

Abstract
Background
Bacteriophages (phages) are widespread in the environment and play a crucial role in the evolution of their bacterial hosts and the emergence of new pathogens.
Results
LSB-1, a reference coliphage strain, was classified as a member of the Podoviridae family with a cystic form (50 ± 5 nm diameter) and short tail (60 ± 5 nm long). The double stranded DNA was about 30 kilobase pairs in length. We identified its host range and determined the gp17 sequences and protein structure using shotgun analysis and bioinformatics technology.
Conclusions
Coliphage LSB-1 possesses a tailspike protein with endosialidase activity which is probably responsible for its specific enteroinvasive E.coli host range within the laboratory.
Background
Bacteriophages (phages) are widespread in the environment and play a crucial role in the evolution of their bacterial hosts and the emergence of new pathogens. They have enormous potential for the development of new drugs, therapies and environmental control technologies, such as natural, non-toxic alternatives for controlling bacterial pathogens. Recent interest in phages has been stimulated by studies that demonstrate the efficacy of phages in preventing and treating infections [1-3].
Phages are readily isolated from water samples in the environment. Some of the isolated phages have shown broad-host range interaction with the bacterial isolates and others have shown either species or strain level specificity. Both polyvalent phages and non-polyvalent phages are morphologically and genetically diverse [4,5]. They are efficient at host recognition but there is no single method of adsorption, and different phages employ different strategies [6,7]. Identification of the mechanisms of adsorption and the host ranges of different phages may allow genetic manipulation to alter the phage host binding profile artificially. To gain a better understanding of the biological properties of phages, we have sequenced the genome of the gp17 from phage LSB-1, which was isolated from sewage samples. We determined its host range and analyzed its 3-dimensional structure to identify possible functional domains.
Results
Coliphage morphology
The LSB-1 phage has a cystic form of 50 ± 5 nm in diameter, with a short noncontractile tail 60 ± 5 nm long (Fig. 1). It is classified as a member of the Podoviridae family [8-10].
Figure 1.

Electron micrograph of phage LSB-1. Electron micrographs of the phages with a short, stout tail. The polyhedral nature of the viral head is shown.
Nucleic acid characterization
LSB-1 coliphage nucleic acid was sensitive to Dnase I, but resistant to Rnase A and S1 nuclease (Fig. 2). It was concluded that all the extracts contained double-stranded linear DNA.
Figure 2.
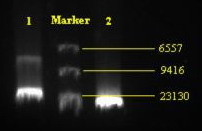
Agarose gel electrophoresis of DNA of phage LSB-1. Lane 1: whole genome DNA digested by Nhe I. Lane 2: whole genome DNA without digestion. Molecular size markers (kb) are HindIII-digested lambda DNA.
Preliminary overview of the coliphages genome
The shotgun sequencing and a primer-walking method was used to assemble the whole linear LSB-1 genome of approximately 30 kb. Fifteen major potential ORFs were identified, all of which could be assigned functions based on homology with corresponding genes in the K1F coliphage in a BLAST search (Fig. 3 and 4).
Figure 3.
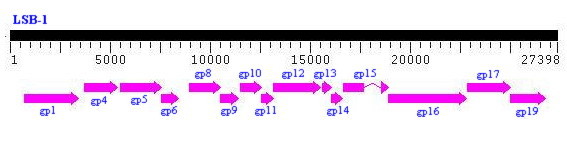
Comparative genomic arrangements of coliphage LSB-1 with K1F. Genome of coliphage LSB-1 is aligned. Arrows indicate functions of the ORFs.
Figure 4.
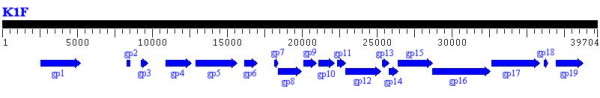
Comparative genomic arrangements of coliphage LSB-1 with K1F. Genome of coliphage K1F is aligned. Arrows indicate functions of the ORFs.
