

Abstract
Tuberoinfundibular peptide of 39 residues (TIP39) synthesizing neurons at the caudal border of the thalamus and in the lateral pons project to areas rich in its receptor, the parathyroid hormone 2 receptor (PTH2R). These areas include many involved in processing nociceptive information. Here we examined the potential role of TIP39 signaling in nociception using a PTH2R antagonist (HYWH) and mice with deletion of TIP39's coding sequence or PTH2R null mutation. Intracerebroventricular (icv) infusion of HYWH significantly inhibited nociceptive responses in tail-flick and hot-plate tests and attenuated the nociceptive response to hindpaw formalin injection. TIP39-KO and PTH2R-KO had increased response latency in the 55 °C hot-plate test and reduced responses in the hindpaw formalin test. The tail-flick test was not affected in either KO line. Thermal hypoalgesia in KO mice was dose-dependently reversed by systemic administration of the cannabinoid receptor 1 (CB1) antagonist rimonabant, which did not affect nociception in wild-type (WT). Systemic administration of the cannabinoid agonist CP 55,940 did not affect nociception in KO mice at a dose effective in WT. WT mice administered HYWH icv, and both KOs, had significantly increased stress-induced analgesia (SIA). Rimonabant blocked the increased SIA in TIP39-KO, PTH2R-KO or after HYWH infusion. CB1 and FAAH mRNA were decreased and increased, respectively, in the basolateral amygdala of TIP39-KO mice. These data suggest that TIP39 signaling modulates nociception, very likely by inhibiting endocannabinoid circuitry at a supraspinal level. We infer a new central mechanism for endocannabinoid regulation, via TIP39 acting on the PTH2R in discrete brain regions.
Keywords: Pain, Nociception, Descending inhibition, Endocannabinoid, Knockout mouse, Supraspinal, Stress-induced analgesia
Introduction
Pain is a compound sensation that indicates real or potential injury. One-way to characterize afferent pain pathways is to distinguish between phylogenetically younger ascending nociceptive pathways that convey sensory-discriminative information from the spinal cord to the ventral posterolateral thalamus and somatosensory cortex, and phylogenetically older pathways that terminate in the medial/intralaminar complex of the thalamus and limbic areas, and are involved in affective and cognitive aspects of pain as well as stress responses (Almeida et al., 2004; Basbaum et al., 2009; Cliffer et al., 1991). Processing of nociceptive information in higher brain structures activates descending pathways that modulate pain. The amygdala, periaqueductal gray (PAG) and rostral ventromedial medulla are major contributors to descending modulation of pain, with endogenous opioids and cannabinoids in these areas playing a significant role in descending inhibition (Fields and Heinricher, 1985; Mason, 2005; Millan, 2002; Willis and Westlund, 1997).
The parathyroid hormone receptor 2 (PTH2R) is expressed by neurons in many areas implicated in processing nociceptive information. This includes cell populations in the spinal cord dorsal horn, PAG, medial and intralaminar thalamic nuclei, amygdala, hypothalamus and somatosensory cortex (Bago et al., 2009; Faber et al., 2007; Wang et al., 2000). This Gs (and Gq in some cells) coupled receptor is expressed by subpopulations of glutamatergic cells in some brain regions, and is activated by tuberoinfundibular peptide of 39 residues (TIP39) (Dobolyi et al., 2006; Usdin et al., 1999a,b). TIP39 is synthesized by neurons in the periventricular gray and posterior intralaminar thalamic nucleus, which are part of the subparafascicular area (SPF) at the caudal border of the thalamus, and by cells of the medial paralemniscal nucleus (MPL) within the posterolateral pons (Dobolyi et al., 2003a,b, 2010). In general, SPF TIP39 neurons send their axons rostrally while the MPL cells project caudally (Dobolyi et al., 2003a,b; Wang et al., 2006), and the distribution of TIP39 immunopositive fibers closely matches the PTH2R expression pattern throughout the CNS (Faber et al., 2007).
