

Abstract
A radiation-induced mouse mutant, Brachyrrhine (Br), exhibits frontonasal dysplasia and renal hypoplasia, two malformations associated with deficiencies in mesenchymal condensation. The purpose of this study was to resolve the Br locus, evaluate possible candidate genes, and identify developmental defects in the mutant chondrocranium. Linkage analysis mapped the Br mutation to a critical region distal to D17Mit76 which contains only one gene, the transcription factor Six2. Sequence analysis of the Six2 gene, including 1.5 kb of the promoter, failed to reveal the Br mutation. However, homozygous Br/Br embryos showed almost complete absence of Six2 mRNA and protein in craniofacial and renal tissues while heterozygous Br/+ embryos displayed intermediate Six2 levels. Mutant embryos displayed malformations of neural crest-derived structures of the anterior cranium where Six2 is normally expressed. These data suggest a mutation in a novel cis-acting regulatory region inhibits Six2 expression and is associated with frontonasal dysplasia and renal hypoplasia.
Keywords: Six2, frontonasal dysplasia, renal hypoplasia, chondrocranium, Br
INTRODUCTION
Craniofacial deformations including ocular hypertelorism, broad nasal root, cleft nose and lip, absence of a nasal tip, and cranium bifidum, have long been recognized as occurring in conjunction (Lehmann-Nitsche, 1901; Gorlin et al., 2001). DeMeyer (1967) was the first to categorize these concomitant features and apply the diagnostic phrase ‘Median Cleft Syndrome.’ Sedano et al. (1970) subsequently formalized the diagnostic process by recognizing four subclassifications among those patients diagnosed with Median Cleft Syndrome and applied the phrase ‘Frontonasal Dysplasia’ (FND) since the authors believed that the primary defect occurred early in morphogenesis affecting the frontonasal prominence. FND occurring alone has a sporadic incidence in humans and reported at 0.43–0.73% (Apesos and Anigian, 1993), but few epidemiological studies exist. Developmental mechanisms responsible for FND remain unclear, but disregulation of median facial prominence fusion must be considered a prominent feature of the disease.
FND is generally considered to be a heritable malformation in humans (DeMyer, 1967; Warkany et al., 1973; Bakken and Aabyholm, 1976; Moreno Fuenmayor, 1980; Fryburg et al., 1993), although various modes of inheritance have been identified. Early studies by Cohen et al. (1971) and Reich et al. (1977) suggested an autosomal dominance pattern of inheritance while Moreno Fuenmayor (1980) reported an autosomal recessive inheritance based on inbred kin. Other studies have suggested the possibility of an X-linked pattern of inheritance (Fryburg et al., 1993; Nevin et al., 1999). De Moor et al. (1987) reported three unrelated children showing a consistent phenotype including hypertelorism, median cleft lip and the absence of a nasal tip and concluded that FND displays a multifactorial inheritance mode. Similarly, penetrance and expression of FND remains problematic. Moreno Fuenmayor (1980) showed that bifid nose had the highest inbreeding coefficient in an inbred pedigree while hypertelorism was less certain, concluding that FND is a genetic disorder with variability of expression. FND also occurs in conjunction with numerous other malformations in syndromic diseases such as trisomy 13, branchio-oto-renal (BOR) syndrome, acromely and others (Guion-Almeida et al., 1996; Gorlin et al., 2001). Specific defects occurring with FND also include defects in eyes (Temple et al., 1990), limbs (Fragoso et al., 1982; Kwee and Lindhout, 1983; Verloes et al., 1992; Slaney et al., 1999), cardiovascular system (De Moor et al., 1987; Meinecke and Blunck, 1989), and kidneys (Roizenblatt et al., 1979). Patterns of inheritance suggest that FND is an inherited phenotypic outcome resulting from numerous potential genetic mutations affecting multiple morphogenetic pathways.
Recent advances in human and experimental teratology have underscored the importance of disturbances in developmental cassettes that result in phenotypic malformations (Jan and Jan, 1993; Epstein et al., 2004). Many of these critical pathways involve transcription factors that can affect development in anatomically remote areas typical of syndromic diseases (Fisher and Scambler, 1994; Seidman and Seidman, 2002). Of particular interest is the co-occurrence of FND in patients with renal hypoplasia (RH) that is characterized by abnormally small kidneys resulting from abnormal morphogenesis (Glassberg et al., 1987). Typically, the hypoplastic kidney is associated with chronic renal failure since an insufficient number of nephrons cannot filter adequately (Brenner and Mackenzie, 1997; Pope et al., 1999; Terzi et al., 2000; Kemper and Muller-Wiefel, 2001; Douglas-Denton et al., 2006). The kidney eventually fails since it becomes unable to excrete wastes, retain electrolytes, and concentrate urine normally. Although numerous molecules are involved in the development of the kidney (Dressler, 2006), specific cellular and genetic defects causing RH remain poorly understood.
We have characterized a mouse mutant, called Brachyrrhine (Br), which arose from X-ray irradiation of the 3H1 strain and carries a semidominant mutation that results in FND and RH (Lozanoff, 1993; Ma and Lozanoff, 1993; Lozanoff et al., 2001). The homozygous mutant mouse (Br/Br) dies at birth due to severely disrupted nephrogenesis and large median facial cleft. The heterozygote mutant (Br/+) survives to adulthood and is fertile, but displays small kidneys and midface retrusion. Previous studies have shown the Br/Br mutant mice demonstrated an absence of major midline internal features of the anterior cranial base, including the nasal septum, presphenoid, and presphenoidal synchondrosis, while the basisphenoid was malformed rostrally. Also in these Br/Br mice, primary and secondary palates failed to form (Singh et al., 1998; McBratney et al., 2003). Although the presphenoid and nasal septum were present in Br/+ mice, both were malformed, and ectopic cartilage was detected in the surrounding regions (Lozanoff, 1993; Lozanoff et al., 1994; Ma and Lozanoff, 1996; McBratney et al., 2003). Experimental evidence suggested that these defects were a result of decreased mesenchymal cellular proliferation during development of these cranial base regions (Lozanoff, 1999; Ma and Lozanoff, 1999; Ma and Lozanoff, 2002). During embryonic kidney development in Br/Br mutant mice, we have shown successful induction of the UB and initial MM condensation (gestational day 11.5, i.e., E11.5), but severe disruption of nephrogenesis shortly afterward (Lozanoff et al., 2001). At E12.0 and E12.5, a greater number of renal vesicles appear in the Br/Br kidney than in the +/+ kidney, but MM condensation around the UB tips can no longer be observed in the Br/Br mice. By E15.0, the +/+ kidney has a clearly defined medulla and cortex, but the Br/Br kidney remains very small and disorganized (Lozanoff et al., 2001). Thus, the Br mutation appears to allow for the initiation of kidney development, but then impedes the process of nephrogenesis, resulting in RH.
Previously, the Br mutation was mapped to an area of distal chromosome 17, but candidate genes for the mutation had not been identified (McBratney et al., 2003). In the current study, we further resolved the physical mapping of the Br locus to a 171 kb critical region on chromosome 17, within which the only gene is Six2. Although extensive sequencing of Six2 gene and immediate promoter region did not reveal any mutations in Br mice, when Six2 mRNA expression during embryonic development was analyzed, we found it greatly downregulated in the mutant mice. Protein analysis confirmed the near absence of Six2 in Br/Br mutant facial prominences, and metanephric mesenchyme in the developing kidney. We also found that the characteristics of FND in postnatal Br mice originate from defects in embryonic chondrocranial morphogenesis, possibly due to fewer mesenchymal cells in the developing craniofacial region. We can conclude that misexpression of Six2 in the Br mouse is associated with mesenchymal defects leading to FND and RH.
RESULTS
Br Outcrosses Display FND and RH
Outbreeding of 3H1-Br/+ mice to Balb and Castaneous (Cast) lines resulted in homozygous dominant mice (Br/Br) with midfacial clefting and renal hypoplasia (Fig. 1) similar to those previously described for the 3H1-Br/Br mice (Ma and Lozanoff, 1993; McBratney et al., 2003). Segregation analysis was performed on outbred lines based on genotypes and phenotypes to ensure that the mutation was inherited as a semidominant lethal mutation. Results showed that offspring from reciprocal 3H1 x Balb F1 heterozygote (Br/+) crosses separated into three groups based on craniofacial morphology with a ratio of 24:51:18 (+/+:Br/+:Br/Br; n=93) that was not significantly different from the expected 1:2:1 ratio (χ2 = 1.65; d.f. = 2; p<0.44). A similar relationship occurred from reciprocal crossings of 3H1 x Cast F1 heterozygote mutant (Br/+) mice, with a ratio of 22:46:20 (+/+:Br/+:Br/Br; n=88) that was not significantly different from the expected relationship (χ2 = 0.27; d.f. = 2, p < 0.87).
Figure 1.

Normal craniofacial and kidney morphology in newborn 3H1 +/+ mice (A,B) compared to representative 3H1 Br/Br (C,D), 3H1 x Cast N2 Br/Br (E,F) and 3H1 x Balb N2 Br/Br (G,H) mutant hybrid mice displaying frontonasal dysplasia and renal hypoplasia. a = adrenal gland; k = kidney. Bar in G = 5 mm; Bar in H = 1 mm.
