

Abstract
The NADPH oxidase (Nox) proteins catalyze the regulated formation of reactive oxygen species (ROS) which play key roles as signaling molecules in several physiological and pathophysiological processes. ROS generation by the Nox1 member of the Nox family is necessary for the formation of extracellular matrix (ECM)-degrading, actin-rich cellular structures known as invadopodia. Selective inhibition of Nox isoforms can provide reversible, mechanistic insights into these cellular processes in contrast to scavenging or inhibition of ROS production. Currently no specific Nox inhibitors have been described. Here, by high-throughput screening, we identify a sub-set of phenothiazines, 2-acetylphenothiazine (here referred to as ML171) (and its related 2-(trifluoromethyl)-phenothiazine) as nanomolar, cell-active and specific Nox1 inhibitors that potently block Nox1-dependent ROS generation, with only marginal activity on other cellular ROS-producing enzymes and receptors including the other Nox isoforms. ML171 also blocks the ROS-dependent formation of ECM-degrading invadopodia in colon cancer cells. Such effects can be reversed by overexpression of Nox1 protein, which is suggestive of a selective mechanism of inhibition of Nox1 by this compound. These results elucidate the relevance of Nox1-dependent ROS generation in mechanisms of cancer invasion, and define ML171 as a useful Nox1 chemical probe and a potential therapeutic agent for inhibition of cancer cell invasion.
Selective chemical inhibition provides a powerful tool for dissecting complex physiological functions mediated by structurally-similar cellular enzymes. In particular, selective inhibition of NADPH oxidase (Nox) family members has the distinct advantage of enabling acute, reversible modulation of molecular function, while circumventing the developmental compensations that can arise in gene deletion studies. We have targeted this approach to elucidate the biological functions mediated by the Nox1 member of the NADPH oxidase family and have identified a novel nanomolar small-molecule Nox1 inhibitor. Importantly, we demonstrate that this chemical probe can be used to clarify the role of Nox1-dependent ROS generation in the pathogenesis of colon cancer.
The NADPH oxidase family, consisting of the homologous enzymes Nox1-4 and the more distantly related Nox5, Duox1 and Duox2, catalyzes the regulated formation of reactive oxygen species (ROS) (1). Among all seven Nox isoforms, the Nox1-4 enzymes share the highest level of structural similarities (2). Their basic catalytic subunit contains a C-terminal dehydrogenase domain featuring a binding site for NADPH and a bound flavin adenine nucleotide (FAD), as well as an N-terminal domain consisting of six transmembrane alpha helices that bind two heme groups. On activation, cytosolic NADPH transfers its electrons to the FAD, which in turn passes electrons sequentially to the two hemes and ultimately to molecular oxygen on the opposing side of the membrane, to form the superoxide anion (3). Although all Nox1-4 isoforms catalyze the reduction of molecular oxygen and are expressed in a complex with p22phox subunit, they differ in both tissue distributions and mechanisms by which their activity is regulated (4). Nox2 is expressed by phagocytic leukocytes and its activity is triggered by inflammatory mediators which induce the assembly of four cytosolic regulatory proteins (p40phox, p47phox, p67phox and Rac2-GTPase) with the Nox2 core enzyme to stimulate superoxide formation. Nox1 and Nox3 are highly expressed in the colon epithelium and in the inner ear respectively and their activity is also regulated by Rac1-GTPase and by related cytosolic adaptors, known as the activator subunit NoxA1 (homologous to p67phox) and the organizer subunits NoxO1, Tks4 and Tks5 (homologues of p47phox). Nox4 is widely distributed in the kidney, bone and vascular cells and its activity is independent of Rac-GTPase.
All Nox enzymes have been implicated in physiological and pathophysiological processes (5). Particularly, Nox1-dependent ROS generation has been shown to play a pivotal role in cell signaling, cell growth, angiogenesis, motility and blood pressure regulation (6–8). Interestingly, ROS generated via Nox1 have been reported to contribute to a growing number of diseases, including cancer, atherosclerosis, hypertension, neurological disorders and inflammation (9–12). In keeping with a role of Nox1 in colon cancer, we have recently shown that in human colon cancer cells Nox1-derived ROS are necessary for the formation of extracellular matrix (ECM)-degrading, actin-rich cellular structures known as invadopodia (13), whose presence directly correlate with the ability of cells to invade the surrounding tissues (14, 15).
Since it has been reported that ROS can also be produced by other cellular enzymes such as xanthine oxidase (XO), cytochrome P450, and mitochondrial oxidases (16–18), dissecting of the contribution to these pathologies of Nox1-derived ROS in comparison to other ROS generators has been complicated by the lack of potent, selective and specific Nox1 inhibitors. Currently, only a few non-specific inhibitors (that also target other Nox isoforms) have been identified (i.e diphenylene iodonium [DPI] and apocynin) (19). Several issues of selectivity, specificity, potency and toxicity limit the value of these compounds as Nox1 inhibitors both as research and clinical tools (20–22). In the case of the most widely used Nox inhibitor DPI, because of its chemical mechanism of inhibition which involves accepting an electron from flavin, followed by covalently reacting with the enzymes or its prosthetic group, DPI rapidly and irreversibly blocks not only all Nox isoforms but also many other flavin-dependent enzymes such as XO (see also Table 1b).