Genome relationship of LSB-1 and other phages
We chose four important proteins, capsid protein (gp10), tail tubular protein (gp12), internal virion protein D (gp16), and endosialidase (gp17), which are present in phage T7 supergroup members, to build phylogenetic trees. As shown in Fig. 5, 6, 7 and 8, trees built using different proteins are broadly similar. Coliphages LSB-1, EcoDs1, and K1F closely clustered towards the end of the group and phages T7, T3, phiYeO3-12 and K11 cluster within another branch of the same group. These data suggest that LSB-1 is most closely related to the K1F-like phages, and should be classified as a new member of the K1F supergroup.
Figure 5.
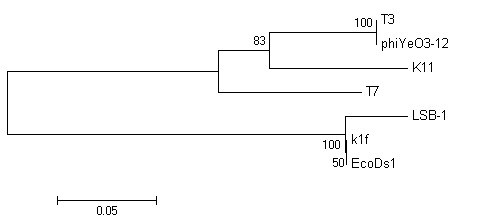
Phylogenetic analysis of the capsid protein (gp10) from seven phages in the T7 supergroup. The alignment of whole sequence was used to construct the neighbor-joining tree. The scale bar represents 0.05 fixed mutations per amino acid position. Bootstrap values based on 1000 computer-generated tree are indicated at the nodes.
Figure 6.
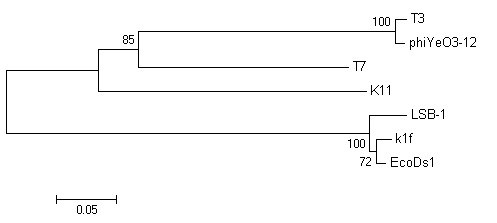
Phylogenetic analysis of the tail tubular protein (gp12) from seven phages in the T7 supergroup. The alignment of whole sequence was used to construct the neighbor-joining tree. The scale bar represents 0.05 fixed mutations per amino acid position. Bootstrap values based on 1000 computer-generated tree are indicated at the nodes.
Figure 7.
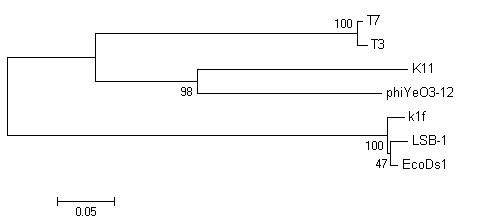
Phylogenetic analysis of the internal virion protein (gp16) from seven phages in the T7 supergroup. The alignment of whole sequence was used to construct the neighbor-joining tree. The scale bar represents 0.05 fixed mutations per amino acid position. Bootstrap values based on 1000 computer-generated tree are indicated at the nodes.
Figure 8.
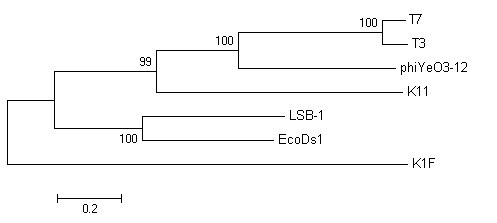
Phylogenetic analysis of the endosialidase (gp17) from seven phages in the T7 supergroup. The alignment of whole sequence was used to construct the neighbor-joining tree. The scale bar represents 0.2 fixed mutations per amino acid position. Bootstrap values based on 1000 computer-generated tree are indicated at the nodes.
Gp17 DNA sequencing and function analysis
As shown in Fig. 9 and 10, the secondary structure of gp17 protein was composed of 33.10% Alpha helix (Hh), 25.25% Extended strand (Ee) and 30.29% Random coil (Cc).
Figure 9.