Segregation analysis of offspring from F1 3H1 x Balb heterozygote mutants (Br/+) backcrossed to inbred 3H1 mice showed that fetuses segregated into two groups (Br/+:+/+) based on phenotypes and corresponding genotypes with a ratio of 58:54 (n = 112) that did not differ significantly from an expected ratio of 1:1 (χ2 = 1.43, d.f.= 1, p < 0.71). Similar results were obtained with offspring from F1 3H1 x Cast heterozygote mutants (Br/+) backcrossed to inbred Cast mice yielding a ratio of (105:98, n = 203) that was not significantly different from the expected 1:1 ratio (χ2 = 0.24, d.f. = 1, p < 0.62).
Chondrocranial Morphology is Defective in Br Mice
Embryonic heads were fixed, stained with Alcian blue, and cleared to characterize abnormal patterns of chondrocranial development at post-conception day 16 (TS24). Wild type crania showed complete and normal chondrocranium morphology (Fig. 2A). The orbitonasal laminae met the central stem in the midline normally, and the central stem was completed by fusion of the trabecular cartilage with the hypophyseal cartilage. The trabecular cartilage increased in width caudally in the region of the presumptive presphenoid bone, fusing laterally with the hypochiasmatic cartilages. The orbital cartilages formed cartilaginous plates adjacent to the eyes, with preoptic and postoptic roots extending towards the central stem and enclosing the optic foramina. (Fig. 2D). The nasal capsule was wider in the Br/+ cranium, which appeared to increase midfacial width (Fig. 2B). The central chondrocranial stem was not fused at the junction of the trabecular and hypophyseal cartilages, while lack of lateral chondrification of the caudal trabecular cartilage left the hypochiasmatic cartilages unfused and isolated. Furthermore, the orbitonasal laminae did not meet the central stem in the midline, and the orbital cartilages were absent resulting in orbitonasal laminae that abut the hypochiasmatic cartilages abnormally due to loss of the preoptic roots (Fig. 2E). The Br/Br cranium had a truncated snout, an underlying wide nasal capsule with a midline cleft, and an incompletely developed anterior cranial base (Fig. 2C). The mutant chondrocranium displayed a severe loss of anterior structures (Fig. 2F). The trabecular cartilages did not fuse in the midline nor did they project caudally into the presumptive presphenoid region resulting in an incomplete central cartilaginous stem. The hypophyseal cartilage projected rostrally as a free spicule. The orbitonasal laminae were laterally displaced and met the hypochiasmatic cartilages abnormally, with the orbital cartilages missing altogether. These features are all consistent with FND.
Figure 2.

Whole mount stained Theiler Stage 24 (E16) crania (A–F). The +/+ head (A,D) displays normal cranial base development while the Br/+ (B,E) cranium shows a fused trabecular cartilage lacking lateral chondrification, and the orbital cartilages are absent (*). Although the hypochiasmatic cartilages are present, they contact the orbitonasal laminae abnormally. The Br/Br cranium (C,F) displays unfused trabecular cartilages that are truncated caudally so that they do not meet the hypohyseal cartilage forming an abnormal rostral point. The orbitonasal laminae are displaced laterally and abnormally abut the reduced hypochiasmatic cartilages while the orbital cartilages are absent. AH = hypochiasmatic cartilage; HC = hypophyseal cartilage; OC = orbital cartilage; OL = orbitonasal lamina; TC = trabecular cartilage.
High resolution linkage analysis
To narrow the critical region for the Br mutation, a total of 720 3H1 x Cast and 250 3H1 x Balb backcross hybrids were analyzed for recombination events occurring in distal chromosome 17. Of the microsatellite markers analyzed, D17Mit76 and D17Mit56 both had only 1 recombination out of 720 mice (LOD score of 213), thereby placing the Br mutation distal to D17Mit76 (Table 1; Fig. 3). Due to an absence of reported microsatellites between D17Mit76 and D17Mit123, we scanned the genomic sequence of this interval for small dinucleotide repeats that could be heterogeneous between 3H1 and Cast mice and identified four novel microsatellites abbreviated as MS3, MS34, MS25, and MS6, located at 86.16 Mb, 86.21 Mb, 86.57 Mb and 86.89 Mb along mouse chromosome 17, respectively (Ensembl genome database, based on NCBI m37 mouse assembly). PCR amplification of each of these novel microsatellites resulted in a size difference between 3H1 and Cast mice large enough to resolve by agarose gel electrophoresis, but not a significant size difference between 3H1 and Balb mice. At MS34, 4 recombinants out of 720 mice were found (LOD score of 206), with zero recombinants for MS3 from the sample out 720 mice (Table 1; Fig. 3). These recombination events define a 171 kb Br critical region between D17Mit76 and MS34. Based on the Ensembl assembly, only one gene exists in this genomic interval, the homeobox transcription factor, Six2 (Accession # NM_011380; MGI # 102778).
Table 1.
Br mutation mapping data using microsatellite markers along mouse chromosome 17. Shown are both Cast and Balb backcrosses, where X is number of recombinants among N total backcrossed mice analyzed. The calculated distance (in centimorgans) and LOD scores are shown on the right.
| Cast | ||||
|---|---|---|---|---|
| Marker | X | N | Distance (cM) | LOD |
| D17Mit122 | 14 | 513 | 2.73 ± 0.72 | 127 |
| D17Mit155 | 7 | 513 | 1.36 ± 0.51 | 138 |
| D17Mit189 | 3 | 513 | 0.58 ± 0.34 | 146 |
| D17Mit56 | 1 | 720 | 0.14 | 213 |
| D17Mit76 | 1 | 720 | 0.14 | 213 |
| D17Mit211 | 0 | 720 | ||
| Br | ||||
| MS3 | 0 | 720 | ||
| MS34 | 4 | 720 | 0.56 | 206 |
| MS25 | 5 | 720 | 0.69 | 204 |
| MS6 | 5 | 720 | 0.69 | 204 |
| D17Mit123 | 17 | 513 | 3.31 ± 0.79 | 122 |
| Balb | ||||
|---|---|---|---|---|
| Marker | X | N | Distance (cM) | LOD |
| D17Mit155 | 5 | 220 | 2.27 ± 1.00 | 56 |
| D17Mit76 | 1 | 248 | 0.40 ± 0.40 | 72 |
| Br | ||||
| D17Mit123 | 18 | 250 | 7.20 ± 1.63 | 47 |
Figure 3.

Schematic of the defined critical region on mouse chromosome 17 for the Br mutation based on the results of the microsatellite linkage analysis. Microsatellite markers analyzed include: D17Mit122, D17Mit155, D17Mit189, D17Mit56, D17Mit76, D17Mit211 D17Mit123, and novel microsatellites MS3, MS34, MS29, MS25, and MS6. Within the 171 kb critical genomic interval, there is only one gene: Six2.
To test if the Br mutation is located in the Six2 gene, PCR primers were designed to amplify the Six2 sequences from genomic DNA for direct sequencing. Sequence analysis was conducted for the entire Six2 coding region (2 exons), the complete sole intron (1.8 kb), the 5′ and 3′ untranslated region, and 1.8 kb upstream from the start codon, which includes the predicted transcription start sites and both of the demonstrated promoter binding sites (Boucher et al., 2000; Brodbeck et al., 2004; Kutejova et al., 2005). However, after comparison of sequences from multiple Br/Br mice and +/+ control mice of the 3H1 strain, no mutation was found in the Six2 gene. Mutations further away from the gene, perhaps in novel cis-acting regulatory regions, were not ruled out by this methodology.
Six2 is Misexpressed in Br embryos
Whole-mount in situ hybridizations with a Six2 cRNA probe showed qualitative differences in expression between +/+, Br/+, and Br/Br mice at various stages of embryonic development (Fig. 4). At day E8.5, which is the earliest point that Six2 expression has been previously detected (Oliver et al., 1995b), wild type mice showed intense staining around the first brachial arch (Fig. 4A), while in Br mutant mice there was an obvious decrease in expression (Fig. 4B,C). Although dramatically decreased at E8.5 in Br/Br mice, Six2 expression did not appear to be completely absent at this point (Fig. 4C). At E11.5, wild type mice displayed a band of expression in the craniofacial region in the area of the forebrain and facial prominences. Staining also was present in the developing axial skeleton and limbs extending caudally to the urogenital region (Fig. 4D). However, Br/+ mice had reduced expression in the same regions relative to the wildtype, while the Br/Br mouse showed an apparent loss of expression of Six2 (Fig. 4E,F). From Six2 in situ hybridizations of E12.5 heads (Fig. 4G-I), the loss of Six2 expression in the Br mutant facial prominences and midface was clearly visible. Similarly, in E12.5 kidneys (Fig. 4J-L), Six2 expression was significantly decreased in Br/+ animals and totally absent in Br/Br mutants. Unexpectedly, we noted that the Br mutant embryos consistently demonstrated activation of Six2 mRNA expression in the lens, which was not observed in wild type embryos. This was the only tissue that demonstrated such activation, which supports the conclusion that the Br mutation alters the transcriptional regulation of the Six2 gene.