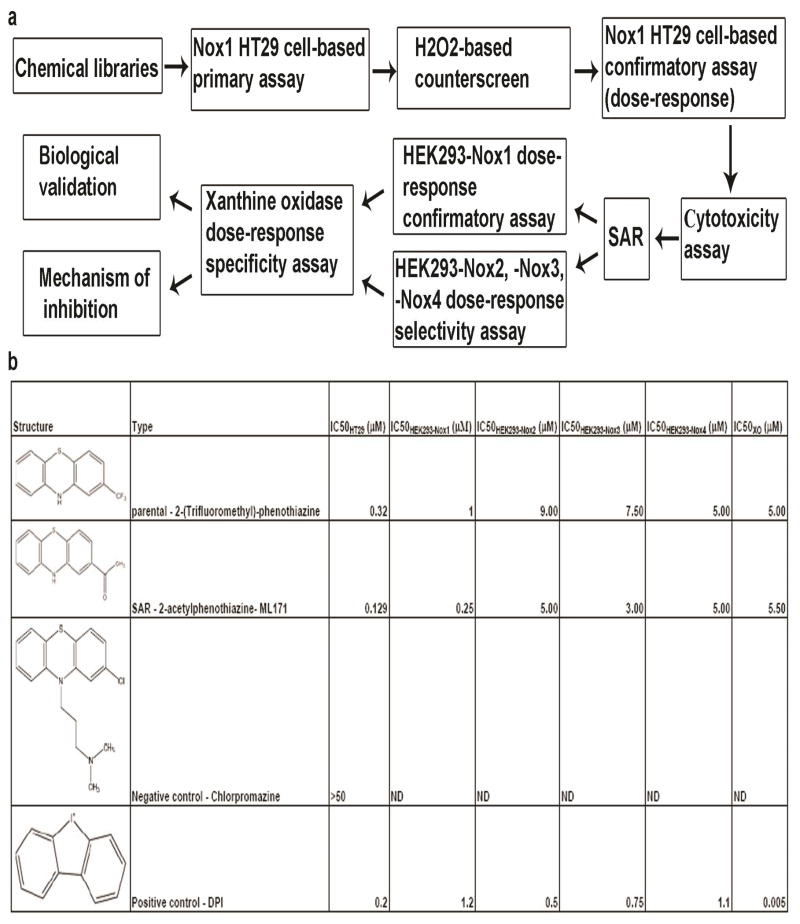
Table 1.
Schematic representation of the screening cascade and table summarizing IC50 values of 2-(trifluoromethyl)-phenothiazine (parental hit), ML171 (after-SAR hit), chlorpromazione (negative control) and DPI (positive control) are reported. (a) A schematic representation of screening cascade for the identification of novel Nox1 inhibitor is illustrated. Compounds which efficiently blocked light emission in our primary assay were counterscreened via i) H2O2-based counterscreen and ii) cytotoxicity assay. Hits were validated in different cell systems (HEK293-Nox1, -Nox2, -Nox3 and -Nox4) for their ability to selectively block Nox1-dependent ROS generation. Selective hits were tested for their ability to non-specifically interfere with XO-dependent ROS formation. Remaining hits were further validated as Nox1 inhibitors in biological systems and their mechanism of action was investigated. (b) IC50 values of 2-(trifluoromethyl)-phenothiazine (parental hit), ML171 (after-SAR hit), chlorpromazine (negative control) and DPI (positive control) are reported from our primary assay (IC50HT29), HEK293-Nox1 confirmatory assay (IC50HEK293-Nox1), HEK293-Nox2, -Nox3, -Nox4 selectivity assay (IC50HEK293-Nox2, IC50HEK293-Nox3 and IC50HEK293-Nox4) and Xanthine oxidase specificity assay (IC50XO).
 |
Here, by using high-throughput screening, we identify a sub-set of phenothiazines, specifically 2-acetylphenothiazine (here referred to as ML171) (and its related 2-(trifluoromethyl)-phenothiazine) as nanomolar, cell-active and specific Nox1 inhibitors that potently block Nox1-dependent ROS generation, with only marginal activity on other cellular ROS-producing enzymes and receptors including the other Nox isoforms. ML171 also blocks the ROS-dependent formation of ECM-degrading invadopodia in colon cancer cells. Such effects can be reversed by over-expression of Nox1 protein, which is possibly suggestive of a selective mechanism of inhibition of Nox1 by this compound. These results elucidate the relevance of Nox1-dependent ROS generation in mechanisms of cancer invasion, and define ML171 as a useful Nox1 chemical probe with potential therapeutic insights.
Results and Discussion
A cell-based high-throughput screening to identify novel small-molecule inhibitor of Nox1
To identify novel specific and selective Nox1 small molecule inhibitors, we developed a cell-based high-throughput screen using a luminol-based chemiluminescence (CL) assay. Luminol has been extensively used as a CL probe to study ROS generation from cell lines and tissues, and its use for this purpose is well-validated (23, 24). The cell line chosen for this screen was human HT29 colon cancer cells, as we had previously shown that these cells endogenously expressed Nox1 as the only members of the Nox family and its cytosolic regulators NoxA1 and NoxO1 (25). In addition, Nox1 was one of the main contributors to ROS generation. A chemical library of 16,000 compounds (Maybridge Hitfinder ®) was screened and 2-(trifluoromethyl)-phenothiazine was among the hits showing the highest potency in blocking ROS generation in our primary assay (Z′=0.48+/−0.08, S:B=18.5+/−4.4, see Supplementary information, Table S1 online). This hit was then validated through a series of counterscreen, secondary and confirmatory screens as illustrated in our screening cascade in Table 1a. Importantly, concentration-response analysis for 2-(trifluoromethyl)-phenothiazine in our confirmatory assay verified its high potency in blocking ROS production in HT29 cells (IC50HT29 = 0.32 μM, see Table 1b). We ruled out the possibility that this compound would act as a false positive or H2O2 scavenger thus reacting non-specifically with H2O2 in our primary assay by performing a cell-free H2O2-based counterscreen. Moreover, 2-(trifluoromethyl)- phenothiazine did not cause cell death when tested at 10 μM in HT29 cells by means of the commonly-used CellTiter Glo toxicity assay (Promega). Further, we tested the ability of 2-(trifluoromethyl)-phenothiazine to selectively block Nox1-dependent ROS generation using the HEK293-Nox cell system reconstituted with protein components required for ROS generation by each individual Nox isoform. As summarized in Table 1b, this molecule blocked Nox1-dependent ROS generation in the HEK293-Nox1 reconstituted system (IC50Nox1 = 1 μM) but had only a marginal activity on the other structurally-related Nox isoforms.