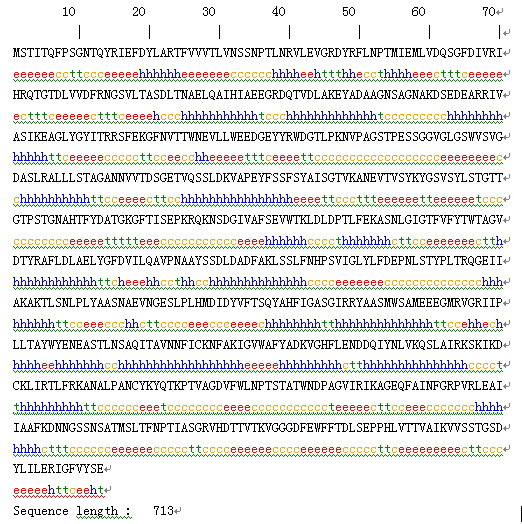
Secondary structure prediction of gp17 protein.
Figure 10.

Supplements and reports of Fig. 9, the secondary structure of gp17 protein comprised 33.1% Alpha helix(Hh), 25.25% Extended strand(Ee) and 30.29% Random coil(Cc).
The PHYRE program generated several possible templates classified as best fit when the gp17 sequence was submitted. All of these templates included the sequences of hydrolase as one of its domains. It also provided the SCOP codes, E-values and estimated precision values for the predicted models. Table 1 shows these parameters provided by different templates.
Table 1.
Parameters provided by different templates
| No | SCOP Code | E-value | Estimated Precision | Classification |
|---|---|---|---|---|
| 1 | c2c1lA_ | 1.1 | 70% | Hydrolase |
| 2 | d7a3ha_ | 4.2 | 45% | glycosidases |
| 3 | c2cksA_ | 4.6 | 40% | Hydrolase |
| 4 | c1wkyA_ | 16 | 10% | Hydrolase |
The endosialidase 3-dimensional structure was chosen to generate coordinates for the Gp17 of LSB-1 based on an E-value of 1.1 and an estimated precision of 70%. Using the PHYRE program, it was difficult to find another closer coordinate template for Gp17 of LSB-1 even though an E-value of less than 2e-6 was considered [11-13]. Gp17 of LSB-1 included endosialidase amino acid residues from Thr290 to Glu713, with the with the exception of forty-one residues (Ser 465 to Trp475, Tyr497 to Val512, and Lys543 to Leu562). These forty-one residues did not match corresponding sequences within the prediction server. The Gp17 3-dimensional structure model predicted two domains: Domain A with hydrolase activity connected by an intervening unstructured sequence to Domain B. The later domain may have a host-anchoring function. A worm algorithm representation of the model is shown in Fig. 11. Domain A, the larger of the two domains, includes residues Thr290 to Phe528 and corresponds to the N-terminal domain of endosialidase. It comprises 4 α helices and 9 β strands with intermittent unstructured intervening regions. The arrangement is characteristic of crystallized hydrolase. Domain B is composed of 1 α helix and 9 β strands with intermittent unstructured intervening regions. It includes residues Gln542 to Glu713 and corresponds to the C-terminal domain of endosialidase. There were no predicted structural constraints in the connecting region between domains A and B which included the residues of Tyr529 to Asp541.
Figure 11.
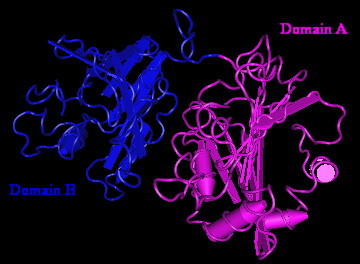
Computer-generated 3-dimensional modeling of gp17 protein. Worm algorithm representation of the 3-dimensional model showing the two main domains. The larger Domain A is homologous to hydrolase sequences. The smaller Domain B is homologous to the sequence of the anchoring domain.
The molecular surface structure of the Gp17 is presented in Fig. 12. It shows the catalytic pocket in Domain A comprising the catalytic triad, Glut405, Arg415, and Arg487, identical to that found in the endosialidase family of enzymes. Domain A is shown topographically in front of Domain B. The connecting region was difficult to see in this representation because of its opaqueness. Fig. 13 and Fig. 14 show the worm algorithm structures of Gp17and the KIF endosialidase respectively.
Figure 12.