Figure 4.

In situ hybridizations for Six2 in whole-mount embryos. In E8.5 (A-C), Six2 was detected primarily in the first brachial arch of wild type embryos (A), with a noticely reduced expression in Br/+ embryos (B), and a further reduction in Br/Br embryos (C). At E11.5 (D-F), Six2 is expressed in the midface, facial prominences, first branchial arch, and the urogenital region of the wild type (D). The Br/+ embryo shows significantly reduced expression (E), and Six2 expression in the Br/Br embryo is nearly absent (F). However, note the activation of Six2 expression in the lens of the Br/+ (E) and more so in Br/Br (F) embryos. At E12.5 (G-L), Six2 expression remains obvious in the cranial base region and facial prominences of wild type heads (G), while reduced in the Br/+ heads (H) and absent in the Br/Br heads (I), with the exception of the lens. Also at E12.5, Six2 expression was strong in wild type kidneys (J), much reduced in Br/+ kidneys (K), and absent in Br/Br kidneys (L).
Histological tissue sections made from E11.5 whole mount in situ hybridizations revealed Six2 expression in the mesenchyme of the medial nasal prominence and mesenchyme dorsal and ventral to the eye of +/+ embryos (Fig. 5A,D). Six2 expression was greatly decreased in these same regions in Br/+ mice and completely lacking in Br/Br mice (Fig. 5B,C; E,F). Six2 was not present in the lens of the +/+ craniofacial tissue (Fig. 5D) but slight expression was detected in the lens of Br/+ mice and strong expression in Br/Br (Fig. 5E,F).
Figure 5.
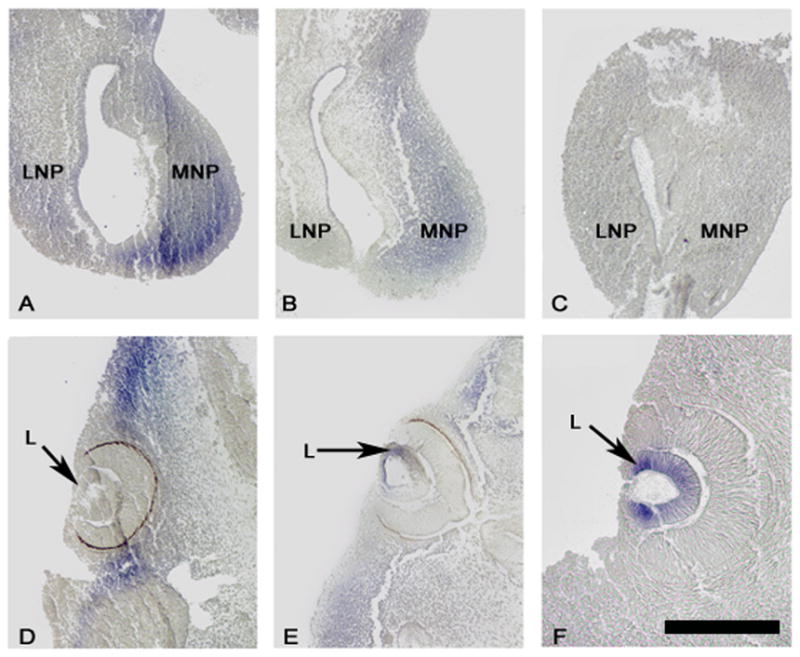
In situ hybridization for Six2 in E11.5 sections. Wild type mice show strong expression in the medial nasal prominence and in the mesenchyme rostral and caudal to the eye (A & D). Six2 expression is reduced in these same regions in Br/+ sections (B & E) and absent in Br/Br sections (C & F). Six2 is detected in the lens of Br/+ mice (E) and very strongly expressed in the Br/Br lens (F), whereas it is not observed in the lens of wild type mice (D). L = lens; LNP = lateral nasal prominence; MNP = medial nasal prominence. Bar = 100 μm.
Results from quantitative real time RT-PCR (qPCR) performed on RNA extracted from E10.5–E11.0 embryos revealed decreased levels of Six2 expression in all Br/+ and Br/Br mice (Fig. 6A). Relative to normalized Six2 expression in wild type embryos, Br/+ heads demonstrated a 55% reduction in Six2 expression and Br/Br heads showed a 92% reduction in expression. The torso also showed reduced Six2 expression with a 40% reduction in Br/+ mice and an 88% reduction in Br/Br mice relative to the normalized wild type mice. Western blot analysis showed that Six2 protein content in E13.5 Br/+ head extracts was approximately 50% of that displayed in wild type samples while Six2 levels in Br/Br head extracts was virtually absent (Fig. 6B). In E13.5 kidney extracts, overall Six2 levels were more variable within each genotype but showed the same pattern of downregulation in Br/+, while Six2 was undetectable in Br/Br kidneys (Fig. 6B). The multiple bands observed on the Western blot for Six2 was possibly due to phosphorylation, since closely related family member Six1 has been shown to be phosphorylated and hyperphosphorylated (Ford et al., 2000). For each sample, Six2 band intensities were quantitated and normalized against β-actin levels (Fig. 6C). In E13.5 head extracts, Six2 protein levels were decreased 53% in Br/+ samples and 96% in Br/Br samples. In E13.5 kidney extracts, Six2 proteins levels were decreased 93% in Br/+ samples, and 100% in Br/Br samples. It is possible that the small, but detectable, level of Six2 in the Br/Br head extracts was associated with the Six2 mRNA expression detected in the lens of Br mutant mice (Figs. 4,5). To test if the Br mutation influences the expression of the Six3 gene, which is located 3.3 kb outside of the Br critical domain (Fig. 3), we used qPCR to measure Six3 expression levels in E11.5 embryonic heads of Br mutant mice. RNA was isolated from three samples from each genotype, and qPCR revealed no significance differences in Six3 expression levels between wild type, Br/+, and Br/Br embryos (Fig. 6D).
Figure 6.

Real time qPCR and Western blot analysis showing that Six2 is significantly down-regulated in Br/+ and Br/Br tissues. qPCR measured Six2 mRNA levels in RNA extracted from the head and torso of E10.5–11.0 +/+, Br/+, and Br/Br mice (A). Expression of Six2 in Br/+ and Br/Br embryos is shown relative to expression of Six2 in +/+ embryos after being normalized against the amount of EF1α1. Error bars correspond to one standard deviation. Western blotting with an anti-Six2 antibody for proteins extracted from the head and kidney of E13.5 +/+, Br/+, and Br/Br mice (B) also shows a descending magnitude of Six2 protein in mutant mice. Quantities of total proteins in each lane were confirmed by probing with an anti-β-actin antibody. The net intensities of each Six2 Western blot band was measured and normalized with the β-actin net intensity for each sample, confirming quantitative decreases in Six2 protein levels in Br/+ and Br/Br mutant embryos (C). Real time qPCR measured no changes in Six3 mRNA expression levels in E11.5 Br/+ and Br/Br mutant embryo heads (D). * = p-value < 0.01.
To verify the absence of Six2 protein in the Br/Br embryos, immunostaining was performed on cryosections from embryos collected at E11.5 and E14.5 (Fig. 7). In the craniofacial region of the E11.5 embryos, Six2 was detected primarily in the mesenchymal cells throughout the medial nasal prominences and a small aggregation of subepithelial cells in the rostral tip of the lateral nasal prominence, as well as in cranial neural crest cells and developing cranial base (Fig. 7A,C). In addition, Six2 was detected in the olfactory epithelium, which gives rise to the olfactory pits where early neurogenesis occurs. In the Br/Br E11.5 embryos, where the developing median cleft was visible as widely spaced medial nasal prominences, Six2 protein was not detected in any of these tissues (Fig. 7B,D). At E11.5 the ureteric bud (UB) has completed its first bifid branch, and in wild type embyros, Six2 was detected in the metanephric mesenchyme (MM) surrounding the growing tubules (Fig. 7E), but was completely absent from MM in the Br/Br embryos (Fig. 7F). By E14.5, the developing kidney has organized so the nephrogenic zone is around the periphery of the kidney. In wild type E14.5 kidneys, Six2 was localized to the outside ring of mesenchymal cells surrounding the tips of branching tubules (Fig. 7G), while in Br/Br kidneys, no Six2 was detected (Fig. 7H).
Figure 7.

Immunofluorescent staining of Six2 in wild type and Br/Br embryos. Six2 staining is shown in green, while nuclei stained with propidium iodide are in red, and areas of overlapping signal are shown as yellow. Six2 only localized in cell nuclei as expected for a transcription factor. In wild type E11.5 heads (A, C), Six2 was localized primarily in the MNP and midline mesenchyme, extending dorsally into the developing chondrocranium. Six2 was also localized to the olfactory epithelium of the nasal pits. In Br/Br embryos, Six2 staining was not detected in any of these tissues (B, D). At E11.5, the UB has begun to branch into the MM, and Six2 staining in wild type embryos was strong in the MM surrounding the UB tubule (E). In the Br/Br E11.5 kidneys, Six2 was not detected (F). In the E14.5 wild type kidney, Six2 was localized around the periphery of the developing kidney in the undifferentiated MM cells (G), while Six2 staining in the disorganized Br/Br E14.5 kidneys is absent (H). LNP = lateral nasal prominence; MNP = medial nasal prominence; OE = olfactory epithelium.
