School of Medicine (to A.M.G. and R.I.).
Supporting Material
References
- 1.Pickford A.R., Campbell I.D. NMR studies of modular protein structures and their interactions. Chem. Rev. 2004;104:3557–3566. doi: 10.1021/cr0304018. [DOI] [PubMed] [Google Scholar]
- 2.Sicheri F., Kuriyan J. Structures of Src-family tyrosine kinases. Curr. Opin. Struct. Biol. 1997;7:777–785. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- 3.Koglin A., Walsh C.T. Structural insights into nonribosomal peptide enzymatic assembly lines. Nat. Prod. Rep. 2009;26:987–1000. doi: 10.1039/b904543k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujita N., Endo S., Ishihama A. Structural requirements for the interdomain linker of α subunit of Escherichia coli RNA polymerase. Biochemistry. 2000;39:6243–6249. doi: 10.1021/bi000020d. [DOI] [PubMed] [Google Scholar]
- 5.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982;104:4546–4570. [Google Scholar]
- 6.Jarymowycz V.A., Stone M.J. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- 7.Palmer A.G., 3rd NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 8.Fushman D., Xu R., Cowburn D. Direct determination of changes of interdomain orientation on ligation: use of the orientational dependence of N-15 NMR relaxation in Abl SH(32) Biochemistry. 1999;38:10225–10230. doi: 10.1021/bi990897g. [DOI] [PubMed] [Google Scholar]
- 9.Copié V., Tomita Y., Torchia D.A. Solution structure and dynamics of linked cell attachment modules of mouse fibronectin containing the RGD and synergy regions: comparison with the human fibronectin crystal structure. J. Mol. Biol. 1998;277:663–682. doi: 10.1006/jmbi.1998.1616. [DOI] [PubMed] [Google Scholar]
- 10.Halle B. The physical basis of model-free analysis of NMR relaxation data from proteins and complex fluids. J. Chem. Phys. 2009;131:224507. doi: 10.1063/1.3269991. [DOI] [PubMed] [Google Scholar]
- 11.Brüschweiler R., Liao X., Wright P.E. Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science. 1995;268:886–889. doi: 10.1126/science.7754375. [DOI] [PubMed] [Google Scholar]
- 12.Barbato G., Ikura M., Bax A. Backbone dynamics of calmodulin studied by N-15 relaxation using inverse detected 2-dimensional NMR-spectroscopy—the central helix is flexible. Biochemistry. 1992;31:5269–5278. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- 13.Baber J.L., Szabo A., Tjandra N. Analysis of slow interdomain motion of macromolecules using NMR relaxation data. J. Am. Chem. Soc. 2001;123:3953–3959. doi: 10.1021/ja0041876. [DOI] [PubMed] [Google Scholar]
- 14.Ryabov Y.E., Fushman D. A model of interdomain mobility in a multidomain protein. J. Am. Chem. Soc. 2007;129:3315–3327. doi: 10.1021/ja067667r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zerbetto M., Polimeno A., Meirovitch E. General theoretical/computational tool for interpreting NMR spin relaxation in proteins. J. Phys. Chem. B. 2009;113:13613–13625. doi: 10.1021/jp9046819. [DOI] [PubMed] [Google Scholar]
- 16.Prompers J.J., Brüschweiler R. General framework for studying the dynamics of folded and nonfolded proteins by NMR relaxation spectroscopy and MD simulation. J. Am. Chem. Soc. 2002;124:4522–4534. doi: 10.1021/ja012750u. [DOI] [PubMed] [Google Scholar]
- 17.Bernadó P., Modig K., Akke M. Structure and dynamics of ribosomal protein L12: an ensemble model based on SAXS and NMR relaxation. Biophys. J. 2010;98:2374–2382. doi: 10.1016/j.bpj.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertini I., Calderone V., Svergun D.I. Evidence of reciprocal reorientation of the catalytic and hemopexin-like domains of full-length MMP-12. J. Am. Chem. Soc. 2008;130:7011–7021. doi: 10.1021/ja710491y. [DOI] [PubMed] [Google Scholar]