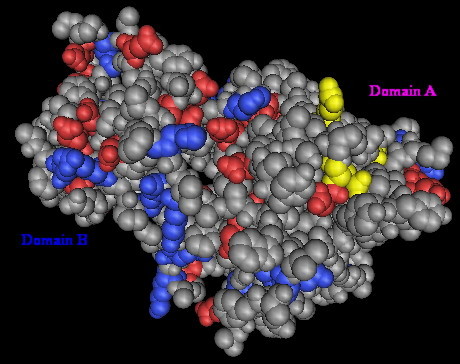
Computer-generated 3-dimensional modeling of gp17 protein. Molecular surface presentation of the predicted catalytic pocket of endosialidase. The active site triad (yellow), Glut405, Arg415 and Arg487, is the amidase reaction site of endosialidase.
Figure 13.
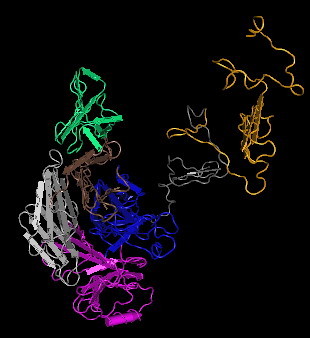
Worm algorithm representation of the model of endosialidase of K1F.
Figure 14.
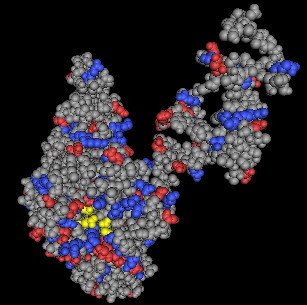
Molecular surface presentation of K1F endosialidase. The active site triad (yellow) is Glut581, Arg596 and Arg647.
Discussion
Previous studies have suggested that gene 17 from the T7-like and K1F-like phage may be playing an important role in host range recognition processes [14,15], but little work has been done on comparing their molecular characterization. K1F-like phages are known to possess tailspike proteins with endosialidase activity that degrades polySia with high substrate specificity. These tailspike-associated enzymatic activities enable the phages to penetrate the capsular layer and are important determinants of the bacteriophage host range [16]. The T7-like phage encodes a tail fiber protein that specifically recognizes and binds to lipopolysaccharide [17] and recognizes E. coli B and many E. coli K-12 strains.
It was expected that molecular characterization would provide evidence for a host adsorbing mechanism of coliphage LSB-1. We found that the LSB-1 has some common features of the phage K1F supergroup. A tailspike protein with endosialidase activity is implicated in allowing a specific enteroinvasive E.coli host range. Insertion of such an endosialidase gene into a non-polyvalent virulent phage may artificially increase its host range to enteroinvasive E.coli. Using manipulation of the phage genome to kill pathogenic bacteria has broad implications for the welfare of both man and animals. Our results may inform new ways of genetic manipulation of phages to alter their host binding profile.
Materials and methods
Purification of phage particles
Bacteriophage LSB-1 was propagated in liquid culture. The EIEC strain 8401 was infected with phages at a multiplicity of infection of 0.1. Following complete lysis of the host cells, cell debris was removed by centrifugation at 4000 × g for 10 min, and phage particles in the supernatant were concentrated by adding polyethylene glycol 6000 and NaCl to achieve final concentrations of 10% and 1.0 M, respectively. Phage particles were collected by centrifugation at 8000 × g for 10 min. Pellets were re-suspended in 0.01× the original volume of sterile SM buffer (5.8 g sodium chloride, 2 g magnesium sulphate, 100 mg gelatin, 50 mL 1 mol·L-1 Tris (pH 7.5) and 945 mL distilled water). For isopycnic centrifugation, the phage suspension was placed on a cesium chloride gradient stepwise using three solutions whose densities were 1.45, 1.50, and 1.70, respectively. After centrifugation for 60 min at 150,000 × g, the phage band was withdrawn and dialyzed against 10 mM Tris-HCl (pH 7.5) containing 10 mM MgSO. The purified phage (approximately 1011PFU/ml) was stored at 4°C until use.