Selectivity was confirmed by measuring the ability of 2-(trifluoromethyl)-phenothiazine to block the activity of other flavoproteins involved in generation of cellular ROS such as XO. This enzyme catalyzes the conversion of xanthine to uric acid with the generation of ROS as a by-product (16). Interestingly, we found that the IC50 of 2-(trifluoromethyl)-phenothiazine towards XO was dramatically higher than the IC50 of DPI towards XO (IC50XO = 5 μM vs IC50XO = 0.005 μM, see Table 1b) indicating that this phenothiazine only slightly inhibits XO-dependent ROS generation compared to DPI. The statistics and additional experimental details about all the assays employed in our screening cascade are reported in Supplementary information, Table S1 online.
Characterization of a sub-set of phenothiazines as potent, specific and selective Nox1 inhibitors
2-(Trifluoromethyl)-phenothiazine belongs to the class of phenothiazines, a scaffold found in various antipsychotic drugs (26), such as chlorpromazine, promazine, and trifluoperazine, inter alia. With the intent of identifying related phenothiazines with higher potency in blocking Nox1-dependent ROS generation, we performed Structure-Activity Relationship (SAR) analysis on several commercially available phenothiazines, including those used as anti-psychotic drugs. These molecules were tested in HT29 cells by luminol-based CL assay for their ability to block ROS production. As summarized in Table 1, we found that chlorpromazine did not inhibit ROS generation in our assay (IC50 > 50 μM). Consistent with this, as indicated in Table 2, we observed that other phenothiazines (i.e. trifluperazine, perphenazine, etc.) used as anti-psychotic drugs were additionally unable to block Nox1-dependent ROS generation. In general, all phenothiazines tested in Table 2 with a substituent on the ring nitrogen were devoid of Nox1 activity. In contrast, several related N-unsubstituted phenothiazines with a substituent para to the ring sulfur were identified which were active in blocking ROS generation. We selected the molecule with the highest potency (2-acetylphenothiazine) and used it for further analyses. In this work we refer to this molecule as ML171, because it is chemical probe #171 from the Molecular Libraries Initiative of the National Institutes of Health. The dose-response analysis in Fig. 1a shows that ML171 strongly blocked ROS generation in HT29 cells (IC50HT29 = 0.129 μM). Moreover, we validated ML171 as an effective inhibitor of ROS generation using a second method of ROS detection. Supplementary Figure S1a shows that ML171 efficiently blocks ROS production measured by carboxy-H2-DCFDA staining as well as DPI used as a positive control. Interestingly, when ML171 was tested in HEK293-Nox1 reconstituted cell system, we observed higher potency in blocking Nox1-dependent ROS generation compared with the parental compound as summarized in Table 1b. Of note, the IC50 values towards Nox2 and Nox3 were also slightly decreased, while this compound was still unable to inhibit XO-dependent ROS generation.
Table 2.
Chemical structures and IC50 values (μM) of all phenothiazines tested in the SAR study are reported. The phenothiazines active in our HT29 cell-based assay are listed according to their decreasing IC50 values starting with our parental compound 2-(trifluoromethyl)-phenothiazine. The phenothiazines unable to block ROS generation in our HT29 cell-based assay are also listed. Among these, several anti-psychotic drugs are also reported.
| Corp ID SID Common Name |
Micromolar IC50 HT29 Assay |
|
|---|---|---|
| SR-01000441858-2 26540425 JFD 03930 |
0.32 | 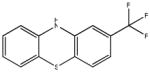 |
| SR-01000597201-3 SID 57287864 (ML171) |
0.129 (AID 2808) | 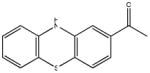 |
| SR-03000000698-1 57287906 |
0.177 (AID 2808) | 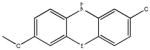 |
| SR-03000000756-1 57287964 |
0.24 (AID 2808) | 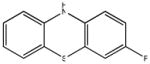 |
| SR-03000000755-1 57287963 |
0.24 (AID 2808) | 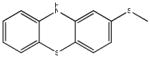 |
| SR-01000596948-2 7287863 |
0.25 | 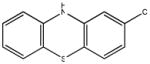 |
| SR-03000000760-1 57287968 |
0.36 (AID 2808) | 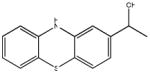 |
| SR-01000721844-3 57287888 |
0.37 (AID 2808) | 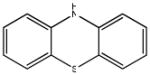 |
| SR-03000000753-1 57287961 |
0.91 (AID 2808) | 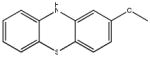 |
| SR-01000477673-3 57287845 |
0.92 (AID 2808) | |
| SR-01000025134-3 57287814 |
0.94 (AID 2808) |  |
| SR-03000000695-1 57287903 |
1.4 (AID2808) | 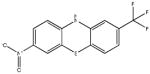 |
| SR-03000000707-1 57287915 |
5.3 (AID 2808) |  |
| SR-01000003048-5 57287813 Fluorophenazine |
Inactive (AID 2808) |  |
| SR-01000003020-6 57287812 |
Inactive (AID 2808) | 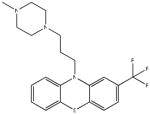 |
| SR-01000000228-4 57288080 promazine |
Inactive (AID 2808) | 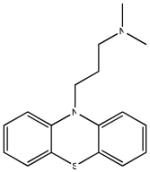 |
| SR-01000000224-4 57288079 Triflupromazine |
Inactive (AID 2808) | 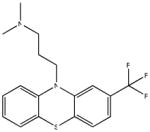 |
| SR-01000000137-4 57287811 |
Inactive (AID 2808) | 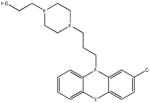 |
| SR-01000000012-5 57287810 chlorpromazine |
Inactive (AID 2808) |  |
Figure 1.

