

Abstract
During the perinatal period, the central nervous system (CNS) is extremely sensitive to metals, including methylmercury (MeHg). Although the mechanism(s) associated with MeHg-induced developmental neurotoxicity remains obscure, several studies point to the glutathione (GSH) antioxidant system as an important molecular target for this toxicant. To extend our recent findings of MeHg-induced GSH dyshomeostasis, the present study was designed to assess the developmental profile of the GSH antioxidant system in the mouse brain during the early postnatal period after in utero exposure to MeHg. Pregnant mice were exposed to different doses of MeHg (1, 3 and 10 mg/L, diluted in drinking water, ad libitum) during the gestational period. After delivery, pups were killed at different time points - postnatal days (PNDs) 1, 11 and 21 - and the whole brain was used for determining biochemical parameters related to the antioxidant GSH system, as well as mercury content and the levels of F2-isoprostane. In control animals, cerebral GSH levels significantly increased over time during the early postnatal period; gestational exposure to MeHg caused a dose-dependent inhibition of this developmental event. Cerebral glutathione peroxidase (GPx) and glutathione reductase (GR) activities significantly increased over time during the early postnatal period in control animals; gestational MeHg exposure induced a dose-dependent inhibitory effect on both developmental phenomena. These adverse effects of prenatal MeHg exposure were corroborated by marked increases in cerebral F2-isoprostanes levels at all time points. Significant negative correlations were found between F2-isoprostanes and GSH, as well as between F2-isoprostanes and GPx activity, suggesting that MeHg-induced disruption of the GSH system maturation is related to MeHg-induced increased lipid peroxidation in the pup brain. In utero MeHg exposure also caused a dose-dependent increase in the cerebral levels of mercury at birth. Even though the cerebral mercury concentration decreased to nearly basal levels at postnatal day 21, GSH levels, GPx and GR activities remained decreased in MeHg-exposed mice, indicating that prenatal exposure to MeHg affects the cerebral GSH antioxidant systems by inducing biochemical alterations that endure even when mercury tissue levels decrease and become indistinguishable from those noted in pups born to control dams. This study is the first to show that prenatal exposure to MeHg disrupts the postnatal development of the glutathione antioxidant system in the mouse brain, pointing to an additional molecular mechanism by which MeHg induces pro-oxidative damage in the developing CNS. Moreover, our experimental observation corroborates previous reports on the permanent functional deficits observed after prenatal MeHg exposure.
Keywords: Methylmercury, prenatal exposure, developmental neurotoxicity, glutathione, oxidative stress, antioxidant enzymes
Introduction
Reactive oxygen species (ROS) are continuously produced in the mammalian central nervous system (CNS) during normal aerobic metabolism, representing a class of biologically active molecules that threaten neuronal survival by their ability to induce lipid peroxidation, protein oxidation and DNA damage (Halliwell 1992). The protective effects against ROS-induced cellular injury in the CNS are elicited by highly complex and integrated defense systems that are composed of both enzymatic and nonenzymatic antioxidants (Cohen 1994).
The glutathione system, which consists of reduced glutathione (GSH - the most important low-molecular sulfhydryl-containing antioxidant) and the GSH-related enzymes, glutathione peroxidase (GPx) and glutathione reductase (GR), is foremost among these antioxidant systems (Dringen et al. 2000). GPx belongs to a class of enzymes that catalyze the reduction of hydrogen peroxide, phospholipid-hydroperoxide and other organic hydroxyperoxides by GSH (Flohé 1997). GSH is converted to its oxidized form (GSSG) after peroxide reduction, and GR catalyzes the NADPH-dependent conversion of GSSG to regenerate GSH, which can act as intracellular reductant (Gul et al. 2000).
Important changes in protein levels and enzyme activities of the antioxidant defense system occur in the mouse brain during the perinatal period (Khan and Black 2003). In fact, gradual increases in the amount of the antioxidant enzyme, GPx, and in GSH levels were observed in the mouse brain during the perinatal period; this phenomenon was proposed to represent a physiologic mechanism by which the brain protects itself from the surge in oxygen concentration encountered after delivery, which results in an increase in oxidative metabolism and, consequently, an increase in ROS generation (Khan and Black 2003).
Methylmercury (MeHg) is an environmental contaminant that has been shown to cause neurological deficits in both animals and humans (Clarkson et al. 2003; Aschner et al. 2007). As a result of the biomethylation of mercury compounds released from anthropogenic sources in waterways, MeHg-containing fish represent a major source of human poisoning (Clarkson et al. 2003). Therefore, populations that rely heavily on fish for food can be exposed to toxic levels of MeHg. Although yet debatable (Grandjean et al. 1997; Weihe et al. 2002; Yokoo et al. 2003; Davidson et al. 2006; Spurgeon 2006), some epidemiological studies have indicated that motor and cognitive impairments are the most common neurological alterations observed in these populations. It is noteworthy that exposure of pregnant women to MeHg can also lead to indirect intoxication of their progeny due to transplacental MeHg transport (Harada 1995; Rice et al. 2003), and some studies have reported that maternal exposure to MeHg causes neurological deficits in their offspring (Grandjean et al. 1997; Murata et al. 2004). Furthermore, exposure to MeHg during early fetal development can be associated with subtle brain damage at levels much lower than those affecting the mature brain (Grandjean and Landrigan 2006).
A number of synchronous mechanisms are likely associated with MeHg-induced neurotoxicity, including impairment of intracellular calcium homeostasis (Sirois and Atchison 2000; Atchison 2005), alteration of glutamate homeostasis (Aschner et al. 2000; Farina et al. 2003a; Mutkus et al. 2005) and oxidative stress (Ou et al. 1999; Manfroi et al. 2004; Yin et al. 2007). MeHg-induced oxidative stress is related to its direct chemical interaction with nonenzymatic antioxidants, such as GSH (Shanker et al. 2005), as well as changes in the activities of antioxidant enzymes (Farina et al. 2005, Carvalho et al., 2007). In this regard, studies from our group have reported that MeHg exposure decreases the levels of GSH and increases the levels of peroxides and thiobarbituric acid reactive substances in the mouse CNS (Manfroi et al. 2004; Franco et al. 2006). In addition, MeHg exposure has been reported to alter the activities of the GSH-related enzymes, GPx and GR, in the mouse CNS (Farina et al. 2003b, 2005; Stringari et al. 2006). Notably, MeHg-induced oxidative stress and MeHg-induced glutamate dyshomeostasis are connected phenomena affecting each other. In fact, MeHg-induced inhibition of astrocyte glutamate transporters leads to increased glutamate concentrations in the extracellular fluid, causing hyperactivation of N-methyl D-aspartate (NMDA)-type glutamate receptors and leading to an increase in Na+ and Ca2+ influx (Choi, 1992). Increased intracellular Ca2+ levels are associated with the generation of reactive oxygen species (Lafon-Cazal et al., 1993). On the other hand, MeHg-induced reactive oxygen species (mainly H2O2) production appears to directly inhibit astrocyte glutamate transporters, leading to increased glutamate concentrations in the extracellular fluid (Allen et al., 2001a,b). Consequently, reactive oxygen species formation resulting from MeHg- and glutamate-induced oxidative stress contributes to mitochondrial dysfunction (Franco et al., 2007a).