Host range determination
Seventeen bacterial strains, listed in Table 2, were tested for sensitivity against the isolated phages. The Spot Test [18] was used to determine the host range of the phage. Lytic activity was examined following overnight incubation at 37°C and recorded on a scale as follows: (N) no plaques, (T) turbid plaques, (C) clear plaques. To obtain an accurate estimate of relative phage lytic activity, all host range determinations were carried out simultaneously using a single high-titer stock of purified bacteriophage and all host cells were incubated at 37°C in LB medium.
Table 2.
Phage and bacterial strains used in the study and host range spectrum of the bacteriophages
| No | Species | Strain | Plaque formation |
|---|---|---|---|
| 1 | Escherichia | 285a | N |
| 2 | Escherichia | ATCC 25922b | N |
| 3 | Escherichia | ATCC 8099b | N |
| 4 | Escherichia | ATCC 10536b | N |
| 5 | Escherichia | JM109c | N |
| 6 | Escherichia | BL21c | N |
| 7 | Escherichia | HDSαc | N |
| 8 | Escherichia | O157H7 44752d | N |
| 9 | Escherichia | EIEC 8401d | C |
| 10 | Escherichia | EIEC ATCC 43893d | C |
| 11 | Staphylococcus aureus | ATCC 6538b | N |
| 12 | Staphylococcus aureus | ATCC 26001b | N |
| 13 | Salmonella typhi | ATCC 50071b | N |
| 14 | Pseudomonas aeruginosa | ATCC 10145b | N |
| 15 | Shigella dysenteriae | ATCC 13313b | N |
| 16 | Α-hemolytic streptococcus | ATCC 32213b | N |
| 17 | Β-hemolytic streptococcus | ATCC 32210b | N |
Obtained from aZhenghui L, Institute of Medical Equipment, Academy of Military Medical Science, China; bBacterial culture center, Institute of Microbiology and Epidemiology, Academy of Military Medical Science, China; cJing a. Department of Microbiology, Third Military Medical University, China; dBacterial culture center, Institute of Epidemiology and Microbiology, Chinese Academy of Preventive Medicine.
C = clear plaque; T = turbid plaque; N = no plaque.
Electron microscopy
To examine phage LSB-1 morphology, 50 μl of purified viral stock solution was fixed by addition of 50 μl of 0.5% glutaraldehyde in 4% paraformaldehyde and a drop of this solution was placed on a carbon coated copper grid After waiting 30 min for settlement, excess liquid was removed and the grid was allowed to dry. A drop of 2% phosphotungstic acid was added for 2 min before excess was removed with filter paper before drying and then examination by TEM (Hitachi, model S-800) at an acceleration voltage of 45 kV.
Nucleic acid characterization
The method used was based upon that described by Sambrook and Russell [19]. Bacteriophage from the concentrated solutions were lysed with the addition of EDTA (final concentration 20 mmol·L-1), proteinase K (final concentration 50 μg·mL-1) and SDS (final concentration 0.5%) and incubation at 56°C for 1 h. The nucleic acid was purified using phenol, phenol/chloroform and chloroform extraction. The final aqueous phase was dialyzed overnight against Tris EDTA buffer (TE). In this method, nucleic acid yield was estimated by agarose gel electrophoresis and comparison of ethidium bromide stain intensity with known DNA standards (Hind III cut lambda phage DNA, New England Biolabs Inc, USA).
The nucleic acid extracts were diluted to a standard concentration of ~20 ng·μL-1. Approximately 250ng of each extract was subjected to digestion with DNase I (Sigma Aldrich), RNase A (Sigma Aldrich) and S1 nuclease (Promega). All reactions were terminated with the addition of EDTA (10 mmol·L-1 final concentration) and analyzed using 0.8% agarose gel electrophoresis at 5 V cm-1.
Gp17 DNA sequencing and analysis
The coliphage genome was sequenced by the shotgun method. Genomic DNA was sheared by sonication, cloned into pUC18 and sequenced with an ABI 3700 automated DNA sequencer, to give 13-fold coverage of the genome. Sequences were assembled into contigs, and gaps linked using a primer-walking technique (Kaczorowski and Szybalski, 1998) [20]. Potential open reading frames (ORFs) were predicted using ORF Finder http://www.ncbi.nlm.nih.gov/projects/gorf/ and manual correction. Translated ORFs were used in a BLAST search against the Swiss-Prot http://us.expasy.org/tools/blast and NCBI protein databases http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastHome. Protein secondary structures were predicted with SOPMA http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html.