ML171 potently blocks Nox1-dependent ROS generation and only increasing over-expression of Nox1 can overcome the blockage of ROS generation caused by ML171 treatment in HEK293 cell system reconstituted with all the components required Nox1-dependent ROS generation. (a) ML171 treatment causes a concentration-dependent inhibition of ROS generation in HT29 cells. Cells were treated for 1 hr with increasing concentration of ML171 and ROS generation was monitored by CL-assay. (b) HEK293 cells were transfected as indicated with increasing concentration of Nox1 and fixed concentration of its regulatory cofactors NoxA1, NoxO1 and constitutive active Rac1 (Rac1-Q61L). After 16hrs, cells were treated as indicated, and ROS generation was measured by CL assay. (c) HEK293 cells were transfected as indicated. After 16hrs, cells were treated as indicated, and ROS generation was measured by CL assay. (d) HEK293 cells were transfected as indicated. After 16hrs, cells were treated as indicated, and ROS generation was measured by CL assay. (e) Dose-response analysis of the effect of ML171 on the ability of recombinant Rac1-GTPase to be loaded with GTP. Recombinant Rac1 was pre-incubated as indicated with increasing concentration of ML171, or with DMSO or 5mM Mg2+ used as negative and positive control respectively. The mixtures were then incubated with 35S-GTPγS as described in the Methods section. Levels of Rac1-35S-GTPγS were measured by scintillation counting. In all panels, one representative experiment from three separate experiments is shown and results are given as mean of triplicates +/− S.D.
Mitochondria have been reported to be an important source of cellular ROS (27). To rule out the possibility that ML171 would non-specifically block mitochondrial ROS generation, we used a mitochondrion-specific hydroethidine-derivative fluorescent dye (MitoSox) to measure ROS generation in DLD1 treated with DMSO control, ML171 or DPI. Supplementary Figure S1b shows that ML171 treatment did not decrease mitochondrial ROS generation compared to DMSO, whereas DPI treatment slightly reduced ROS generation in mitochondria.
ML171 specifically targets Nox1
The mechanism of inhibition of some long-used Nox inhibitors has never been rigorously proven (19, 20). We next sought to clarify whether our newly-identified Nox1 inhibitor blocked Nox1-dependent ROS generation by acting on Nox1 or its regulatory subunits NoxA1, NoxO1 and Rac1-GTPase. Unfortunately, the absence of a crystal structure of Nox1 and most of its regulators prevents us from directly approaching this issue by protein crystallography. Nevertheless, to define key protein-subunit components necessary for the Nox1 inhibitory activity, we tested the ability of increasing concentration of Nox1, NoxA1 or NoxO1 to overcome the inhibition of ROS generation caused by ML171 treatment in HEK293-Nox1 reconstituted cell system. As shown in Fig. 1b, only increasing over-expression of Nox1 could rescue ROS generation in this system, while increasing over-expression of either NoxA1 (Fig. 1c) or NoxO1 (Fig. 1d) did not overcome the inhibition of superoxide formation caused by treatment with ML171.
GTP-bound Rac1 has been described to be required for full Nox1 activation (28, 29). To rule out the possibility that ML171 might affect Rac1 ability to be loaded with GTP, this molecule was tested in vitro in a Rac1-loading assay. As reported in the concentration-response analysis shown in Fig. 1e, we found that this compound could only marginally block Rac1 loading compared with Mg2+ used as a positive control. Similar results were obtained using the parental molecule (2-trifluoromethyl)-phenothiazine (not shown). Since both Nox1 and Nox3 are regulated by active Rac1 (30), these results are in agreement with the data shown in Table 1 indicating that ML171 only inhibited Nox1 (and not Nox3) activity (IC50HEK293-Nox1= 0.25 μM vs IC50HEK293-Nox3= 3 μM). These data suggest that Nox1 (and not its cytosolic regulators or Rac1-GTPase) is the protein targeted by these phenothiazines.
ML171 does not affect the activity of Nox2 or other CNS-expressed G-protein coupled receptors (GPCRs)
We sought to identify a selective Nox1 inhibitor with marginal effects on other Nox isoforms which could be used as a tool in the study of Nox1-dependent biological functions and as a therapeutic agent. Chronic granulomatous disease (CGD) is a hereditary disease due to mutation in Nox2 or its regulatory subunits and characterized by susceptibility to certain fungal and bacterial infections because of impaired defense against microorganisms (31). This is certainly a concern for the use of Nox inhibitors in humans. The IC50 values reported in Table 1b indicate that compared with DPI both parental and after-SAR phenothiazine compounds have higher specificity and selectivity towards Nox1 than other Nox isoforms (including Nox2). To further verify that the ML171 had only a marginal effect on Nox2 activity, we tested its ability to block Nox2-dependent ROS generation using both the well-validated Nox2 cell-free system, and human neutrophils treated with Formyl-Methionyl-Leucyl-Phenylalanine (fMLP) (32). As shown in the dose-response analysis illustrated in Fig. 2a (cell free system) and Fig. 2b (fMLP-treated human neutrophils) online, increasing concentrations of ML171 did not affect ROS generation via Nox2 (IC50Nox2 > 10 μM) in both cases, while a DPI concentration of 10 μM used as a positive control was sufficient to dramatically inhibit superoxide formation.
Figure 2.