- 19.Borsi V., Luchinat C., Parigi G. Global and local mobility of apocalmodulin monitored through fast-field cycling relaxometry. Biophys. J. 2009;97:1765–1771. doi: 10.1016/j.bpj.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertini I., Gupta Y.K., Yuan J. Paramagnetism-based NMR restraints provide maximum allowed probabilities for the different conformations of partially independent protein domains. J. Am. Chem. Soc. 2007;129:12786–12794. doi: 10.1021/ja0726613. [DOI] [PubMed] [Google Scholar]
- 21.Ryabov Y., Fushman D. Structural assembly of multidomain proteins and protein complexes guided by the overall rotational diffusion tensor. J. Am. Chem. Soc. 2007;129:7894–7902. doi: 10.1021/ja071185d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clore G.M., Szabo A., Gronenborn A.M. Deviations from the simple 2-parameter model-free approach to the interpretatation of N-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990;112:4989–4991. [Google Scholar]
- 23.Chang S.L., Szabo A., Tjandra N. Temperature dependence of domain motions of calmodulin probed by NMR relaxation at multiple fields. J. Am. Chem. Soc. 2003;125:11379–11384. doi: 10.1021/ja034064w. [DOI] [PubMed] [Google Scholar]
- 24.Gronenborn A.M., Filpula D.R., Clore G.M. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 25.Ruckert M., Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J. Am. Chem. Soc. 2000;122:7793–7797. [Google Scholar]
- 26.Wittekind M., Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α-carbon and β-carbon resonances in proteins. J. Magn. Reson. B. 1993;101:201–205. [Google Scholar]
- 27.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 28.Keller R. CANTINA Verlag, Goldau; Switzerland: 2004. The Computer Aided Resonance Tutorial. [Google Scholar]
- 29.Ottiger M., Delaglio F., Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 30.Zweckstetter M. NMR: prediction of molecular alignment from structure using the PALES software. Nat. Protoc. 2008;3:679–690. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 31.Ulmer T.S., Ramirez B.E., Bax A. Evaluation of backbone proton positions and dynamics in a small protein by liquid crystal NMR spectroscopy. J. Am. Chem. Soc. 2003;125:9179–9191. doi: 10.1021/ja0350684. [DOI] [PubMed] [Google Scholar]
- 32.Palmer A.G., 3rd NMR probes of molecular dynamics: overview and comparison with other techniques. Annu. Rev. Biophys. Biomol. Struct. 2001;30:129–155. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- 33.Farrow N.A., Zhang O., Kay L.E. Spectral density function mapping using 15N relaxation data exclusively. J. Biomol. NMR. 1995;6:153–162. doi: 10.1007/BF00211779. [DOI] [PubMed] [Google Scholar]
- 34.Loria J.P., Rance M., Palmer A.G. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J. Am. Chem. Soc. 1999;121:2331–2332. [Google Scholar]
- 35.Grzesiek S., Bax A. The importance of not saturating H2O in protein NMR—application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 1993;115:12593–12594. [Google Scholar]
- 36.Dosset P., Hus J.C., Marion D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 37.Tjandra N., Feller S.E., Pastor R.W., Bax A. Rotational diffusion anisotropy of human ubiquitin from N-15 NMR relaxation. J. Am. Chem. Soc. 1995;117:12562–12566. [Google Scholar]
- 38.Miller W.G., Brant D.A., Flory P.J. J. Mol. Biol. 1967;23:67–80. [Google Scholar]
- 39.Evers T.H., van Dongen E.M., Merkx M. Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry. 2006;45:13183–13192. doi: 10.1021/bi061288t. [DOI] [PubMed] [Google Scholar]
- 40.Zweckstetter M., Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Am. Chem. Soc. 2000;122:3791–3792. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.