DISCUSSION
In this study, we have demonstrated that the mutation of the Br mouse is associated with misexpression of the Six2 gene. The mutation was previously shown to be inherited as an autosomal dominant trait in the 3H1 mouse strain (McBratney et al., 2003), and we have further demonstrated that animals derived from outcrosses with Balb and Castaneus strains also show similar phenotypic frequencies. Thus, background did not affect expression of FND and RH in these outcross strains and the morphogenetic pathway did not appear to be modified by different backgrounds. This finding suggests that the Br morphogenetic pathway is likely inherited without modifying or multifactorial gene effects.
Previous analysis revealed a normal karyotype in the Br mutant mice, and mapped the Br mutation to the distal region of chromosome 17 (Beechey et al., 1997; McBratney et al., 2003). In this study, we used a large backcross sample to narrow the candidate region to a 171 kb critical region between D17Mit76 and the novel microsatellite MS34. Within this genomic interval there is only one gene, the homeobox transcription factor Six2, which has been previously shown to be expressed in developing mesenchymal tissue including head and urogenital system at the time of overt midfacial and renal differentiation (Oliver et al., 1995b). Based on the spatiotemporal expression pattern, as well as the linkage analysis data, we considered Six2 an excellent candidate gene to be the target of the Br mutation. Although direct sequencing did not identify the Br mutation within the exons, intron, untranslated regions, or immediate 5′ promoter region of the Six2 gene, further analysis demonstrated the Br mutation dramatically alters Six2 expression.
The Six gene family is becoming recognized as a set of critical transcription factors involved in embryonic morphogenesis (Kawakami et al., 2000). Members of this gene family in the mouse include six genes divided into three subgroups based on amino acid sequence similarities including Six1/2, Six3/6 and Six4/5. These genes function in a conserved gene regulatory network involving mammalian orthologues of drosophila genes ey, so, eya and dach (Kawakami et al., 2000; Brodbeck and Englert, 2004). Developmental pathways involving Six genes are recognized as important for the development of both the cranium and renal system and when disturbed, contribute to syndromes involving the head and kidney, such as branchio-oto-renal disease (Abdelhak et al., 1997; Kumar et al., 1998; Xu et al., 1999; Buller et al., 2001).
Whole mount in situ hybridizations and immunostaining detecting regions of Six2 expression in wild type mice were consistent with studies of Six2 previously published (Oliver et al., 1995a; Ohto et al., 1998; Brodbeck et al., 2004). Beginning early in mouse development (E8.5), Six2 was detected in the rostral craniofacial region, primarily in mesenchymal cells of neural crest origin. By E11.5, Six2 expression localized to the mesenchymal cells of the nasal prominences, midline, and developing skull vault, as well as the olfactory epithelium. At E11.5, when the kidney first begins to develop, Six2 was expressed in urogenital region, primarily in the condensing MM surrounding the growing ureteric bud. In each of these tissues, Six2 expression was significantly decreased in Br/+ heterozygous mutants, and largely absent in Br/Br homozygous mutants. Measured by qPCR, there was less Six2 mRNA present in the head and torso of Br mice with evidence of a dosage effect on expression, since one copy of the mutant allele reduces the amount of Six2 by approximately half (44–60%) in the head and torso compared to the wild type. Two copies of the mutant Br allele greatly reduced Six2 in the head and torso (8–12%) of relative expression compared to normal mice. These lower levels were confirmed by Western blotting and immunostaining, further suggesting that defective Six2 production is associated with the craniofacial and renal defects characterized in Br mice.
This study provides further evidence that the FND phenotype seen in Br mice is associated with the disruption of chondrocranial morphogenesis and alterations in the neural crest-derived mesenchyme destined for the anterior cranial base. Haploinsufficiency of Six2 is reflected in the chondrocranial morphology of the Br/+ mice, as specific cartilages of the chondrocranium were concurrently malformed or absent. The orbital cartilages did not form, and the trabecular cartilages showed delayed and reduced chondrification in Br/+ mutant mice, while in Br/Br mice, the trabecular cartilages never fused, resulting in the complete absence of the presphenoid. Also, the relative positions of the hypochiasmatica cartilages and orbitonasal laminae were mildly altered in Br/+ mice and then further displaced in Br/Br mice. If Six2 affects migration and proliferation of neural crest cells of the anterior cranial base, this would not directly affect the mesoderm derived hypochiasmatic cartilages (unpublished data, B. McBratney-Owen), so their modified placement could be a secondary alteration to the primary changes in orbital and trabecular cartilage development. Similarly, the lateral displacement of the orbitonasal lamina might be a secondary affect of the unfused trabecular cartilages.
In the cranium, the Pax/Six/Eya/Dach network has been studied in placode development (Schlosser, 2006), and individual family members function in critical morphogenetic steps including optic, nasal and otic placode formation (Zou et al., 2004; Purcell et al., 2005). The Six transcription factors appear to work in conjunction with Eya proteins (Zou et al., 2004; Purcell et al., 2005); however, this network may not require Pax expression as an upstream regulator in all tissues (Xu et al., 1999; Ozaki et al., 2004; Purcell et al., 2005). Also, while Six1 and Six3 are expressed in tissues with a neuronal fate, Six2 expression in the head appears to be mainly located to neural crest-derived mesenchymal tissue (Oliver et al., 1995b). This cell population presumably contributes to the morphogenesis of the frontonasal ectodermal zone known to establish a proximodistal growth of the frontonasal prominence (Marcucio et al., 2005). Based on observations reported here, neural crest-derived head mesenchyme destined to form the interior cartilages of the anterior chondrocranium requires expression of Six2. We observed a decrease in proliferation among this sub-population of neural crest cells (Ma and Lozanoff, 1999) and subsequently found less mesenchymal tissue in the presumptive trabecular cartilage, suggesting Six2 may promote cellular proliferation in neural crest cells that form the trabecular and orbital cartilages. Therefore, while other Pax/Six/Eya/Dach network defects have been linked with malformations of the otic placodes (Zou et al., 2004) and lens and nasal placodes (Purcell et al., 2005), the Br mouse serves as a model for linking a defect in the expression of a member of this network with abnormal anterior cranial base and medial facial prominence development, subsequently leading to FND.
Early in development, Br/Br mice showed normal kidney initiation events with initial branching of the ureteric buds and condensation of the primitive nephrogenic zone occurring between E11.0 and E11.5 (Lozanoff et al., 2001). However, subsequent branching of the duct system diminished at E12.0 with failure of further nephrogenic zone condensation. The role of the Pax/Six/Eya/Dach network in the kidney has been relatively well studied and shows that members of this network converge to up-regulate Gdnf (glial-cell-line-derived neurotrophic factor), which induces ureteric buds to grow into the metanephric mesenchyme (Xu et al., 1999; Ribes et al., 2003; Brodbeck et al., 2004; Brodbeck and Englert, 2004). Although Six2 has been shown to be able to activate Gdnf expression by binding to two sites on the Gdnf promoter (Brodbeck et al., 2004), in the absence of Six2, shown in Lozanoff et al (2001) and in Self et al (2006), intial budding of the ureteric bud was still successful. This may be a result of the recently discovered Hox11-Pax2-Eya complex which activates both the Six2 and Gdnf transcription (Gong et al., 2007), supported by a study showing loss of Pax2 in the MM leads to an absence of Gdnf expression (Brophy et al., 2001). Lozanoff and colleagues (2001) reported that Pax2 protein is expressed normally in condensing kidney mesenchyme of Br mice at E11.5; however, Pax2 protein expression is erratically expressed around renal tubules from E13 onwards in Br mutants. In analysis of the Six2-null kidney, Self et al. (2006) showed that lack of Six2 affected the MM condensation and differentiation around the UB tips, resulting in premature and ectopic epithelial differentiation. Each of these previous studies support our hypothesis that the Br kidney phenotype results from defective Six2 pathways.