ML171 does not block Nox2-dependent ROS generation. (a) Dose-response analysis of the effect of ML171 on Nox2 activity using Nox2-cell free system. ROS formation is measured continuously for 8 minutes and it is determined as the rate of superoxide dismutase inhibitable cytochrome c reduction. (b) Dose-response analysis of the effect of ML171 on Nox2 activity using human neutrophils isolated from the blood of healthy donors. Human neutrophils were incubated with the indicated concentration of ML171 and then stimulated with 5 μM fMLP as described in the Method section. ROS formation is measured continuously for 40 minutes by CL-assay. In (a) and (b) one representative experiment from three separate experiments is shown.
Another possible side-effect of the use of this sub-set of phenothiazines as Nox1 inhibitors in the clinic is the potential anti-psychotic effect due to the presence of the phenothiazine structure. SAR analysis in Table 2 indicates that several phenothiazines with anti-psychotic effect did not alter Nox1-dependent ROS generation. In addition, to further investigate the possibility that ML171 might exert an unwanted anti-psychotic effect, we tested the ability of this molecule to competitively bind a large battery of human or rodent G-protein coupled receptors (GPCRs), channels and transporters expressed in the CNS. As shown in Supplementary information, Table S2 online, ML171 did not significantly bind most of the receptors tested in the binding assays with the exception of serotonin (5-HT2B and 5-HT2C), and adrenergic (alpha2C) receptors (% of inhibition >60%). Of note, secondary concentration-response analysis revealed Ki values in the micromolar range which is suggestive of low affinity of our newly-identified low nanomolar Nox1 inhibitor for these GPCR receptors. Although previous studies have reported inhibitory effects of phenothiazines on the activity of some members of Nox family (33, 34), our data suggest that ML171 does not inhibit Nox2 function in the immune system nor likely exert unwanted anti-psychotic effects.
ML171 blocks the formation of functional invadopodia in human colon cancer cells
Invadopodia are dynamic phospho-tyrosine-rich structures with an actin core and abundant actin regulatory proteins (e.g. cortactin) capable of proteolytically degrading the ECM (35). They appear as actin protrusions of the ventral plasma membrane and their formation in human cancer cells correlates with their invasiveness both in vitro and in vivo (36). Activation of the tyrosine kinase c-Src is required for the formation of functional invadopodia (37). Evidence has recently demonstrated that the localized ROS generation by Nox1 is necessary for the formation of extracellular matrix (ECM)-degrading invadopodia in human DLD1 colon cancer cells, which endogenously express Nox1 (13). Therefore, we next sought to validate our newly-identified Nox1 inhibitor to determine whether it could block the invadopodia formation in DLD1 cells. To this aim, DLD1 cells were transfected with empty vector or with constitutive active Src (SrcYF) to trigger the formation of invadopodia and then treated for 1hr with ML171, or DMSO or DPI which were used as negative or positive controls respectively. After staining with invadopodia markers phalloidin and cortactin, we found that ML171 treatment strongly decreased SrcYF-induced invadopodia formation as much as DPI, as shown by confocal microscopy in Fig. 3a and in the quantification of three independent experiments in Fig. 3b. To further confirm the specificity of ML171 in blocking Nox1-dependent invadopodia formation, we assayed the effect of its analog chlorpromazine, which does not interfere with Nox1 activity (IC50HT29 > 50 μM, see Table 1), on invadopodia formation. Consistent with this, when DLD1 cells were transfected with SrcYF and treated for 1hr with chlorpromazine or DMSO control, we did not observe a decreased ability of DLD1 cells to form cortactin-positive, F-actin-rich invadopodia, as shown by confocal microscopy in Fig 4a and in the quantification of three independent experiments in Fig. 4b.
Figure 3.

ML171 treatment significantly blocks invadopodia formation in DLD1 cells. (a) DLD1 cells were plated on glass coverslips and after 24hrs cells were transfected with active SrcYF or empty vector (mock). 48hrs after transfection, cells were treated as indicated for 1hr with 10 μM of ML171, or with DMSO or 10 μM DPI. Afterwards, cells were fixed and stained (see Methods section) and visualized by confocal miscroscopy (100X). White arrows indicate phalloidin-positive (right column) or cortactin-positive (left column) invadopodia. Scale bars, 5 μm. One representative picture from three separate experiments is shown. (b) Quantification from three independent biological experiments shown in (a) is given: the number of phalloidin positive-invadopodia (left panel) or cortactin positive-invadopodia (right panel) was counted and averaged from 50 cells/condition for each experiment. Error bars represent SEM. *p<0.001
Figure 4.

Chlorpromazine treatment does not affect invadopodia formation in DLD1 cells. (a) DLD1 cells were plated on glass coverslips and after 24hrs cells were transfected with active SrcYF. 48hrs after transfection, cells were treated as indicated. Afterwards, cells were stained (see Methods) and visualized by confocal miscroscopy (100X). White arrows indicate phalloidin-positive (right column) or cortactin-positive (left column) invadopodia. Scale bars, 5 μm. One representative picture from three separate experiments is shown. (b) Quantification from three independent biological experiments shown in (a) is given: the number of phalloidin positive-invadopodia (left panel) or cortactin positive-invadopodia (right panel) was counted and averaged from 50 cells/condition for each experiment. Error bars represent SEM.
The formation of functional invadopodia relates to the ability of cells to degrade the ECM (36, 38). To further confirm that ML171 blocks the ROS-dependent, Nox1-mediated formation of functional invadopodia, we tested its effect on ECM degradation in DLD1 cells. As expected, SrcYF overexpression induced ECM degradation when cells were treated with DMSO control, while DPI treatment blocked this effect (Fig. 5a). Moreover, we observed that ML171 treatment strongly reduced the ability of SrcYF-transfected DLD1 cells to degrade the ECM. Importantly, the overexpression of Nox1 at least partially restored the capacity of these cells to degrade the ECM when treated with ML171. The quantification of three independent experiments is shown in Fig. 5b. These results support the validity of the use of ML171 as a specific and selective Nox1 inhibitor.