Despite evidence favoring the GSH antioxidant system as a potential molecular target of MeHg-induced neurotoxicity, developmental studies on the interaction between MeHg and the GSH antioxidant system are scarce. Accordingly, the present study was aimed at investigating the effects of prenatal MeHg exposure on the postnatal development of the GSH antioxidant system in the mouse brain. Several important points were considered in the design of the experimental protocol: (i) the developing CNS is highly susceptible to the toxic effects of xenobiotics, including MeHg; (ii) prenatal exposure to MeHg is a common phenomenon observed in the human population; and (iii) antioxidant activity is particularly important during the postnatal period due to the increase in ROS generation after delivery.
Materials and Methods
Chemicals
Methylmercury (II) chloride was obtained from Aldrich Chemical Co. (Milwaukee, WI). β-nicotinamide adenine dinucleotide phosphate sodium salt - reduced form, 5-5′-dithio-bis (2-nitrobenzoic) acid, glutathione reductase from baker’s yeast, oxidized glutathione and reduced glutathione were obtained from Sigma (St. Louis, Mo., USA). All other chemicals were of the highest grade commercially available.
Animals
Adult Swiss Albino mice (male and female), 90 days old, from our breeding colony were maintained at 22 ± 2 °C, on a 12:12 h light/dark cycle, with free access to food (Nuvital, PR, Brazil) and water. The breeding regimen consisted of grouping three virgin females with one male during two days. The presence of a vaginal plug was evaluated twice a day (at 8:00 a.m. and 5:00 p.m.), and was used as an indicator of pregnancy (gestational day 1 - GD 1). Pregnant mice at the GD 1 were selected and housed individually in opaque plastic cages.
Treatment
Thirty two pregnant mice at the GD 1 were randomly assigned to one of four groups of eight animals each. Three groups were exposed to MeHg solutions (1, 3 or 10 mg of methylmercury (II) chloride/L, diluted in tap water) ad libitum as their sole liquid source from GD1 to parturition. Pregnant mice from the control group received tap water ad libitum as their sole liquid source from GD1 to parturition. Maternal liquid and solid ingestions were monitored daily. Because each mother was allocated in an individual cage, the amount of water and food consumption was calculated as the difference between the amount (water or food) available and the remaining amount after a 24 h period. After parturition (postnatal day 1 - PND 1), dams from all groups received tap water ad libitum as their sole liquid source until the end of the weaning period (PND 21). At this time-point, all litters were normalized to a number of 8 pups per litter: litters with 9 or a higher number of animals were humanely culled to 8 pups. Throughout this period, the pups were maintained with their respective mothers (eight per litter; see scheme 1). At different time points (PND 1, 11 and 21), two pups from each litter were sacrificed by decapitation, and the whole brains (minus the cerebella) were removed and used for biochemical analyses. All experiments were conducted in accordance with the Guiding Principles in the Use of Animals in Toxicology, adopted by the Society of Toxicology in July 1989, and all experiments were approved by our ethics committee for animal use at the Universidade Federal de Santa Catarina (PP00060/CEUA e 23080.024292/2006-22/UFSC).
Scheme 1. treatment schedule.
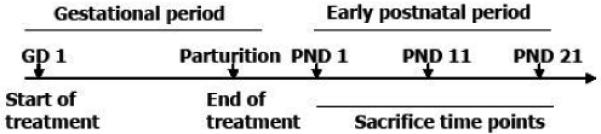
Pregnant mice were exposed to MeHg solutions (0, 1, 3 and 10 mg/L, diluted in tap water) from gestational day (GD) 1 until delivery. MeHg exposure was ended after parturition and, thereafter, lactating dams received only tap water ad libitum. One pup from each litter was sacrificed at postnatal days (PNDs) 1, 11 and 21 for the biochemical analysis.
Tissue preparation
One whole brain (derived from one pup per litter for each treatment/time point) was homogenized (1:10 w/v) in HEPES [N-(2-hydroxyethyl) piperazine-N’-(2-ethanesulfonic acid)] 25 mM, pH 7.4 buffer. An aliquot of 10 μL was removed from the tissue homogenate for Hg determinations. The remaining volume was centrifuged at 20,000 x g, at 4°C for 30 minutes to obtain a cerebral supernatant, which was used to determine glutathione levels and enzyme activity. Another whole brain (derived from the second pup per litter for each treatment/time point) was homogenized in Folch solution for F2-isoprostanes determination (Milne et al., 2007). Briefly, evaporated lipid extract was hydrolyzed using KOH and the internal standard, [2H4]-15-F2t-IsoP added. Following extraction using C-18 and silica Sep-Pac cartridges, purification by thin layer chromatography, and conversion to pentafluorobenzyl ester trimethylsilyl ether derivatives, F2-isoprostanes was dissolved in dry undecane.
Biochemical determinations
The determinations of glutathione levels and enzyme activities were carried out in a Varian Cary 50 spectrophotometer (Varian Inc., Palo Alto, CA, USA). The GR activity was determined based on the protocol developed by Carlberg and Mannervik (1985). Briefly, GR reduces GSSG to GSH at the expense of NADPH, the disappearance of which can be followed at 340 nm. GPx activity was determined based on the protocol developed by Wendel (1981) by indirectly measuring the consumption of NADPH at 340 nm. The GPx uses GSH to reduce the tert-butyl hydroperoxide, producing GSSG, which is readily reduced to GSH by GR using NADPH as a reducing equivalent donor. Glutathione was measured as non-protein thiols based on the protocol developed by Ellman (1959) with minor modifications (Farina et al. 2003c). Briefly, samples were precipitated in cooled trichloroacetic acid 10% and centrifuged at 15,000 x g for 2 minutes, and the supernatant was incubated with DTNB in a 0.5 M phosphate buffer, pH 8.0. Absorbances were measured at 412 nm. Protein concentrations were determined according to Bradford (1976), using a bovine serum albumin as the standard. The cerebral levels of F2-isoprostanes (F2-IsoPs), prostaglandin-like molecules produced by free radical-mediated peroxidation of arachidonic acid, were measured according to the method of Milne et al. (2007), using gas chromatography/mass spectrometry (GC/MS) with selective ion monitoring. Cerebral mercury content was measured by cold vapor atomic absorption spectrometry according to a method previously described by our group (Franco et al. 2007b).