Phylogenetic trees were constructed using Mega 4 software following published protocols [21,22].
The amino acid sequence of endosialidase was determined and compared with the protein structure family databases PDB [23], SCOP [24,25], and PFAM [26] to identify the most suitable template structure. The eventual template structure was taken from PDB. Three-dimensional models were created using PHYRE http://www.sbg.bio.ic.ac.uk/~phyre/ by mapping the coordinates of the template structure with aligned residues of the endosialidase. Computer-generated three-dimensional models were viewed and analyzed using CN3 D 4.1 application software programs obtained from http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml.
Conclusions
In summary, a typical Podoviridae morphology and the double-stranded nature of its DNA give the coliphage LSB-1 some common features with the phage K1F supergroup. It possesses a tailspike protein with endosialidase activity which is probably responsible for its specific enteroinvasive E.coli host range within the laboratory.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YC and HX conceived of the study, carried out the experimental work, analysis and drafted the manuscript. XM and LC participated in its design and coordination and helped to draft the manuscript. GH and ZR participated in its design and experimental work. LZ participated in coordination of the study. All authors read and approved the final manuscript.
Contributor Information
Yaming Chai, Email: charming1982@yahoo.cn.
Hongyan Xiong, Email: hongyanxiong@sohu.com.
Xiangyu Ma, Email: lzluna22@126.com.
Liqing Cheng, Email: linhpf1@163.com.
Guorong Huang, Email: yollowrong@sina.com.
Zhonglin Rao Lin Zhang, Email: wang.qingqing1985@hotmail.com.
Acknowledgements
The authors would like to thank Jinxin Ke and Wenqi Huang for their help in electron microscopy. The project was supported by National Natural Science Foundation of China (Grant No.30571637), Scientific Research Project of the Chongqing China (Grant No. CSTC, 2008AC5005) and National Key Technology R&D Program of China (Grant No. 2008BAD96B06-05).
References
- Hermoso JL, García. Taking aim on bacterial pathogens: from phage therapy to enzybiotics. Curr Opin Microbiol. 2007;10:461–472. doi: 10.1016/j.mib.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Mann. The potential of phages to prevent MRSA infections. Res Microbiol. 2008;159:400–405. doi: 10.1016/j.resmic.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Sulakvelidze. Phage therapy: an attractive option for dealing with antibiotic-resistant bacterial infections. Drug Discov Today. 2005;10:807–809. doi: 10.1016/S1359-6446(05)03441-0. [DOI] [PubMed] [Google Scholar]
- Hyman ST. Bacteriophage Host Range and Bacterial Resistance. Adv Appl Microbiol. 2010;70:217–248. doi: 10.1016/S0065-2164(10)70007-1. full_text. [DOI] [PubMed] [Google Scholar]
- Song HY, Xu XM, Zhang. Isolation of a novel polyvalent virulent bacteriophage of E. coli. J Med Coll PLA. 2007;22:261–267. doi: 10.1016/S1000-1948(07)60054-9. [DOI] [Google Scholar]
- Konopa KT. Isolation of coliphage lambda ghosts able to adsorb onto bacterial cells. Biochim Biophys Acta. 1975;399:460–467. doi: 10.1016/0304-4165(75)90273-1. [DOI] [PubMed] [Google Scholar]
- Paranchych PM, Bradley. Stages in phage R17 infection. Virology. 1970;41:465–473. doi: 10.1016/0042-6822(70)90168-6. [DOI] [PubMed] [Google Scholar]
- DudaIcosahedral Tailed dsDNA Bacterial Viruses. Encyclopedia of Virology. 2008. pp. 30–37. full_text.
- MolineuxT7-Like Phages (Podoviridae) Encyclopedia of Virology. 2004. pp. 1722–1729. full_text.