Recently, a Six2 knockout mouse was created and characterized (Self et al., 2006), but displayed several differences compared to the Br mice. The Six2-null displayed RH, but no craniofacial abnormalities, while heterozygous-null mice were not reported to exhibit any obvious abnormalities. The kidney phenotype of the Br/Br mice is very similar to the Six2-null mice, in that although there is an early increase in tubule differentiation, nephrogenesis rapidly ceases (Lozanoff et al., 2001; Self et al., 2006). However, we have observed that Br/+ mice have smaller than normal kidneys (Lozanoff et al., 2001), indicating that haploinsufficiency of Six2 may also affect nephrogenesis. The major difference with the Br mice is the Six2 knockout’s absence of any abnormal craniofacial phenotype (Self et al., 2006), which was unexpected given the strong expression of Six2 in the first brachial arch, midface mesenchyme, and facial prominences. We have previously described various abnormalities in the Br mice occuring in tissues derived from these Six2-expressing cells, such as median orofacial clefting (McBratney et al., 2003), absence of the presphenoid and nasal septum (McBratney et al., 2003), and decreases in cellular proliferation in the chondrocranium of the cranial base (Ma and Lozanoff, 1999). In their study, Self et al. did not report whether Six2 expression was indeed missing in the Six2-null embryos’ craniofacial tissues, or if other Six family members were perhaps activated in these mice as potential compensatory mechanisms. Since almost nothing is known regarding the biological role of the Six2 transcription factor in craniofacial development, it is difficult to hypothesize the cause of this discrepency between the two mutant mouse models. Further analysis of both the Br mice and the Six2 knockout mice will hopefully clarify the reason for the variation, and yield more insight into the role of Six2 during mammalian development.
The data presented in this study suggest that the Br mutation affects the transcriptional regulation of Six2 rather than the Six2 mRNA molecule itself. This hypothesis was additionally supported by the detection of Six2 RNA in the lens of the Br mutants, which does not occur in wild type mice. However, the expression levels of the neighboring Six3 gene, normally expressed in the forebrain and developing eye, was unaltered in Br mutant embryos. A growing number of studies of developmentally important genes are establishing the significance of distant cis-acting elements in regulating their complex spatiotemporal expression pattern. These include genes such as Sox9 (Wunderle et al., 1998; Bien-Willner et al., 2007), Sox10 (Deal et al., 2006), shh (Roessler et al., 1997; Epstein et al., 1999; Lettice et al., 2003; Sagai et al., 2005; Jeong et al., 2006), Pax6 (Kleinjan et al., 2001), WT1 (Moore et al., 1998), Myf5 (Hadchouel et al., 2000), Mrf4 (Carvajal et al., 2001), Dach (Nobrega et al., 2003), and members of the BMP/GDF family of signaling molecules (DiLeone et al., 2000; Mortlock et al., 2003). Transgenic mice were created using ~900 bp of the Six2 promoter cloned upstream to a lacZ reporter gene, and lacZ expression in those embryos appeared in the MM and the 1st branchial arch, but not in the craniofacial region (Brodbeck et al., 2004; Kutejova et al., 2005). This suggests that the ~900 bp region can drive some basal level of Six2 expression in these tissues, but does not contain other critical Six2 regulatory elements. This region was sequenced in the Br mutant mice and no mutations were detected. Therefore, it is likely that the Br mutation creates or interrupts an uncharacterized cis-acting regulatory sequence upstream or downstream of the Six2 gene that silences Six2 in the tissues in which it is normally expressed. Future work will be directed at identifing the mutation responsible for FND and RD in the Br mouse model, and further characterizing the biological function of Six2 for embryonic craniofacial and kidney development and disease.
EXPERIMENTAL PROCEDURES
Animals
Adult mice were housed under standard conditions with a 12-hour light cycle. Females mated with a male overnight were examined for a vaginal plug the following morning and constituted gestational day E0.5 if present. Embryos were obtained between days E10.5 and E18.5 and were subsequently staged using Theiler staging criteria (TS) to ensure the developmental stage of each embryo was similar to the conception day (E) designation (Theiler, 1989). Only animals of the same E designation and TS were compared. To provide a control of chondrocranial morphology, 3H1 mice were inbred without any mixture to mice with the Br mutation and compared with mice obtained from reciprocal mating of 3H1-Br/+ animals. For physical mapping of the Br mutation and to determine whether the mutation could be transferred to a different genetic background and produce the same phenotype deficiencies, 3H1-Br/+ mice were outbred with inbred lines of Castaneous (3H1-Br/+ x Cast) and Balb (3H1-Br/+ x Balb) mice. Offspring from these crosses were reciprocally backcrossed and associated whole mount specimens were compared.
Segregation Analysis
To test whether Br exhibits a similar inheritance pattern in outcross animals, segregation analysis was performed as described by McBratney et al. (2003). Reciprocal matings between 3H1 x Cast or 3H1 x Balb F1 heterozygote (Br/+) hybrids were performed. Embryos were collected between E11.5 and birth, phenotypically scored, and genotyped using D17Mit76 as a PCR marker based on the recombination results derived from microsatellite data. A χ2 test was used to determine whether the incidence of Br differed significantly from the expected 1:2:1 (heterozygous crosses) or 1:1 (heterozygous x homozygous normal crosses) ratios.
Whole-Mount Cartilage Staining
Whole embryos from E13.5 to E18.5 were dissected from the uterus in PBS and stored in 70% EtOH. Because the chondrocranium is composed only of cartilage as it begins to form, crania stained in Alcian blue alone revealed individual cartilages of the chondrocranium most clearly. Following fixation in Bouin’s fixative overnight, embryos were washed in 0.1% NH4OH, 70% EtOH until embryos appeared bleached. Embryos were then equilibrated in 5% acetic acid twice for 1 hour each time and left overnight in 0.05% Alcian blue 8GX (Fisher) in fresh 5% acetic acid. This was followed by washing twice for 1 hour each in 5% acetic acid, soaking in methanol twice for 1 hour each and clearing in 1:2 benzyl alcohol:benzyl benzoate (BABB). Crania were then removed and heads dissected to reveal the chondrocranium.
Microscopy
Whole embryos at E14 were dissected from the uterus in PBS and stored in 70% EtOH. Specimens were dehydrated, cleared in xylene, and mounted in paraffin. Wax chucks were sectioned at 10 μm. Slides were cleared in xylene, rehydrated, soaked in Gill Modified Hematoxylin Harlecohematoxylin (Harleco), Clarifier 2 (Richard-Allan Scientific), Bluing reagent (Richard-Allan Scientific), Eosin Y (Richard-Allan Scientific), followed by dehydration and a final clearing in xylene. Slides were mounted with cover slips and viewed on a microscope and patterns of cell density and morphology were evaluated qualitatively.
Microsatellite Linkage Analysis and Genotyping
The Br mutation was previously mapped to a 4.9 Mb interval on mouse chromosome 17 between microsatellite markers D17Mit189 and D17Mit221 (McBratney et al., 2003). To refine the current map, we expanded the interspecific reciprocal backcross of 3H1 Br/+ males and Cast and Balb using a total of 720 3H1 x Cast N2 and 250 3H1 x Balb N2 backcross hybrids utilizing an interspecific backcross strategy. Mice were scored for sex at weaning, killed, and genomic DNA was extracted from either blood or tail tissue samples. Seven microsatellite markers on distal chromosome 17 (D17Mit122, D17Mit155, D17Mit189, D17Mit56, D17Mit76, D17Mit211, and D17Mit123) were scored for the Castaneus backcross while three microsatellites (D17Mit155, D17Mit76, and D17Mit123) were scored for the Balb backcross to confirm results from the larger Cast sample. Microsatellite primers were synthesized based on sequences listed at http://www.informatics.jax.org and optimized. Pairs of oligonucleotides were used to amplify each marker using a Thermo Electron thermocycler with a PCR profile consisting of an initial denaturation at 94°C for 4 minutes, then 35 cycles of 30 seconds at 94°C (denaturation), 30 seconds at 55–60°C (annealing), and 30 seconds at 72°C (extension), with a final extension at 72°C for 4 minutes. The PCR products were separated by electrophoresis in 2–4% agarose gels and stained with ethidium bromide. The gels were photographed (Kodak Gel Logic 200) and genotypes were scored for recombination. Data were analyzed with Map Manager software (http://www.mapmanager.org). A two-point LOD score analysis was conducted between all pairs of markers and the probable position of the Br locus was calculated relative to surrounding microsatellite markers. The marker informative for both crosses and with the lowest recombination frequency (D17Mit76) was used to genotype mice in subsequent analyses, as it likely to be nearest to the site of the Br mutation.
Four additional microsatellites not previously reported were identified and utilized in this study, microsatellite 3 (MS3), 6 (MS6), 25 (MS25), and 34 (MS34). Using the Ensembl Database, novel microsatellites were identified by dinucleotide repeats of 80 basepairs or more. Primers flanking the repeat region were designed following conventional methods. The primers were tested utilizing genomic DNA samples from the inbred parental strains to ensure adequate separation.
DNA Sequencing
Intronic primers were designed to amplify and sequence the two exons of Six2 by PCR from genomic DNA extracted from Br/Br mice and +/+ siblings. In addition, overlapping pairs of primers were designed to amplify and sequence over 1 kb of the Six2 promoter region, including the reported transcription factor binding sites (Brodbeck et al., 2004; Kutejova et al., 2005) and the predicted transcription start sites (Boucher et al., 2000). Overlapping pairs of primers were also designed to amplify and sequence the 1.8 kb intron of Six2. PCR was performed with standard amplification conditions using Amplitaq Gold polymerase (Applied Biosystems). Due to GC-rich sequence at the 5′ end of exon 1, we included 2 M Betaine in amplification and sequencing reactions for this region. After amplification, the DNA fragments were agarose gel purified using GeneClean Spin Kit (Q-Biogene), and the sequencing was performed by the Center for Genomic, Proteomic, and Bioinformatic Research facility at the University of Hawaii at Manoa.