Statistical analysis
Differences between groups (treatment and ontogenetic effects) were evaluated by one-way analysis of variance, followed by Duncan’s multiple range tests when appropriate. Two-way analysis of variance was also performed: different treatments versus different postnatal periods. Correlation between the analyzed biochemical parameters was evaluated by two-tailed Pearson correlation. For studies on the potential correlation between F2-isoprostane levels and the glutathione related parameters (GSH levels, GR and GPx activities), litter-mate pups were used.
Results
Mortality and general health
No maternal mortality was observed throughout the experimental period. Maternal body weight at delivery and liquid and solid consumption throughout the treatment period were not significantly different between groups (Table 1). Values of liquid and solid consumption are in agreement with previous studies (Manfroi et al., 2004) and with the high metabolic rate of a pregnant mouse. Based on the maternal daily liquid ingestions, daily MeHg doses of 0.63, 1.9 and 6.3 mg/kg were calculated for mothers treated with 1, 3 and 10 mg/L MeHg solutions, respectively. Even though the higher dose would be highly toxic, pup body weights were not different between groups from birth until PND 21 (Table 2).
Table 1.
Maternal body weight at delivery; liquid and solid consumption throughout the gestational period. The amount of maternal water and food consumption was calculated as the difference between the amount (water or food) available and the remaining amount after a 24 h period. The values are presented as mean ± SEM. n = 8 mothers per treatment. No significant differences between groups were found by one-way ANOVA
| Groups | Body weight at delivery (g) |
Liquid consumption (mL/day) |
Solid consumption (g/day) |
|---|---|---|---|
| Control | 45.8 ± 3.1 | 28.8 ± 1.1 | 21.8 ± 2.1 |
|
| |||
| MeHg 1 mg/L | 43.2 ± 2.0 | 29.2 ± 0.9 | 23.2 ± 1.7 |
|
| |||
| MeHg 3 mg/L | 44.7 ± 3.5 | 28.4 ± 1.8 | 22.4 ± 1.8 |
|
| |||
| MeHg 10 mg/L | 42.8 ± 4.1 | 27.4 ± 1.9 | 21.4 ± 2.2 |
Table 2.
Pup body weights between birth and postnatal day (PND) 21. The values are expressed as grams and presented as mean ± SEM. n = 8 litters per treatment (1 pup per litter for each time point). No significant differences between groups were found by one-way ANOVA
| Groups | PND 1 | PND 11 | PND 21 |
|---|---|---|---|
| Control | 1.7 ± 0.1 | 7.7 ± 0.4 | 14.3 ± 0.8 |
|
| |||
| MeHg 1 mg/L | 1.9 ± 0.1 | 7.4 ± 0.2 | 13.8 ± 0.6 |
|
| |||
| MeHg 3 mg/L | 1.8 ± 0.1 | 7.7 ± 0.4 | 13.2 ± 1.2 |
|
| |||
| MeHg 10 mg/L | 1.7 ± 0.1 | 7.4 ± 0.4 | 13.7 ± 0.8 |
Biochemical parameters
The developmental profile of cerebral glutathione (GSH) levels during the early postnatal period after in utero exposure to MeHg is depicted in Figure 1. On PND 1, cerebral GSH levels were similar for all groups. In control animals, cerebral GSH levels were ~2.5 fold higher on PND 11 and PND 21 compared to PND 1. However, in utero exposure to MeHg caused a dose-dependent inhibition of the developmental increase in GSH. Two-way analysis of variance showed a significant [F11,55 = 3.92; p = 0.003] interaction between treatment and time for cerebral GSH levels.
Figure 1.
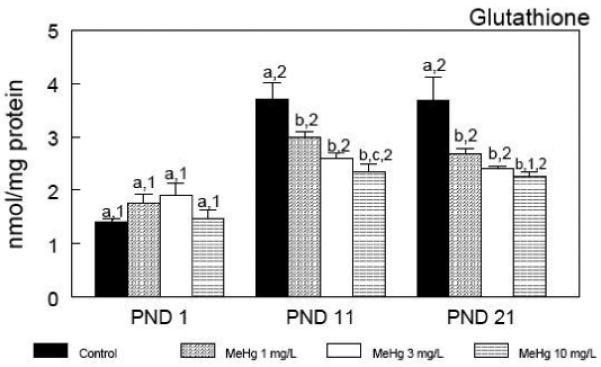
Developmental profile of cerebral glutathione levels during the early postnatal period after in utero exposure to MeHg. Pregnant mice were exposed during the gestational period to different doses of MeHg diluted in their drinking water. After parturition, three pups per litter were randomly selected and killed at different time points - postnatal days (PNDs) 1, 11 and 21. Data are represented as mean ± SEM. n = 8 litters per treatment (1 pup per litter for each time point). Symbols not sharing the same number at the same treatment (ontogenetic effect) are statistically different (P < 0.05) as measured by one-way ANOVA followed by the Duncan’s multiple range test. Symbols not sharing the same letter at the same ontogenetic period (treatment effect) are statistically different (P < 0.05) as measured by one-way ANOVA followed by the Duncan’s multiple range test.
The developmental profile of cerebral glutathione peroxidase (GPx) activity (Figure 2) closely paralleled that of cerebral GSH levels (Figure 1). Indeed, cerebral GPx activity increased during the early postnatal period, and a positive correlation (Pearson coefficient = 0.786; P < 0.001) was found between cerebral GSH levels and cerebral GPx activity in control animals. MeHg induced a dose-dependent decrease in cerebral GPx activity at all time points. Moreover, MeHg caused a dose-dependent inhibitory effect on the physiological increase of GPx activity during the early postnatal period (Figure 2). Cerebral glutathione reductase (GR) activity showed a developmental increase in control animals during the early postnatal period (Figure 3). A positive correlation (Pearson coefficient = 0.795; P < 0.001) was found between cerebral GSH levels and cerebral GR activity in control animals. On PND 1, cerebral GR activity was similar for all groups. Similar to GSH levels and GPx activity, the normal temporal increase in cerebral GR activity during the early postnatal period was inhibited by in utero MeHg exposure (Figure 3).
Figure 2.