- Rothman-DenesEnterobacteria Phage N4 (Podoviridae) Encyclopedia of Virology. 2004. pp. 450–454. full_text.
- Bujnicki AE, Fischer LR. LiveBench-2: large-scale automated evaluation of protein structure prediction servers. Proteins. 2001. pp. 184–191. [DOI] [PubMed]
- Bujnicki AE, Fischer LR. Structure prediction meta server. Bioinformatics. 2001;17:750–751. doi: 10.1093/bioinformatics/17.8.750. [DOI] [PubMed] [Google Scholar]
- Cristobal AZ, Fischer. et al. A study of quality measures for protein threading models. BMC Bioinformatics. 2001;2:5. doi: 10.1186/1471-2105-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida KM, Hattori SY, Kawase SI. Structure and function of a novel coliphage-associated sialidase. FEMS Microbiol Lett. 2000;182:333–337. doi: 10.1111/j.1574-6968.2000.tb08917.x. [DOI] [PubMed] [Google Scholar]
- Schulz DS, Frank KS, Mühlenhoff AD, Gerardy-Schahn RF. Structural Basis for the Recognition and Cleavage of Polysialic Acid by the Bacteriophage K1F Tailspike Protein EndoNF. J Mol Biol. 2010;397:341–351. doi: 10.1016/j.jmb.2010.01.028. [DOI] [PubMed] [Google Scholar]
- Bull ER, Molineux. A tale of tails: Sialidase is key to success in a model of phage therapy against K1-capsulated Escherichia coli. Virology. 2010;398:79–86. doi: 10.1016/j.virol.2009.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven BL, Maizel MU, Parry JS, Hainfeld FW. Molecular substructure of a viral receptor-recognition protein: The gp17 tail-fiber of bacteriophage T7. J Mol Biol. 1988;200:351–365. doi: 10.1016/0022-2836(88)90246-X. [DOI] [PubMed] [Google Scholar]
- Champagne NG. The spot test method for the in-plant enumeration of bacteriophages with paired cultures of Lactobacillus delbrueckii subsp. bulgaricus and Streptococcus salivarius subsp. thermophilus. Int Dairy J. 1995;5:417–425. doi: 10.1016/0958-6946(95)00011-Q. [DOI] [Google Scholar]
- Sambrook DW. A laboratory manual. CSHL Press. New York; 2001. Molecular cloning. [Google Scholar]
- Kaczorowski WS. Genomic DNA sequencing by SPEL-6 primer walking using hexamer ligation. Gene. 1998;223:83–91. doi: 10.1016/S0378-1119(98)00241-8. [DOI] [PubMed] [Google Scholar]
- Heres DV. Phylogenetic analysis of the pathogenic bacteria Spiroplasma penaei based on multilocus sequence analysis. J Invertebr Pathol. 2010;103:30–35. doi: 10.1016/j.jip.2009.10.004. [DOI] [PubMed] [Google Scholar]
- El Ossmani BB, Aboukhalid MB, Gazzaz DZ, Chafik JT. Assessment of phylogenetic structure of Berber-speaking population of Azrou using 15 STRs of Identifiler kit. Leg Med. 2010;12:52–56. doi: 10.1016/j.legalmed.2009.10.004. 22. Ossmani, B.B., boukhalid, M.B., Gazzaz, D.Z. & Chafik, J.T. (2010). Assessment of phylogenetic structure of Berber-speaking population of Azrou using 15 STRs of Identifiler kit. Leg Med. 12, 52-56. [DOI] [PubMed] [Google Scholar]
- Berman KH, Nakamura. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Andreeva DH, Brenner. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 2004;32:D226–D229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner CC, Hubbard AG. Understanding protein structure: using scop for fold interpretation. Methods Enzymol. 1996;266:635–643. doi: 10.1016/s0076-6879(96)66039-x. full_text. [DOI] [PubMed] [Google Scholar]
- Bateman EB, Durbin. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 1999;27:260–262. doi: 10.1093/nar/27.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]