In Situ Hybridization
Embryos at E11.5 derived from reciprocal 3H1 x Balb Br/+ or 3H1 x Cast Br/+ crosses were collected. The yolk sac was dissected free, trypsinized, and DNA extracted for genotyping using D17Mit76 as a PCR-based marker. Only litters containing all three genotypes (+/+, Br/+ and Br/Br) were retained. A total of 28 embryos from 4 litters were subjected to in situ hybridization to detect Six2 and Six3 expression following Belo et al. (1997). The probe for Six2 was provided by Dr. Pin Xu (McLaughlin Research Institute)(Xu et al., 2003), and the probe for Six3 was provided by Dr. Guillermo Oliver (St. Judes Children’s Research Hospital)(Oliver et al., 1995b). The labeled RNA probe was synthesized, using digoxygenin-11-UTP (Roche) and T7 RNA polymerase (Promega). Subsequently, specimens were embedded in paraffin, sectioned at 7 μm, cover-slipped and examined for staining in the developing craniofacial and urogenital regions utilizing a BX-41 Olympus compound microscope.
Real-time Quantitative PCR (qPCR)
A total of 26 embryos (E10.5–11.0) from four litters of embryos derived from reciprocal 3H1 x Balb Br/+ or 3H1 x Cast Br/+ crosses were collected. Each litter contained +/+, Br/+ and Br/Br embryos, as determined by genotyping the yolk sack (as above). Tissues were collected by separating the head from the torso at the level just caudal to the first branchial arch. Each tissue component was placed immediately in 300 μL of RNAlater (Ambion) and stored at 4°C for one to three weeks before processing. Tissues were transferred into TRI Reagent (Molecular Research Center) to extract total RNA. Total RNA was reverse transcribed with oligo dT (University of Hawaii Biotech Core, Honolulu, HI), dNTP mix (Sigma) and MMLV-Reverse Transcriptase (Promega). The resulting cDNA was amplified by using the iQ SYBR Green Supermix reaction procedure with the MyiQ iCycler thermal cycler and single color real-time PCR detection system (Bio-Rad). Primer sets specific for Six2 (F) 5′-GCC TGC GAG CAC CTC CAC AAG AAT-3′, (R) 5′-CAC CGA CTT GCC ACT GCC ATT GAG-3′; Six3 (F) 5′-GCT ATG CTG AAA CTC TGT CC-3′, (R) 5′-GGA TGT TAC TCC TCA AAC GG-3′; eukaryotic elongation factor 1 alpha (EF1α1) (F) 5′-CTG GCA TGG TGG TTA CCT TTG CTC-3′, (R) 5′-GGT AGT CAG AGA AGC TCT CAA CAC-3′; and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (F) 5′-TGC ACC ACC AAC TGC TTA GC-3′, (R) 5′-GGC ATG GAC TGT GGT CAT GAG-3′ were designed with the aid of Primer Designer program (Scientific and Educational Software). The PCR condition consisted of an initial 5 minutes denaturation at 94°C, followed by reactions cycled through denaturation for 15 sec at 94°C, annealing for 20 sec at 60°C, and extension for 40 sec at 72°C. After 50 cycles of amplification, the PCR products were resolved on an agarose gel to confirm that a single band of predicted size was amplified. The expression levels of Six2 and Six3 in each embryo sample was calibrated against housekeeping genes EF1α1 and GAPDH RNA expression with the average relative ratio set to 1, as described previously (Alarcon and Marikawa, 2004; Marikawa et al., 2004). The amounts of Six2 and Six3 mRNA from the heterozygous and homozygous mutant embryos were compared with the normalized amounts from the wild-type embryos.
Western Blotting
Embryonic kidneys and whole heads were collected from embryos at E13.5 and immediately frozen, while additional tissue from each embryo was collected for genomic DNA extraction for genotyping. Proteins were extracted by incubating the two kidneys from each embryo in 25 microliters of RIPA buffer (10 mM Tris, pH 7.2, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% Triton X-100, 1% Deoxycholate) with protease inhibitors for 20 min on ice, then adding 25 μl of 2X Laemmli buffer and boiling for 5 minutes. The whole heads were homogenized in 1.5 ml of RIPA buffer and incubated on ice for 30 min. Lysates were centrifuged (20,000 × g) for 20 min at 4° C and the soluble proteins were collected. The concentration of proteins was measured using the Bradford reagent (Biorad). For Western blots, 10 μl of kidney extracts and 10 μg of whole head lysates from 3 embryos each of Br/Br, Br/+, and +/+ were loaded in each lane of a 10% SDS- PAGE gel. Immunoblotting was performed following standard procedures using anti- Six2 primary antibody (CeMines), anti-β-actin primary antibody (Abcam) as a loading control, and anti-rabbit secondary antibody conjugated to HRP (Jackson Immunoresearch). Detection of bound antibodies was accomplished using ECL reagents (Pierce). Blot films were scanned with a flatbed scanner and band intensities were quantified with Kodak 1D Image Analysis Software (Eastman Kodak Company).
Immunofluorescent microscopy
Whole embryos were collected after timed matings at E11.5 and E14.5 and snap frozen. Cryosections of the craniofacial and kidney regions were cut at 10 μm and fixed with methanol for 10 min at −20° C. After blocking with 5% normal goat serum (Jackson ImmunoResearch), sections were incubated with rabbit polyclonal anti-Six2 antibodies (Proteintech Group, Inc.) diluted in 5% normal goat serum with 0.1% triton X-100. After washing away unbound antibody, sections were incubated with goat anti-rabbit antibodies labeled with Alexa 488 (Invitrogen), counterstained with propidium iodide, and mounted in 50% glycerol in PBS. Images were taken on an Olympus BX41 fluorescent microscope.
Acknowledgments
Grant sponsor: NIH/NIDDK; Grant number: R01 DK064752-02 (SL).
Supported by R01 DK064752-02 (SL), INBRE student fellowship P20 RR16467 (AAS), and a Meiji Studentship Award (LM). B.M.-O was generously supported by NIH grants AR36819 and T32 DE017544 (to B.R. Olsen).
References
- Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15:157–164. doi: 10.1038/ng0297-157. [DOI] [PubMed] [Google Scholar]
- Alarcon VB, Marikawa Y. Molecular study of mouse peri-implantation development using the in vitro culture of aggregated inner cell mass. Mol Reprod Dev. 2004;67:83–90. doi: 10.1002/mrd.20015. [DOI] [PubMed] [Google Scholar]
- Apesos J, Anigian GM. Median cleft of the lip: its significance and surgical repair. Cleft Palate Craniofac J. 1993;30:94–96. doi: 10.1597/1545-1569_1993_030_0094_mcotli_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Bakken AF, Aabyholm G. Frontonasal dysplasia. Possible hereditary connection with other congenital defects. Clin Genet. 1976;10:214–217. doi: 10.1111/j.1399-0004.1976.tb00036.x. [DOI] [PubMed] [Google Scholar]
- Beechey C, Boyd Y, Searle AG. Brachyrrhine, Br, a mouse craniofacial mutant maps to distal mouse chromosome 17 and is a candidate for midline cleft syndrome. Mouse Genome. 1997;95:692–694. [Google Scholar]
- Bien-Willner GA, Stankiewicz P, Lupski JR. SOX9cre1, a cis-acting regulatory element located 1.1 Mb upstream of SOX9, mediates its enhancement through the SHH pathway. Hum Mol Genet. 2007;16:1143–1156. doi: 10.1093/hmg/ddm061. [DOI] [PubMed] [Google Scholar]
- Boucher CA, Winchester CL, Hamilton GM, Winter AD, Johnson KJ, Bailey ME. Structure, mapping and expression of the human gene encoding the homeodomain protein, SIX2. Gene. 2000;247:145–151. doi: 10.1016/s0378-1119(00)00105-0. [DOI] [PubMed] [Google Scholar]
- Brenner BM, Mackenzie HS. Nephron mass as a risk factor for progression of renal disease. Kidney Int Suppl. 1997;63:S124–127. [PubMed] [Google Scholar]
- Brodbeck S, Besenbeck B, Englert C. The transcription factor Six2 activates expression of the Gdnf gene as well as its own promoter. Mech Dev. 2004;121:1211–1222. doi: 10.1016/j.mod.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Brodbeck S, Englert C. Genetic determination of nephrogenesis: the Pax/Eya/Six gene network. Pediatr Nephrol. 2004;19:249–255. doi: 10.1007/s00467-003-1374-z. [DOI] [PubMed] [Google Scholar]
- Brophy PD, Ostrom L, Lang KM, Dressler GR. Regulation of ureteric bud outgrowth by Pax2-dependent activation of the glial derived neurotrophic factor gene. Development. 2001;128:4747–4756. doi: 10.1242/dev.128.23.4747. [DOI] [PubMed] [Google Scholar]