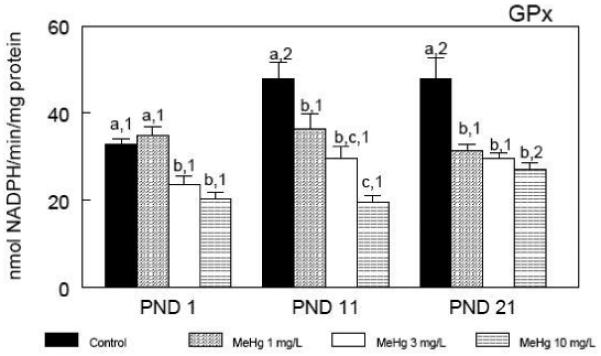
Developmental profile of cerebral glutathione peroxidase (GPx) activity during the early postnatal period after in utero exposure to MeHg. Pregnant mice were exposed during the gestational period to different doses of MeHg diluted in their drinking water. After parturition, three pups per litter were randomly selected and killed at different time points - postnatal days (PNDs) 1, 11 and 21. Data are represented as mean ± SEM. n = 8 litters per treatment (1 pup per litter for each time point). Symbols not sharing the same number at the same treatment (ontogenetic effect) are statistically different (P < 0.05) as measured by one-way ANOVA followed by the Duncan’s multiple range test. Symbols not sharing the same letter at the same ontogenetic period (treatment effect) are statistically different (P < 0.05) as measured by one-way ANOVA followed by the Duncan’s multiple range test.
Figure 3.
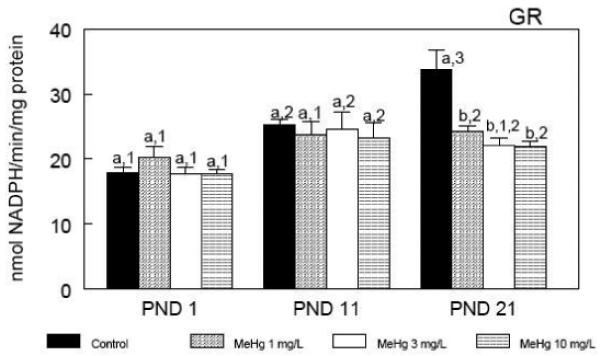
Developmental profile of cerebral glutathione reductase (GR) activity during the early postnatal period after in utero exposure to MeHg. Pregnant mice were exposed during the gestational period to different doses of MeHg diluted in their drinking water. After parturition, three pups per litter were randomly selected and killed at different time points - postnatal days (PNDs) 1, 11 and 21. Data are represented as mean ± SEM. n = 8 litters per treatment (1 pup per litter for each time point). Symbols not sharing the same number at the same treatment (ontogenetic effect) are statistically different (P < 0.05) as measured by one-way ANOVA followed by the Duncan’s multiple range test. Symbols not sharing the same letter at the same ontogenetic period (treatment effect) are statistically different (P < 0.05) as measured by one-way ANOVA followed by the Duncan’s multiple range test.
The cerebral levels of F2-isoprostanes, accurate and reliable indicators of lipid peroxidation in vivo (Roberts and Morrow 2002), are depicted in Figure 4. Importantly, F2-isoprostanes level was the only biochemical brain parameter that did not vary over time after birth until PND 21. Corroborating the adverse effects of MeHg on the GSH antioxidant system, the prenatal exposure to MeHg induced a dose-dependent increase in cerebral lipid peroxidation. It is noteworthy that such increases were inherent to all the time points. Consistent with the above observations, in utero MeHg exposure caused a dose-dependent increase in the cerebral levels of mercury at PND1 (Figure 5). Interestingly, cerebral mercury levels decreased after birth until PND 21 at a very rapid rate, reaching basal levels at weaning.
Figure 4.
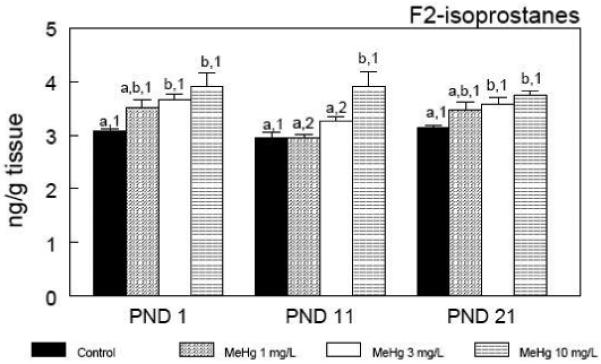
Cerebral F2-isoprostane levels during the early postnatal period after in utero exposure to MeHg. Pregnant mice were exposed during the gestational period to different doses of MeHg diluted in their drinking water. After parturition, three pups per litter were randomly selected and killed at different time points - postnatal days (PNDs) 1, 11 and 21. Data are represented as mean ± SEM. n = 5 litters per treatment (1 pup per litter for each time point). Symbols not sharing the same number at the same treatment (ontogenetic effect) are statistically different (P < 0.05) as measured by one-way ANOVA followed by the Duncan’s multiple range test. Symbols not sharing the same letter at the same ontogenetic period (treatment effect) are statistically different (P < 0.05) as measured by one-way ANOVA followed by the Duncan’s multiple range test.
Figure 5.
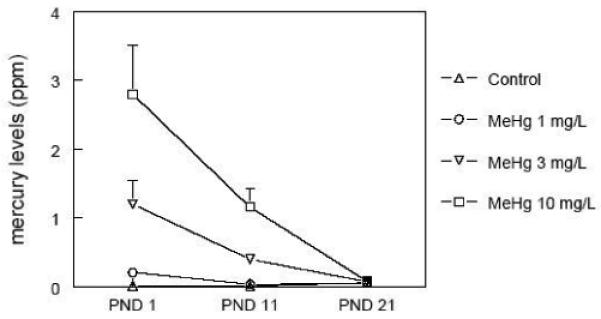
Cerebral mercury levels during the early postnatal period after in utero exposure to MeHg. Pregnant mice were exposed during the gestational period to different doses of MeHg diluted in their drinking water. After parturition, three pups per litter were randomly selected and killed at different time points - postnatal days (PNDs) 1, 11, and 21. Data are represented as mean ± SEM. n = 5 litters per treatment (1 pup per litter for each time point).