Figure 5.

ML171 treatment significantly blocks ECM degradation in DLD1 cells. (a) DLD1 cells were transfected and treated as indicated. After 48 hrs, cells were stained with Alexa-Fluor-568 phalloidin and visualized by epifluorescence microscopy (60X). The white arrows indicate areas in which cells (in red) degrade the ECM (in green). The merge is shown in the right column. Scale bars, 10 μm. One representative image from three separate experiments is shown. (b) Quantification from three independent biological experiments shown in (a) is given: for each experiment, the total degradation area was obtained as sum of degradation areas calculated using Metamorph software from 25 random fields and reported as percentage (SrcYF+DMSO set as 100%). In the graph, error bars represent SEM. *p <0.01 ** p<0.05 (Mann-Whitney U test).
Strong evidence points to a pivotal role of the Nox enzymes as a predominant source of ROS in many human diseases (5). Specific inhibition of Nox isoforms can provide reversible, mechanistic insights into Nox-dependent biological functions and pathological states in contrast to scavenging or inhibition of ROS production by other ROS-generating cellular enzymes. Unfortunately, no specific and selective Nox inhibitors have been described previously. Using our screening cascade, we have identified by high-throughput screening a sub-set of phenothiazines, 2-acetylphenothiazine (ML171) (and its related 2-(Trifluoromethyl)-phenothiazine) as nanomolar, cell-active and specific Nox1 inhibitors that potently block Nox1-dependent ROS generation, with only marginal activity on other cellular ROS-producing enzymes and receptors including the other Nox isoforms (see Table 1). We show that our sub-set of phenothiazines specifically blocks Nox1-dependent ROS generation with almost no effect on the formation of ROS by xanthine oxidase, which on the other hand is strongly inhibited by the most currently used Nox inhibitor (i.e. DPI). Our findings possibly suggest that Nox1 (and not its cytosolic regulators NoxO1, NoxA1 or Rac1) is the protein targeted by these phenothiazines (Fig. 1a–d), which provides a molecular basis for their mechanism of inhibition. Future studies should possibly clarify the three dimensional features of this interaction using crystal structure information about Nox1. Nevertheless, in this work we have validated ML171 as an effective Nox1 inhibitor. Consistent with this, treatment with this molecule (and not with its non-functional analog chloropromazine) strongly decreased Nox1-dependent invadopodia formation (Fig. 3a–b) and ECM degradation (Fig. 5a–b) in DLD1 cells.
We believe that this compound would be extremely useful in dissecting out the contributions of Nox1 in biological processes and pathological states. Additionally, this inhibitor has the potential as a novel therapeutic approach for treatment of various redox stress-dependent diseases and/or cancer metastasis.
Methods
Description of all assays employed in our screening cascade is provided in Supplementary information as Supplementary Methods. All assays are listed in Supplementary Table S1.
Reagents
Cell culture medium, fetal bovine serum, supplements and Hank’s Balanced Salt Solution (HBSS, Cat# 24020-117) were from Invitrogen. Nox1, NoxA1, NoxO1 and Rac1-Q61L expression plasmids were previously described (13). Plasmids for transfection were purified using the Qiagen Qiafilter system. DLD1 and HT29 colonic adenocarcinoma cells (Cat# CCL-221 and HTB-38) were purchased from ATCC. 35S-GTPγS (Cat# NEG030H) was obtained from Perkin Elmer. Chlorpromazine (C0982), DPI (D2926), horseradish peroxidase (HRP) (77330), fMet-Leu-Phe (fMLP) (F3506), NADPH (N5130), FAD (F6625), cytochrome c (105201) and luminol (09253) were purchased from Sigma. 2-(Trifluoromethyl)-phenothiazine (CID 7082, product code JFD03930), compound library (HitFinder version 4, Maybridge) and other positive hits from primary assay were bought from Maybridge. 2-acetylphenothiazine (CID 81131, product code STK301831) was bought from Vitas-M Laboratories. SAR compounds were synthesized or obtained through the Scripps Research Institute Molecular Screening Center. All structures, sources and data can be found in PubChem database within PubMed under Assay Identifiers (AID) 1792, 1796, 1823, 2538 and 2541. The following antibodies were purchased as indicated: mouse monoclonal cortactin antibody 4F10 from Millipore. Anti-mouse Alexa-Fluor-568 and Alexa-Fluor-568 phalloidin were purchased from Molecular Probes.
Cell culture and transfections
Human HEK293, human DLD1 and HT29 cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen) containing 10% heat-inactivated fetal bovine serum (Invitrogen), 2mM glutamine, and antibiotics (100 units/ml penicillin and 100 g/ml streptomycin) at 37 °C in 5% CO2. HEK293 cells grown in 6-well plates (Falcon) were transfected by Lipofectamine 2000 (InVitrogen) using 5 μg of total DNA and 10 μl of Lipofectamine reagent per well. DLD1 cells were transfected by Lipofectamine 2000 following manufacturer’s instruction. 24–48 hrs after transfection cells were processed for further analysis.
Rac loading assay
Rac1 protein was expressed and purified as GST fusion proteins in E. coli using standard isolation protocols, and the GST tag was cleaved with thrombin prior to use in in vitro loading assays, following the manufacturer’s directions. Purified Rac1 protein was quantified by the binding of 35S-GTPγS. 20 μg of recombinant Rac1 protein was incubated with increasing concentration of ML171, DMSO control or 5 mM MgCl2 for 10 min at room temperature. Rac1 was then loaded with 25 μCi of 35S-GTPγS in a mixture with 0.1 mM of unlabeled GTPγS for 20 minutes in ice. The reaction was stopped by addition of 25mM MgCl2 and subjected to filter binding to nitrocellulose and quantified by scintillation counting. The ability of ML171 to inhibit Rac1 loading with GTP was revealed as loss of radioactivity bound to Rac1 (% of inhibition of Rac1 loading due to DMSO and Mg2+ are set 0% and 100% respectively).