- Buller C, Xu X, Marquis V, Schwanke R, Xu PX. Molecular effects of Eya1 domain mutations causing organ defects in BOR syndrome. Hum Mol Genet. 2001;10:2775–2781. doi: 10.1093/hmg/10.24.2775. [DOI] [PubMed] [Google Scholar]
- Carvajal JJ, Cox D, Summerbell D, Rigby PW. A BAC transgenic analysis of the Mrf4/Myf5 locus reveals interdigitated elements that control activation and maintenance of gene expression during muscle development. Development. 2001;128:1857–1868. doi: 10.1242/dev.128.10.1857. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Jr, Sedano HO, Gorlin RJ, Jirasek JE. Frontonasal dysplasia (median cleft face syndrome): comments on etiology and pathogenesis. Birth Defects Orig Artic Ser. 1971;7:117–119. [PubMed] [Google Scholar]
- De Moor MM, Baruch R, Human DG. Frontonasal dysplasia associated with tetralogy of Fallot. J Med Genet. 1987;24:107–109. doi: 10.1136/jmg.24.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal KK, Cantrell VA, Chandler RL, Saunders TL, Mortlock DP, Southard-Smith EM. Distant regulatory elements in a Sox10-beta GEO BAC transgene are required for expression of Sox10 in the enteric nervous system and other neural crest-derived tissues. Dev Dyn. 2006;235:1413–1432. doi: 10.1002/dvdy.20769. [DOI] [PubMed] [Google Scholar]
- DeMyer W. The median cleft face syndrome. Differential diagnosis of cranium bifidum occultum, hypertelorism, and median cleft nose, lip, and palate. Neurology. 1967;17:961–971. doi: 10.1212/wnl.17.10.961. [DOI] [PubMed] [Google Scholar]
- DiLeone RJ, Marcus GA, Johnson MD, Kingsley DM. Efficient studies of long-distance Bmp5 gene regulation using bacterial artificial chromosomes. Proc Natl Acad Sci U S A. 2000;97:1612–1617. doi: 10.1073/pnas.97.4.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas-Denton RN, McNamara BJ, Hoy WE, Hughson MD, Bertram JF. Does nephron number matter in the development of kidney disease? Ethn Dis. 2006;16:S2–40–45. [PubMed] [Google Scholar]
- Dressler GR. The cellular basis of kidney development. Annu Rev Cell Dev Biol. 2006;22:509–529. doi: 10.1146/annurev.cellbio.22.010305.104340. [DOI] [PubMed] [Google Scholar]
- Epstein CJ, Erickson RP, Wynshaw-Boris A. Inborn Errors of Development. New York: Oxford University Press; 2004. [Google Scholar]
- Epstein DJ, McMahon AP, Joyner AL. Regionalization of Sonic hedgehog transcription along the anteroposterior axis of the mouse central nervous system is regulated by Hnf3-dependent and -independent mechanisms. Development. 1999;126:281–292. doi: 10.1242/dev.126.2.281. [DOI] [PubMed] [Google Scholar]
- Fisher E, Scambler P. Human haploinsufficiency--one for sorrow, two for joy. Nat Genet. 1994;7:5–7. doi: 10.1038/ng0594-5. [DOI] [PubMed] [Google Scholar]
- Ford HL, Landesman-Bollag E, Dacwag CS, Stukenberg PT, Pardee AB, Seldin DC. Cell cycle-regulated phosphorylation of the human SIX1 homeodomain protein. J Biol Chem. 2000;275:22245–22254. doi: 10.1074/jbc.M002446200. [DOI] [PubMed] [Google Scholar]
- Fragoso R, Cid-Garcia A, Hernandez A, Nazara Z, Cantu JM. Frontonasal dysplasia in the Klippel-Feil syndrome: a new associated malformation. Clin Genet. 1982;22:270–273. doi: 10.1111/j.1399-0004.1982.tb01444.x. [DOI] [PubMed] [Google Scholar]
- Fryburg JS, Persing JA, Lin KY. Frontonasal dysplasia in two successive generations. Am J Med Genet. 1993;46:712–714. doi: 10.1002/ajmg.1320460623. [DOI] [PubMed] [Google Scholar]
- Glassberg KI, Stephens FD, Lebowitz RL, Braren V, Duckett JW, Jacobs EC, King LR, Perlmutter AD. Renal dysgenesis and cystic disease of the kidney: a report of the Committee on Terminology, Nomenclature and Classification, Section on Urology, American Academy of Pediatrics. J Urol. 1987;138:1085–1092. doi: 10.1016/s0022-5347(17)43510-5. [DOI] [PubMed] [Google Scholar]
- Gong KQ, Yallowitz AR, Sun H, Dressler GR, Wellik DM. A Hox-Eya-Pax complex regulates early kidney developmental gene expression. Mol Cell Biol. 2007;27:7661–7668. doi: 10.1128/MCB.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Cohen MM, Jr, Hennekam RCM. Syndromes of the head and neck. New York: Oxford University Press; 2001. [Google Scholar]
- Guion-Almeida ML, Richieri-Costa A, Saavedra D, Cohen MM., Jr Frontonasal dysplasia: analysis of 21 cases and literature review. Int J Oral Maxillofac Surg. 1996;25:91–97. doi: 10.1016/s0901-5027(96)80048-8. [DOI] [PubMed] [Google Scholar]
- Hadchouel J, Tajbakhsh S, Primig M, Chang TH, Daubas P, Rocancourt D, Buckingham M. Modular long-range regulation of Myf5 reveals unexpected heterogeneity between skeletal muscles in the mouse embryo. Development. 2000;127:4455–4467. doi: 10.1242/dev.127.20.4455. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Functional gene cassettes in development. Proc Natl Acad Sci U S A. 1993;90:8305–8307. doi: 10.1073/pnas.90.18.8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong Y, El-Jaick K, Roessler E, Muenke M, Epstein DJ. A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers. Development. 2006;133:761–772. doi: 10.1242/dev.02239. [DOI] [PubMed] [Google Scholar]
- Kawakami K, Sato S, Ozaki H, Ikeda K. Six family genes--structure and function as transcription factors and their roles in development. Bioessays. 2000;22:616–626. doi: 10.1002/1521-1878(200007)22:7<616::AID-BIES4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Kemper MJ, Muller-Wiefel DE. Renal function in congenital anomalies of the kidney and urinary tract. Curr Opin Urol. 2001;11:571–575. doi: 10.1097/00042307-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, Seawright A, Schedl A, Quinlan RA, Danes S, van Heyningen V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum Mol Genet. 2001;10:2049–2059. doi: 10.1093/hmg/10.19.2049. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kimberling WJ, Weston MD, Schaefer BG, Berg MA, Marres HA, Cremers CW. Identification of three novel mutations in human EYA1 protein associated with branchio-oto-renal syndrome. Hum Mutat. 1998;11:443–449. doi: 10.1002/(SICI)1098-1004(1998)11:6<443::AID-HUMU4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Kutejova E, Engist B, Mallo M, Kanzler B, Bobola N. Hoxa2 downregulates Six2 in the neural crest-derived mesenchyme. Development. 2005;132:469–478. doi: 10.1242/dev.01536. [DOI] [PubMed] [Google Scholar]
- Kwee ML, Lindhout D. Frontonasal dysplasia, coronal craniosynostosis, pre- and postaxial polydactyly and split nails: a new autosomal dominant mutant with reduced penetrance and variable expression? Clin Genet. 1983;24:200–205. doi: 10.1111/j.1399-0004.1983.tb02240.x. [DOI] [PubMed] [Google Scholar]
- Lehmann-Nitsche R. Ein seltener Fall von angeborener medianer Spaltung der oberen Gesichtschalfte. Virchow Arch. 1901;163:26–134. [Google Scholar]
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- Lozanoff S. Midfacial retrusion in adult brachyrrhine mice. Acta Anat (Basel) 1993;147:125–132. doi: 10.1159/000147492. [DOI] [PubMed] [Google Scholar]
- Lozanoff S. Sphenoethmoidal growth, malgrowth and midfacial profile. In: Chaplain MAJ, Singh GD, McLachlan J, editors. On growth and form: spatio-temporal patterning in biology. New York: John Wiley and Sons, Inc; 1999. pp. 357–372. [Google Scholar]
- Lozanoff S, Johnston J, Ma W, Jourdan-Le Saux C. Immunohistochemical localization of Pax2 and associated proteins in the developing kidney of mice with renal hypoplasia. J Histochem Cytochem. 2001;49:1081–1097. doi: 10.1177/002215540104900903. [DOI] [PubMed] [Google Scholar]
- Lozanoff S, Jureczek S, Feng T, Padwal R. Anterior cranial base morphology in mice with midfacial retrusion. Cleft Palate Craniofac J. 1994;31:417–428. doi: 10.1597/1545-1569_1994_031_0417_acbmim_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Ma W, Lozanoff S. External craniofacial features, body size, and renal morphology in prenatal brachyrrhine mice. Teratology. 1993;47:321–332. doi: 10.1002/tera.1420470409. [DOI] [PubMed] [Google Scholar]