Discussion
Several experimental studies with rodents and primates have established that prenatal exposure to MeHg can cause neurotoxicity (Gilbert et al. 1996; Kakita et al. 2000; Weiss et al. 2005; Reed et al. 2006; Johansson et al., 2007). While prenatal exposure to MeHg in fish-eating populations is a well described event, its role in triggering neurotoxicity is still under debate (Grandjean et al. 1997; Weihe et al., 2002; Yokoo et al., 2003; Davidson et al., 2006; Spurgeon 2006). Even though the molecular mechanisms associated with MeHg-induced developmental neurotoxicity are not fully understood, its pro-oxidative properties have been linked to the neuropathological sequalae of perinatal exposures (Vicente et al. 2004). In this regard, the glutathione (GSH) antioxidant system is a significant molecular target involved with the neurotoxic effects of MeHg during the early postnatal period, as evidenced by decreased GSH levels, as well as decreased activities of GSH-related enzymes (Manfroi et al. 2004; Franco et al. 2006; Stringari et al. 2006). The results presented herein showed that in utero exposure to MeHg, which did not change cerebral GSH levels and glutathione reductase (GR) activity at birth, effectively inhibited the developmental profile of the cerebral GSH antioxidant system during the early postnatal period. In fact, cerebral GSH levels, GPx and GR activities in MeHg-treated pups did not parallel the increases observed in the control animals during the early postnatal period. From a toxicological point of view, these results represent an important finding, adding a new molecular mechanism by which MeHg could cause neurotoxicity in the developing CNS, where the inhibition of the physiological maturation of an important antioxidant system might contribute to the oxidative damage observed after prenatal MeHg exposure.
Cerebral GSH levels were measured as non-protein thiols based on the protocol developed by Ellman (1959), which does not discriminate between reduced glutathione and other non-protein thiols. Because GSH accounts for approximately 90 % of the total non-protein thiols in cells (Cooper et al., 1998; Franco and Cidlowski, 2006), one could suppose that the high increase (180 %) in non-protein thiols levels from PND 1 to PND 11/21 in control animals is predominantly related to ontogenetic increases in GSH levels. This finding is in agreement with a previous study by Rice and Russo-Menna (1998), which shows a gradual increase in GSH synthesis from PND 3 to PND 23 in rodent cerebral cortex. On the other hand, it is important to consider that the Ellman’s reagent detects the reduced form of glutathione (GSH), but is unable to detect its oxidized (GSSG) form. Taking into account the decrease of GSH/GSSG under pro-oxidative conditions (Cooper and Kristal, 1997) and the direct chemical interaction between MeHg and GSH, forming GS-HgCH3 complexes (Dutczak and Ballatori, 1994), it is likely that the observed decrease in cerebral GSH levels in MeHg-exposed mice is related, at least in part, to the formation of GSSG and/or GS-HgCH3.
A gradual increase in protein levels and enzyme activities of the antioxidant defense system have been reported to occur in the mouse brain during the perinatal period (Khan and Black 2003). This phenomenon represents a physiological mechanism by which the brain protects itself from the surge in oxygen concentration encountered after delivery, which results in an increase in oxidative metabolism and, consequently, an increase in ROS generation (Khan and Black 2003). In agreement with these observations, we also noted a gradual increase in cerebral GSH levels, as well as in the GPx and GR activities during the early postnatal period. It is noteworthy that the maturation of different components of the GSH antioxidant system occurs simultaneously. In fact, the cerebral GSH levels were positively correlated to cerebral GR (Pearson coefficient = 0.795; P < 0.001) and GPx (Pearson coefficient = 0.786; P < 0.001) activities in control animals. In addition, cerebral GR and GPx activities were also positively and significantly correlated (Pearson coefficient = 0.687; P = 0.001) in these animals, reinforcing the idea of the simultaneous maturation of GSH levels, GR and GPx activities during the early postnatal period. This developmental balance was significantly perturbed by in utero exposure to MeHg, affecting the maturation of this important molecular process for protecting against pro-oxidative damage in the CNS. Indeed, when all experimental groups were analyzed together, significant positive correlations were also found between cerebral GSH levels and GPx activity (Pearson coefficient = 0.631; P < 0.001), GSH levels and GR activity (Pearson coefficient = 0.701; P < 0.001), and GPx and GR activities (Pearson coefficient = 0.560; P < 0.001). Accordingly, we conclude that prenatal exposure to MeHg renders the brain more susceptible to the deleterious effects of reactive oxygen species generated during cerebral aerobic metabolism.
While the GSH antioxidant system has a myriad of functions (Dringen and Harrlinger 2003), one of its most important roles is the detoxification of endogenously generated peroxides. Indeed, GPx catalyzes the reduction of hydrogen peroxide, phospholipid-hydroperoxide and other organic hydroxyperoxides by GSH (Flohé 1997), yielding oxidized glutathione (GSSG) which, in turn, is reduced back to GSH in a NADPH-dependent reaction catalyzed by glutathione reductase (Gul et al. 2000). Taking into account the deleterious effects of peroxide and peroxide-derived radicals in the CNS (Dringen et al. 2005), it is not surprising that the disruption of the GSH antioxidant system can result in oxidative damage to the brain. Here, we have observed that the cerebral levels of F2-isoprostanes were significantly increased in response to prenatal MeHg exposure. From a mechanistic point of view, the evidence of such a phenomenon is important as it represents the consequence of MeHg-induced disruption of the GSH oxidant system as well as suboptimal ability to combat ROS generated within the CNS. This idea is further reinforced by the fact that the disruption of the cerebral GSH antioxidant system was inversely correlated to the increased levels of cerebral F2-isoprostanes. Indeed, significant negative correlations were found between F2-isoprostanes and GSH (Pearson coefficient = −0.392; P = 0.003) as well as between F2-isoprostanes and GPx activity (Pearson coefficient = −0.434; P = 0.001). Moreover, the correlation between cerebral F2-isoprostanes and GR activity was almost significant (Pearson coefficient = −0.221; P = 0.096). These results show quantitative relationships among the different endpoints, suggesting that MeHg-induced disruption of the GSH system maturation is related, at least in part, to the observed MeHg-induced increased lipid peroxidation in the pup brain. In agreement with these observations, MeHg exposure (Vendrell et al., 2007) and deficient GPx activity (Cheng et al., 1999) have been associated with increased isoprostane levels. Moreover, cerebral isoprostanes have been previously demonstrated to be formed in increased amounts in neuropathological conditions related to oxidative stress (Musiek et al., 2007).
The increased cerebral mercury levels observed after the in utero exposure to MeHg are in agreement with other studies on the rapid placental transfer of MeHg from mothers to offspring (Watanabe et al. 1999). An intriguing phenomenon observed in our study was the extremely rapid decline of brain mercury levels from birth to weaning (Figure 5; half-life ≅ 10 days). Ballatori and Clarkson (1982) showed that MeHg has a long half-time in neonatal rodents (rats), which was ascribed to the inability of the neonatal liver to excrete MeHg into the bile, a key step for its elimination in adults. These authors also showed that the ability of the rat to excrete MeHg into the bile develops between 2 and 4 weeks of age, and that it is correlated with the ability of the developing liver to excrete GSH into the bile. On the other hand, Fair et al. (1987) observed the rapid elimination of MeHg from the mouse CNS after a single intracerebral injection (half-life = 1.6 days in cerebellum). Interestingly, these authors showed that cerebral MeHg is distributed to erythrocytes, the kidneys and the liver. Since we did not evaluate the inter-organ MeHg distribution and the rates of MeHg excretion through urine or bile, this topic represents an unresolved question that warrants additional investigation. Nonetheless, data reported in the literature indicate that the kinetics of MeHg distribution/elimination depend upon the developmental period, as well as the strain, sex, route of administration and dose (Ballatori and Clarkson 1982; Nielsen and Andersen 1991a,b).