Luminol-based chemiluminescence (CL) assay
This assay was performed as previously described (25). Briefly, 16hrs after transfection, 5*105 HEK293 cells per assay were dispensed in white 96-well plate (Berthold) and mixed with 250 μM luminol and 1U horseradish peroxidase HRP (final concentration) in 200 μl total final volume in each well. Chemiluminescence was recorded using 96-well plate luminometer (Berthold) 5 min after the addition of HRP/luminol mixture for 30 min at room temperature without any stimulation.
Confocal and epifluorescence microscopy
24 hrs after transfection, DLD1 cells plated on glass or FITC-labeled gelatin-coated coverslips were fixed in 4% paraformaldehyde (PFA) at room temperature for 10 minutes. Successively, cells were permeabilized in 0.5% Triton for 10 minutes and blocked in 2%BSA in PBS for 45 minutes at room temperature. Cells were then immunolabeled as indicated in the figure legends with appropriate primary and Alexa-Fluor 568-conjugated secondary antibodies. F-actin was detected by using Alexa-Fluor 568-conjugated phalloidin. Cells were mounted on slides with Mowiol mounting medium (Calbiochem) according to the manufacturer’s instructions. Epifluorescence and confocal images of fixed cells were acquired using the same setup described in (13).
ECM degradation assays
Fluorescently labeled gelatin-coated coverslips were prepared as previously described (39). 24hrs after transfection, cells were trypsinized and plated on FITC-labeled gelatin-coated coverslips. After 2hrs, cells were treated as indicated with 10 μM of ML171 or DPI and control DMSO for 1.5hrs. 24 hrs later, cells were fixed in 4% PFA, stained with Alexa-Fluor-568 phalloidin and visualized by epifluorescence microscopy (60X).
Isolation and maintenance of human neutrophils
Fresh human blood was collected through the normal blood donor program at the Scripps Research Institute in accordance with an institutional review board-approved protocol as described (40).
Determination of superoxide production in human neutrophils
For human neutrophils in suspension, the cells were treated for 10 minutes with increasing concentration of ML171, DMSO control or 10 μM DPI. Superoxide production of neutrophils stimulated with the final concentration of 5*10−6 M fMLP was measured by CL assay continuously for 40 minutes.
Determination of superoxide production in Nox2-cell free assay
Human neutrophil membrane fractions (GSP) and neutrophil cytosolic fractions (GSS) were prepared as described (41).10 μg of purified neutrophil membrane and 100 μg of purified neutrophil cytosol were used for each reaction and the determination of superoxide production was performed as described (42).
Statistical analysis
In this study overall, representative experiments from at least three independent experiments are shown. Results for each experiment are given as the mean of triplicates +/− standard deviation (S.D.). Statistically significant differences between sample groups were determined using two-tailed t-tests (Microsoft Excel, Redmond WA) unless differently indicated (see Figure legend 5b). p values for differences observed in figures were always <0.001 unless differently indicated (see Figure legend 4b). All IC50 values were calculated using a 4 parameter fit.
Supplementary Material
Acknowledgments
The authors thank Bruce Fowler, Jackie Chapman, Benjamin Bohl and Ryan Mitchell for technical help and Michael Howell for helpful discussion. % of inhibition values and Ki determinations for CNS receptors and channels were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. We would like to dedicate this paper to the memory of our mentor and colleague Gary M. Bokoch. This work was supported by NIH grant HL48008 (to GMB and CDM) and the National Institutes of Health Molecular Library Screening Center Network (5U54MH074404, to HR) which supported the research efforts of WRR, SJB and HR. This is manuscript IMM20805 from The Scripps Research Institute
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org. In addition, because this work was performed in collaboration with an NIH Comprehensive Screening Center in the Molecular Libraries Initiative, the primary data will be deposited in PubChem under the assay identifiers (AID) listed in the paper and online supporting information and can be accessed at http://www.ncbi.nlm.nih.gov.