- Ma W, Lozanoff S. Morphological deficiency in the prenatal anterior cranial base of midfacially retrognathic mice. J Anat. 1996;188 (Pt 3):547–555. [PMC free article] [PubMed] [Google Scholar]
- Ma W, Lozanoff S. Spatial and temporal distribution of cellular proliferation in the cranial base of normal and midfacially retrusive mice. Clin Anat. 1999;12:315–325. doi: 10.1002/(SICI)1098-2353(1999)12:5<315::AID-CA2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Ma W, Lozanoff S. Differential in vitro response to epidermal growth factor by prenatal murine cranial-base chondrocytes. Arch Oral Biol. 2002;47:155–163. doi: 10.1016/s0003-9969(01)00105-4. [DOI] [PubMed] [Google Scholar]
- Marcucio RS, Cordero DR, Hu D, Helms JA. Molecular interactions coordinating the development of the forebrain and face. Dev Biol. 2005;284:48–61. doi: 10.1016/j.ydbio.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Marikawa Y, Fujita TC, Alarcon VB. An enhancer-trap LacZ transgene reveals a distinct expression pattern of Kinesin family 26B in mouse embryos. Dev Genes Evol. 2004;214:64–71. doi: 10.1007/s00427-003-0377-x. [DOI] [PubMed] [Google Scholar]
- McBratney BM, Margaryan E, Ma W, Urban Z, Lozanoff S. Frontonasal dysplasia in 3H1 Br/Br mice. Anat Rec A Discov Mol Cell Evol Biol. 2003;271:291–302. doi: 10.1002/ar.a.10034. [DOI] [PubMed] [Google Scholar]
- Meinecke P, Blunck W. Frontonasal dysplasia, congenital heart defect, and short stature: a further observation. J Med Genet. 1989;26:408–409. doi: 10.1136/jmg.26.6.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AW, Schedl A, McInnes L, Doyle M, Hecksher-Sorensen J, Hastie ND. YAC transgenic analysis reveals Wilms’ tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech Dev. 1998;79:169–184. doi: 10.1016/s0925-4773(98)00188-9. [DOI] [PubMed] [Google Scholar]
- Moreno Fuenmayor H. The spectrum of frontonasal dysplasia in an inbred pedigree. Clin Genet. 1980;17:137–142. doi: 10.1111/j.1399-0004.1980.tb00122.x. [DOI] [PubMed] [Google Scholar]
- Mortlock DP, Guenther C, Kingsley DM. A general approach for identifying distant regulatory elements applied to the Gdf6 gene. Genome Res. 2003;13:2069–2081. doi: 10.1101/gr.1306003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevin NC, Leonard AG, Jones B. Frontonasal dysostosis in two successive generations. Am J Med Genet. 1999;87:251–253. doi: 10.1002/(sici)1096-8628(19991126)87:3<251::aid-ajmg10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302:413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- Ohto H, Takizawa T, Saito T, Kobayashi M, Ikeda K, Kawakami K. Tissue and developmental distribution of Six family gene products. Int J Dev Biol. 1998;42:141–148. [PubMed] [Google Scholar]
- Oliver G, Mailhos A, Wehr R, Copeland NG, Jenkins NA, Gruss P. Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development. 1995a;121:4045–4055. doi: 10.1242/dev.121.12.4045. [DOI] [PubMed] [Google Scholar]
- Oliver G, Wehr R, Jenkins NA, Copeland NG, Cheyette BN, Hartenstein V, Zipursky SL, Gruss P. Homeobox genes and connective tissue patterning. Development. 1995b;121:693–705. doi: 10.1242/dev.121.3.693. [DOI] [PubMed] [Google Scholar]
- Ozaki H, Nakamura K, Funahashi J, Ikeda K, Yamada G, Tokano H, Okamura HO, Kitamura K, Muto S, Kotaki H, Sudo K, Horai R, Iwakura Y, Kawakami K. Six1 controls patterning of the mouse otic vesicle. Development. 2004;131:551–562. doi: 10.1242/dev.00943. [DOI] [PubMed] [Google Scholar]
- Pope JCt, Brock JW, 3rd, Adams MC, Stephens FD, Ichikawa I. How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J Am Soc Nephrol. 1999;10:2018–2028. doi: 10.1681/ASN.V1092018. [DOI] [PubMed] [Google Scholar]
- Purcell P, Oliver G, Mardon G, Donner AL, Maas RL. Pax6-dependence of Six3, Eya1 and Dach1 expression during lens and nasal placode induction. Gene Expr Patterns. 2005;6:110–118. doi: 10.1016/j.modgep.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Reich EW, Cox RP, McCarthy JG, Becker MH, Genieser NB, Converse J. A new heritable syndrome with frontonasal dysplasia and associated extracranial anomalies. Birth Defects. 1977;88 [Google Scholar]
- Ribes D, Fischer E, Calmont A, Rossert J. Transcriptional control of epithelial differentiation during kidney development. J Am Soc Nephrol. 2003;14(Suppl 1):S9–15. doi: 10.1097/01.asn.0000067647.05964.9f. [DOI] [PubMed] [Google Scholar]
- Roessler E, Ward DE, Gaudenz K, Belloni E, Scherer SW, Donnai D, Siegel-Bartelt J, Tsui LC, Muenke M. Cytogenetic rearrangements involving the loss of the Sonic Hedgehog gene at 7q36 cause holoprosencephaly. Hum Genet. 1997;100:172–181. doi: 10.1007/s004390050486. [DOI] [PubMed] [Google Scholar]
- Roizenblatt J, Wajntal A, Diament AJ. Median cleft face syndrome or frontonasal dysplasia: a case report with associated kidney malformation. J Pediatr Ophthalmol Strabismus. 1979;16:16–20. doi: 10.3928/0191-3913-19790101-05. [DOI] [PubMed] [Google Scholar]
- Sagai T, Hosoya M, Mizushina Y, Tamura M, Shiroishi T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 2005;132:797–803. doi: 10.1242/dev.01613. [DOI] [PubMed] [Google Scholar]
- Schlosser G. Induction and specification of cranial placodes. Dev Biol. 2006 doi: 10.1016/j.ydbio.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Sedano HO, Cohen MM, Jr, Jirasek J, Gorlin RJ. Frontonasal dysplasia. J Pediatr. 1970;76:906–913. doi: 10.1016/s0022-3476(70)80374-2. [DOI] [PubMed] [Google Scholar]
- Seidman JG, Seidman C. Transcription factor haploinsufficiency: when half a loaf is not enough. J Clin Invest. 2002;109:451–455. doi: 10.1172/JCI15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. Embo J. 2006;25:5214–5228. doi: 10.1038/sj.emboj.7601381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh GD, Johnston J, Ma W, Lozanoff S. Cleft palate formation in fetal Br mice with midfacial retrusion: tenascin, fibronectin, laminin, and type IV collagen immunolocalization. Cleft Palate Craniofac J. 1998;35:65–76. doi: 10.1597/1545-1569_1998_035_0065_cpfifb_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Slaney SF, Goodman FR, Eilers-Walsman BL, Hall BD, Williams DK, Young ID, Hayward RD, Jones BM, Christianson AL, Winter RM. Acromelic frontonasal dysostosis. Am J Med Genet. 1999;83:109–116. [PubMed] [Google Scholar]
- Temple IK, Brunner H, Jones B, Burn J, Baraitser M. Midline facial defects with ocular colobomata. Am J Med Genet. 1990;37:23–27. doi: 10.1002/ajmg.1320370107. [DOI] [PubMed] [Google Scholar]
- Terzi F, Burtin M, Friedlander G. Using transgenic mice to analyze the mechanisms of progression of chronic renal failure. J Am Soc Nephrol. 2000;11(Suppl 16):S144–148. [PubMed] [Google Scholar]
- Theiler K. The house mouse: atlas of embryonic development. New York: Springer-Verlag; 1989. [Google Scholar]
- Verloes A, Gillerot Y, Walczak E, Van Maldergem L, Koulischer L. Acromelic frontonasal “dysplasia”: further delineation of a subtype with brain malformation and polydactyly (Toriello syndrome) Am J Med Genet. 1992;42:180–183. doi: 10.1002/ajmg.1320420209. [DOI] [PubMed] [Google Scholar]
- Warkany J, Bofinger MK, Benton C. Median facial cleft syndrome in half-sisters. Dilemmas in genetic counseling. Teratology. 1973;8:273–285. doi: 10.1002/tera.1420080307. [DOI] [PubMed] [Google Scholar]
- Wunderle VM, Critcher R, Hastie N, Goodfellow PN, Schedl A. Deletion of long-range regulatory elements upstream of SOX9 causes campomelic dysplasia. Proc Natl Acad Sci U S A. 1998;95:10649–10654. doi: 10.1073/pnas.95.18.10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23:113–117. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003;130:3085–3094. doi: 10.1242/dev.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou D, Silvius D, Fritzsch B, Xu PX. Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development. 2004;131:5561–5572. doi: 10.1242/dev.01437. [DOI] [PMC free article] [PubMed] [Google Scholar]