Even though the cerebral mercury concentration decreased nearly to basal levels at postnatal day 21, GSH levels, GPx and GR activities remained decreased in MeHg-exposed mice when compared to control animals. These results indicate that prenatal exposure to MeHg affects cerebral GSH antioxidant systems by inducing biochemical alterations that endure even when mercury decreases nearly to basal levels. The apparently “paradoxical” long-lasting effect of in utero MeHg exposure on the GSH system, which was observed even when Hg levels were nearly undetectable, should be responsible for the permanent damage (increased brain lipid peroxidation) observed latter in the suckling period. Our experimental observation appears to be closely related to permanent functional deficits observed after prenatal MeHg exposure (Grandjean 2007).
Another important phenomenon worth noting was the effectiveness of the lower dose tested (1 mg/L) in causing the blockage of normal development of GSH system. At this dosage, levels of Hg deposited in the brains of suckling mice were indistinguishable from those observed in control animals. These results support the notion that exposure to MeHg during early fetal development can be associated with subtle brain damage at levels much lower than those expected to affect the mature brain (Dalgard et al., 1994; Costa et al., 2004). Furthermore, these results indicate the potential occurrence of a dangerous but silent pandemic of subclinical MeHg neurotoxicity (Grandjean and Landrigan 2006).
This study is the first to show that the prenatal exposure to MeHg disrupts the postnatal development of the GSH antioxidant system in the mouse brain, pointing to an additional molecular mechanism by which MeHg induces pro-oxidative damage in the developing CNS, thereby rendering the brain more susceptible to the deleterious effects of ROS normally produced by aerobic metabolism. Furthermore, the delayed neurochemical changes observed during the suckling period following gestational MeHg exposure (even in the presence of extremely low levels of mercury in the brain), indicates that exposure to MeHg during critical periods of development can trigger long-lasting biochemical changes associated with oxidative stress in developing organisms. Although it is difficult to extrapolate the results from animal models to humans, our results may indicate that the neurodevelopmental deleterious effects of perinatal MeHg exposure observed in epidemiological studies with children can be related, at least in part, to permanent neurochemical changes triggered by low levels of mercury during critical periods of brain development.
Acknowledgements
This study was supported by the FINEP research grant “Rede Instituto Brasileiro de Neurociência (IBN-Net)” # 01.06.0842-00 and by grants from CNPq to M. Farina (475329/2004-0 and 474796/2006-0) and US Public Health Service Grant NIEHS 07331 to M. Aschner.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen JW, Mutkus LA, Aschner M. Methylmercury-mediated inhibition of 3H-D-aspartate transport in cultured astrocytes is reversed by the antioxidant catalase. Brain Res. 2001a;902:92–100. doi: 10.1016/s0006-8993(01)02375-7. [DOI] [PubMed] [Google Scholar]
- Allen JW, Shanker G, Aschner M. Methylmercury inhibits the in vitro uptake of the glutathione precursor, cystine, in astrocytes, but not in neurons. Brain Res. 2001b;894:131–140. doi: 10.1016/s0006-8993(01)01988-6. [DOI] [PubMed] [Google Scholar]
- Aschner M, Syversen T, Souza DO, Rocha JB, Farina M. Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Braz J Med Biol Res. 2007;40:285–291. doi: 10.1590/s0100-879x2007000300001. [DOI] [PubMed] [Google Scholar]
- Aschner M, Yao CP, Allen JW, Tan KH. Methylmercury alters glutamate transport in astrocytes. Neurochem Int. 2000;37:199–206. doi: 10.1016/s0197-0186(00)00023-1. [DOI] [PubMed] [Google Scholar]
- Atchison WD. Is chemical neurotransmission altered specifically during methylmercury-induced cerebellar dysfunction? Trends Pharmacol Sci. 2005;26:549–557. doi: 10.1016/j.tips.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Clarkson TW. Developmental changes in the biliary excretion of methylmercury and glutathione. Science. 1982;216:61–63. doi: 10.1126/science.7063871. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol. 1985;113:484–490. doi: 10.1016/s0076-6879(85)13062-4. [DOI] [PubMed] [Google Scholar]
- Carvalho MC, Franco JL, Ghizoni H, Kobus K, Nazari E, Rocha JBT, Nogueira CW, Dafre AL, Muller YMR, Farina M. Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomotor deficits and cerebellar toxicity in mice. Toxicology. 2007;239:195–203. doi: 10.1016/j.tox.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Cheng W, Fu YX, Porres JM, Ross DA, Lei XG. Selenium-dependent cellular glutathione peroxidase protects mice against a pro-oxidant-induced oxidation of NADPH, NADH, lipids, and protein. FASEB J. 1999;13:1467–1475. doi: 10.1096/fasebj.13.11.1467. [DOI] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Clarkson TW, Magos L, Myers GJ. The toxicology of mercury--current exposures and clinical manifestations. N Engl J Med. 2003;349:1731–1737. doi: 10.1056/NEJMra022471. [DOI] [PubMed] [Google Scholar]
- Cohen G. Enzymatic/nonenzymatic sources of oxyradicals and regulation of antioxidant defenses. Ann N Y Acad Sci. 1994;738:8–14. doi: 10.1111/j.1749-6632.1994.tb21784.x. [DOI] [PubMed] [Google Scholar]
- Cooper AJL. Role of astrocytes in maintaining cerebral glutathione homeostasis and in protecting the brain against xenobiotics and oxidative stress. In: Shaw CA, editor. The Role of Glutathione in the Nervous System. Taylor and Francis; Washington: 1998. pp. 91–115. [Google Scholar]
- Cooper AJ, Kristal BS. Multiple roles of glutathione in the central nervous system. Biol Chem. 1997;378:793–802. [PubMed] [Google Scholar]