References
- 1.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological reviews. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 2.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nature reviews. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 3.Bokoch GM, Knaus UG. NADPH oxidases: not just for leukocytes anymore! Trends in biochemical sciences. 2003;28:502–508. doi: 10.1016/S0968-0004(03)00194-4. [DOI] [PubMed] [Google Scholar]
- 4.Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free radical biology & medicine. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambeth JD, Krause KH, Clark RA. NOX enzymes as novel targets for drug development. Seminars in immunopathology. 2008;30:339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- 6.Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, Lambeth JD. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:715–720. doi: 10.1073/pnas.022630199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 8.Sadok A, Bourgarel-Rey V, Gattacceca F, Penel C, Lehmann M, Kovacic H. Nox1-dependent superoxide production controls colon adenocarcinoma cell migration. Biochimica et biophysica acta. 2008;1783:23–33. doi: 10.1016/j.bbamcr.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 9.Block ML. NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC neuroscience. 2008;9(Suppl 2):S8. doi: 10.1186/1471-2202-9-S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cifuentes ME, Pagano PJ. Targeting reactive oxygen species in hypertension. Current opinion in nephrology and hypertension. 2006;15:179–186. doi: 10.1097/01.mnh.0000214776.19233.68. [DOI] [PubMed] [Google Scholar]
- 11.Nauseef WM. Nox enzymes in immune cells. Seminars in immunopathology. 2008;30:195–208. doi: 10.1007/s00281-008-0117-4. [DOI] [PubMed] [Google Scholar]
- 12.Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer letters. 2008;266:37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gianni D, Diaz B, Taulet N, Fowler B, Courtneidge SA, Bokoch GM. Novel p47(phox)-related organizers regulate localized NADPH oxidase 1 (Nox1) activity. Science signaling. 2009;2:ra54. doi: 10.1126/scisignal.2000370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buccione R, Caldieri G, Ayala I. Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer metastasis reviews. 2009;28:137–149. doi: 10.1007/s10555-008-9176-1. [DOI] [PubMed] [Google Scholar]
- 15.Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends in cell biology. 2007;17:107–117. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Faggioni R, Gatti S, Demitri MT, Delgado R, Echtenacher B, Gnocchi P, Heremans H, Ghezzi P. Role of xanthine oxidase and reactive oxygen intermediates in LPS- and TNF-induced pulmonary edema. The Journal of laboratory and clinical medicine. 1994;123:394–399. [PubMed] [Google Scholar]
- 17.Gottlieb RA. Cytochrome P450: major player in reperfusion injury. Archives of biochemistry and biophysics. 2003;420:262–267. doi: 10.1016/j.abb.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Jaeschke H, Mitchell JR. Mitochondria and xanthine oxidase both generate reactive oxygen species in isolated perfused rat liver after hypoxic injury. Biochemical and biophysical research communications. 1989;160:140–147. doi: 10.1016/0006-291x(89)91632-x. [DOI] [PubMed] [Google Scholar]
- 19.Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxidants & redox signaling. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 20.Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Current drug metabolism. 2008;9:686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- 21.O’Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. The Biochemical journal. 1993;290(Pt 1):41–49. doi: 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Donnell VB, Smith GC, Jones OT. Involvement of phenyl radicals in iodonium inhibition of flavoenzymes. Molecular pharmacology. 1994;46:778–785. [PubMed] [Google Scholar]
- 23.Dahlgren C, Karlsson A, Bylund J. Measurement of respiratory burst products generated by professional phagocytes. Methods Mol Biol. 2007;412:349–363. doi: 10.1007/978-1-59745-467-4_23. [DOI] [PubMed] [Google Scholar]
- 24.DeChatelet LR, Long GD, Shirley PS, Bass DA, Thomas MJ, Henderson FW, Cohen MS. Mechanism of the luminol-dependent chemiluminescence of human neutrophils. J Immunol. 1982;129:1589–1593. [PubMed] [Google Scholar]
- 25.Gianni D, Bohl B, Courtneidge SA, Bokoch GM. The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1. Molecular biology of the cell. 2008;19:2984–2994. doi: 10.1091/mbc.E08-02-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitchell SC. Phenothiazine: the parent molecule. Current drug targets. 2006;7:1181–1189. doi: 10.2174/138945006778226552. [DOI] [PubMed] [Google Scholar]
- 27.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free radical biology & medicine. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. The Journal of biological chemistry. 2006;281:17718–17726. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- 29.Miyano K, Ueno N, Takeya R, Sumimoto H. Direct involvement of the small GTPase Rac in activation of the superoxide-producing NADPH oxidase Nox1. The Journal of biological chemistry. 2006;281:21857–21868. doi: 10.1074/jbc.M513665200. [DOI] [PubMed] [Google Scholar]
- 30.Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Molecular and cellular biology. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cross AR, Noack D, Rae J, Curnutte JT, Heyworth PG. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (first update) Blood cells, molecules & diseases. 2000;26:561–565. doi: 10.1006/bcmd.2000.0333. [DOI] [PubMed] [Google Scholar]
- 32.Sklar LA, Hyslop PA, Oades ZG, Omann GM, Jesaitis AJ, Painter RG, Cochrane CG. Signal transduction and ligand-receptor dynamics in the human neutrophil. Transient responses and occupancy-response relations at the formyl peptide receptor. The Journal of biological chemistry. 1985;260:11461–11467. [PubMed] [Google Scholar]
- 33.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. The Biochemical journal. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Traykov T, Hadjimitova V, Goliysky P, Ribarov S. Effect of phenothiazines on activated macrophage-induced luminol-dependent chemiluminescence. General physiology and biophysics. 1997;16:3–14. [PubMed] [Google Scholar]
- 35.Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Current opinion in cell biology. 2008;20:235–241. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 36.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clinical & experimental metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 37.Lowe C, Yoneda T, Boyce BF, Chen H, Mundy GR, Soriano P. Osteopetrosis in Src-deficient mice is due to an autonomous defect of osteoclasts. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:4485–4489. doi: 10.1073/pnas.90.10.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldassarre M, Pompeo A, Beznoussenko G, Castaldi C, Cortellino S, McNiven MA, Luini A, Buccione R. Dynamin participates in focal extracellular matrix degradation by invasive cells. Molecular biology of the cell. 2003;14:1074–1084. doi: 10.1091/mbc.E02-05-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blouw B, Seals DF, Pass I, Diaz B, Courtneidge SA. A role for the podosome/invadopodia scaffold protein Tks5 in tumor growth in vivo. European journal of cell biology. 2008;87:555–567. doi: 10.1016/j.ejcb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Sun C, Glogauer M, Bokoch GM. Human neutrophils coordinate chemotaxis by differential activation of Rac1 and Rac2. J Immunol. 2009;183:2718–2728. doi: 10.4049/jimmunol.0900849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curnutte JT, Kuver R, Scott PJ. Activation of neutrophil NADPH oxidase in a cell-free system. Partial purification of components and characterization of the activation process. The Journal of biological chemistry. 1987;262:5563–5569. [PubMed] [Google Scholar]
- 42.Kao YY, Gianni D, Bohl B, Taylor RM, Bokoch GM. Identification of a conserved Rac-binding site on NADPH oxidases supports a direct GTPase regulatory mechanism. The Journal of biological chemistry. 2008;283:12736–12746. doi: 10.1074/jbc.M801010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.