- Costa LG, Aschner M, Vitalone A, Syversen T, Soldin OP. Developmental neuropathology of environmental agents. Annu Rev Pharmacol Toxicol. 2004;44:87–110. doi: 10.1146/annurev.pharmtox.44.101802.121424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgard C, Grandjean P, Jorgensen PJ, Weihe P. Mercury in umbilical cord: Implications for risk assessment for Minamata Disease. Environ Health Perspect. 1994;102:548–550. doi: 10.1289/ehp.94102548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson PW, Myers GJ, Cox C, Wilding GE, Shamlaye CF, Huang LS, Cernichiari E, Sloane-Reeves J, Palumbo D, Clarkson TW. Methylmercury and neurodevelopment: longitudinal analysis of the Seychelles child development cohort. Neurotoxicol Teratol. 2006;28:529–535. doi: 10.1016/j.ntt.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Davidson PW, Myers GJ, Weiss B, Shamlaye CF, Cox C. Prenatal methyl mercury exposure from fish consumption and child development: a review of evidence and perspectives from the Seychelles Child Development Study. Neurotoxicology. 2006;27:1106–1109. doi: 10.1016/j.neuro.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- Dringen R, Pawlowski PG, Hirrlinger J. Peroxide detoxification by brain cells. J Neurosci Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- Dutczak WJ, Ballatori N. Transport of the glutathione-methylmercury complex across liver canalicular membranes on reduced glutathione carriers. J Biol Chem. 1994;269:9746–9751. [PubMed] [Google Scholar]
- Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Fair PH, Balthrop JE, Braddon-Galloway S. The toxicity, distribution and elimination of methylmercury in mice following intracerebral injection. Neurotoxicology. 1987;8:281–289. [PubMed] [Google Scholar]
- Farina M, Brandao R, Lara FS, Soares FA, Souza DO, Rocha JB. Mechanisms of the inhibitory effects of selenium and mercury on the activity of delta-aminolevulinate dehydratase from mouse liver, kidney and brain. Toxicol Lett. 2003c;139:55–66. doi: 10.1016/s0378-4274(02)00454-x. [DOI] [PubMed] [Google Scholar]
- Farina M, Dahm KC, Schwalm FD, Brusque AM, Frizzo ME, Zeni G, Souza DO, Rocha JB. Methylmercury increases glutamate release from brain synaptosomes and glutamate uptake by cortical slices from suckling rat pups: modulatory effect of ebselen. Toxicol Sci. 2003a;73:135–140. doi: 10.1093/toxsci/kfg058. [DOI] [PubMed] [Google Scholar]
- Farina M, Franco JL, Ribas CM, Meotti FC, Missau FC, Pizzolatti MG, Dafre AL, Santos AR. Protective effects of Polygala paniculata extract against methylmercury-induced neurotoxicity in mice. J Pharm Pharmacol. 2005;57:1503–1508. doi: 10.1211/jpp.57.11.0017. [DOI] [PubMed] [Google Scholar]
- Farina M, Frizzo ME, Soares FA, Schwalm FD, Dietrich MO, Zeni G, Rocha JB, Souza DO. Ebselen protects against methylmercury-induced inhibition of glutamate uptake by cortical slices from adult mice. Toxicol Lett. 2003b;144:351–357. doi: 10.1016/s0378-4274(03)00242-x. [DOI] [PubMed] [Google Scholar]
- Flohe L. Selenium in peroxide metabolism. Med Klin (Munich) 1997;92(Suppl 3):5–7. doi: 10.1007/BF03041948. [DOI] [PubMed] [Google Scholar]
- Franco JL, Braga H,C, Nunes AK, Ribas CM, Stringari J, Silva AP, Garcia Pomblum SC, Moro AM, Bohrer D, Santos AR, Dafre AL, Farina M. Lactational exposure to inorganic mercury: evidence of neurotoxic effects. Neurotoxicol Teratol. 2007b;29:360–367. doi: 10.1016/j.ntt.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Franco JL, Braga HC, Stringari J, Missau FC, Posser T, Mendes BG, Leal RB, Santos ARS, Dafre AL, Pizzolatti MG, Farina M. Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: protective effects of quercetin. Chem Res Toxicol. 2007a doi: 10.1021/tx7002323. in press. [DOI] [PubMed] [Google Scholar]
- Franco JL, Teixeira A, Meotti FC, Ribas CM, Stringari J, Garcia Pomblum SC, Moro AM, Bohrer D, Bairros AV, Dafre AL, Santos AR, Farina M. Cerebellar thiol status and motor deficit after lactational exposure to methylmercury. Environ Res. 2006;102:22–28. doi: 10.1016/j.envres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Franco R, Cidlowski JA. SLCO/OATP-like transport of glutathione in FasL-induced apoptosis: glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J Biol Chem. 2006;281:29542–29557. doi: 10.1074/jbc.M602500200. [DOI] [PubMed] [Google Scholar]
- Gilbert SG, Rice DC, Burbacher TM. Fixed interval/fixed ratio performance in adult monkeys exposed in utero to methylmercury. Neurotoxicol Teratol. 1996;18:539–546. doi: 10.1016/0892-0362(96)00081-5. [DOI] [PubMed] [Google Scholar]
- Grandjean P. Methylmercury toxicity and functional programming. Reprod Toxicol. 2007;23:414–420. doi: 10.1016/j.reprotox.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Landrigan PJ. Developmental neurotoxicity of industrial chemicals. Lancet. 2006;368:2167–2178. doi: 10.1016/S0140-6736(06)69665-7. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Weihe P, White RF, Debes F, Araki S, Yokoyama K, Murata K, Sorensen N, Dahl R, Jorgensen PJ. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol Teratol. 1997;19:417–428. doi: 10.1016/s0892-0362(97)00097-4. [DOI] [PubMed] [Google Scholar]
- Gul M, Kutay FZ, Temocin S, Hanninen O. Cellular and clinical implications of glutathione. Indian J Exp Biol. 2000;38:625–634. [PubMed] [Google Scholar]
- Halliwell B. Oxygen radicals as key mediators in neurological disease: fact or fiction? Ann Neurol. 1992;32(Suppl):S10–15. doi: 10.1002/ana.410320704. [DOI] [PubMed] [Google Scholar]
- Harada M. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit Rev Toxicol. 1995;25:1–24. doi: 10.3109/10408449509089885. [DOI] [PubMed] [Google Scholar]
- Johansson C, Castoldi AF, Onishchenko N, Manzo L, Vahter M, Ceccatelli S. Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotox Res. 2007;11:241–260. doi: 10.1007/BF03033570. [DOI] [PubMed] [Google Scholar]
- Kakita A, Wakabayashi K, Su M, Sakamoto M, Ikuta F, Takahashi H. Distinct pattern of neuronal degeneration in the fetal rat brain induced by consecutive transplacental administration of methylmercury. Brain Res. 2000;859:233–239. doi: 10.1016/s0006-8993(00)01964-8. [DOI] [PubMed] [Google Scholar]
- Khan JY, Black SM. Developmental changes in murine brain antioxidant enzymes. Pediatr Res. 2003;54:77–82. doi: 10.1203/01.PDR.0000065736.69214.20. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Manfroi CB, Schwalm FD, Cereser V, Abreu F, Oliveira A, Bizarro L, Rocha JB, Frizzo ME, Souza DO, Farina M. Maternal milk as methylmercury source for suckling mice: neurotoxic effects involved with the cerebellar glutamatergic system. Toxicol Sci. 2004;81:172–178. doi: 10.1093/toxsci/kfh201. [DOI] [PubMed] [Google Scholar]
- Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ. Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- Murata K, Sakamoto M, Nakai K, Weihe P, Dakeishi M, Iwata T, Liu XJ, Ohno T, Kurosawa T, Kamiya K, Satoh H. Effects of methylmercury on neurodevelopment in Japanese children in relation to the Madeiran study. Int Arch Occup Environ Health. 2004;77:571–579. doi: 10.1007/s00420-004-0542-1. [DOI] [PubMed] [Google Scholar]
- Murata K, Weihe P, Budtz-Jorgensen E, Jorgensen PJ, Grandjean P. Delayed brainstem auditory evoked potential latencies in 14-year-old children exposed to methylmercury. J Pediatr. 2004;144:177–183. doi: 10.1016/j.jpeds.2003.10.059. [DOI] [PubMed] [Google Scholar]
- Musiek ES, McLaughlin B, Morrow JD. Electrophilic cyclopentenone isoprostanes in neurodegeneration. J Mol Neurosci. 2007;33:80–86. doi: 10.1007/s12031-007-0042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutkus L, Aschner JL, Syversen T, Aschner M. Methylmercury alters the in vitro uptake of glutamate in GLAST- and GLT-1-transfected mutant CHO-K1 cells. Biol Trace Elem Res. 2005;107:231–245. doi: 10.1385/BTER:107:3:231. [DOI] [PubMed] [Google Scholar]
- Nielsen JB, Andersen O. Methyl mercuric chloride toxicokinetics in mice. I: Effects of strain, sex, route of administration and dose. Pharmacol Toxicol. 1991a;68:201–207. doi: 10.1111/j.1600-0773.1991.tb01223.x. [DOI] [PubMed] [Google Scholar]
- Nielsen JB, Andersen O. Methyl mercuric chloride toxicokinetics in mice. II: Sexual differences in whole-body retention and deposition in blood, hair, skin, muscles and fat. Pharmacol Toxicol. 1991b;68:208–211. doi: 10.1111/j.1600-0773.1991.tb01224.x. [DOI] [PubMed] [Google Scholar]
- Ou YC, White CC, Krejsa CM, Ponce RA, Kavanagh TJ, Faustman EM. The role of intracellular glutathione in methylmercury-induced toxicity in embryonic neuronal cells. Neurotoxicology. 1999;20:793–804. [PubMed] [Google Scholar]
- Reed MN, Paletz EM, Newland MC. Gestational exposure to methylmercury and selenium: effects on a spatial discrimination reversal in adulthood. Neurotoxicology. 2006;27:721–732. doi: 10.1016/j.neuro.2006.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DC, Schoeny R, Mahaffey K. Methods and rationale for derivation of a reference dose for methylmercury by the U.S. EPA. Risk Anal. 2003;23:107–115. doi: 10.1111/1539-6924.00294. [DOI] [PubMed] [Google Scholar]
- Roberts LJ, 2nd, Morrow JD. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808–820. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanker G, Syversen T, Aschner JL, Aschner M. Modulatory effect of glutathione status and antioxidants on methylmercury-induced free radical formation in primary cultures of cerebral astrocytes. Brain Res Mol Brain Res. 2005;137:11–22. doi: 10.1016/j.molbrainres.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Sirois JE, Atchison WD. Methylmercury affects multiple subtypes of calcium channels in rat cerebellar granule cells. Toxicol Appl Pharmacol. 2000;167:1–11. doi: 10.1006/taap.2000.8967. [DOI] [PubMed] [Google Scholar]
- Spurgeon A. Prenatal methylmercury exposure and developmental outcomes: review of the evidence and discussion of future directions. Environ Health Perspect. 2006;114:307–312. doi: 10.1289/ehp.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringari J, Meotti FC, Souza DO, Santos AR, Farina M. Postnatal methylmercury exposure induces hyperlocomotor activity and cerebellar oxidative stress in mice: dependence on the neurodevelopmental period. Neurochem Res. 2006;31:563–569. doi: 10.1007/s11064-006-9051-9. [DOI] [PubMed] [Google Scholar]
- Vendrell I, Carrascal M, Vilaro MT, Abian J, Rodriguez-Farre E, Sunol C. Cell viability and proteomic analysis in cultured neurons exposed to methylmercury. Hum Exp Toxicol. 2007;26:263–272. doi: 10.1177/0960327106070455. [DOI] [PubMed] [Google Scholar]
- Vicente E, Boer M, Netto C, Fochesatto C, Dalmaz C, Rodrigues Siqueira I, Goncalves CA. Hippocampal antioxidant system in neonates from methylmercury-intoxicated rats. Neurotoxicol Teratol. 2004;26:817–823. doi: 10.1016/j.ntt.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Watanabe C, Kasanuma Y, Dejima Y, Satoh H. The effect of prenatal methylmercury exposure on the GSH level and lipid peroxidation in the fetal brain and placenta of mice. Tohoku J Exp Med. 1999;187:121–126. doi: 10.1620/tjem.187.121. [DOI] [PubMed] [Google Scholar]
- Weihe P, Hansen JC, Murata K, Debes F, Jorgensen P, Steuerwald U, White RF, Grandjean P. Neurobehavioral performance of Inuit children with increased prenatal exposure to methylmercury. Int J Circumpolar Health. 2002;61:41–49. doi: 10.3402/ijch.v61i1.17404. [DOI] [PubMed] [Google Scholar]
- Weiss B, Stern S, Cox C, Balys M. Perinatal and lifetime exposure to methylmercury in the mouse: behavioral effects. Neurotoxicology. 2005;26:675–690. doi: 10.1016/j.neuro.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Wendel A. Glutathione peroxidase. Methods Enzymol. 1981;77:325–333. doi: 10.1016/s0076-6879(81)77046-0. [DOI] [PubMed] [Google Scholar]
- Yin Z, Milatovic D, Aschner JL, Syversen T, Rocha JB, Souza DO, Sidoryk M, Albrecht J, Aschner M. Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Res. 2007;1131:1–10. doi: 10.1016/j.brainres.2006.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoo EM, Valente JG, Grattan L, Schmidt SL, Platt I, Silbergeld EK. Low level methylmercury exposure affects neuropsychological function in adults. Environ Health. 2003;2:8. doi: 10.1186/1476-069X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]