Functions attributed to the PTH2R/TIP39 system include modulation of the stress response, anxiety, and hypothalamic neuroendocrine factor secretion (Fegley et al., 2008; LaBuda et al., 2004; Palkovits et al., 2009; Sugimura et al., 2003; Ward et al., 2001). Intrathecal or peripheral administration of TIP39 has pro-nociceptive effects (Dobolyi et al., 2002; LaBuda and Usdin, 2004; Matsumoto et al., 2010), and it may modulate an affective component of pain (LaBuda and Usdin, 2004). PTH2Rs are present in essentially all of the brain areas that receive nociceptive information via the phylogenetically older ascending nociceptive pathways (Faber et al., 2007; Wang et al., 2000). This led us to hypothesize that TIP39 regulates some aspects of nociceptive signaling at a supraspinal level, potentially by affecting endogenous opioid and/or cannabinoid circuits within limbic pathways involved in pain modulation, pain affect, or stress responses to pain.
In this study we evaluate the contribution of TIP39 signaling to acute thermo-nociception using tail-flick and hot-plate assays, and to tonic nociception by the response to hindpaw formalin injection. We first performed nociceptive tests following intracerebroventricular (icv) and intrathecal administration of Histidine3-Tyrosine4-Tryptophan5-Histidine6-TIP39 (HYWH), a PTH2R antagonist. HYWH is an analog of the thirty-nine amino acid TIP39, in which four residues in the amino terminal domain that is involved in receptor activation are replaced by more bulky amino acids, such that the peptide retains high affinity for, but does not activate, the PTH2R (Kuo and Usdin, 2007). Second, we used global TIP39 (Fegley et al., 2008) and PTH2R (Dimitrov and Usdin, in press) knockout mouse lines in a similar set of experiments. Third, we examined the effects on nociception of a cannabinoid receptor 1 (CB1) antagonist, and a cannabinoid agonist, in combination with manipulation of TIP39 signaling and stress. Also, we compared the expression patterns of mRNA encoding CB1 and fatty acid amide hydrolase (FAAH), one of the enzymes responsible for endocannabinoid metabolism, in TIP39-KO and WT mice using in situ hybridization histochemistry. Finally, we examined colocalization in the amygdala between the PTH2R and vesicular glutamate transporter 2 (VGlut2), a marker for glutamatergic synapses. These experiments led us to infer that endogenous TIP39 modulates nociception at a supraspinal level, likely through effects on endocannabinoid signaling.
Materials and methods
Animals
All procedures were approved by the National Institute of Mental Health Animal Care and Use Committee and in accordance with the Institute for Laboratory Animal Research Guide for the Care and Use of Laboratory Animals which is consistent with EC Directive 86/609/EEC. Mice were housed under a 12/12 h light/dark cycle (lights on at 6:00 am) with free access to food and water. Males between 80 and 140 days of age and weighing from 28 to 32 g were used for all experiments and were singly housed for 1 – 3 weeks before the start of an experiment. WT and KO mice from the same Het X Het breedings were used in each experiment. Each mouse was used in not more than three different experiments with at least seven days elapsing between the experiments. Testing was conducted in the morning with at least 1 h for the animal's acclimatization to one of the two rooms used for testing. Development and genotyping of TIP39- (Fegley et al., 2008) and PTH2R-KO (Dimitrov and Usdin, in press) mice have been previously described. C57Bl/6J mice used for icv cannulation experiments were obtained from Jackson Laboratories (Bar Harbor, ME).
Nociception tests
The tail-flick, hot-plate and hindpaw formalin tests were adapted from “Current Protocols in Neuroscience” (Bannon and Malmberg, 2007).
Tail-flick
A standard apparatus (Analgesia Meter Model 390, Life Science, Woodland Hills, CA) with adjustable light intensity was used for the tail-flick. While gently restraining the animal by hand, a 100% or 40% intensity light beam was focused on the dorsal part of the tail, 2 cm from its end. Cut-off time was set at 10 s. The average of three consecutive readings was used as the tail-flick response latency for each animal. The high intensity light produced an average response latency under 2 s and the low intensity light about 3 s. Accuracy of the measurements was validated by video recording some of the experiments and counting the frames from tail illumination to tail-flick. Immersion of the tail was also used in some experiments to measure the thermal withdrawal response. Holding a mouse in a plastic tube restrainer, the distal half of its tail was submerged in a beaker of water at the selected temperature and the time until vigorous tail withdrawal measured.
Hot-plate
The hot-plate (Hot Plate Analgesia Meter, Columbus Instruments, OH) was set at 42°, 48° or 55 °C and the cut-off times used were 200, 150 and 100 s, respectively. Mice were placed on the hot surface over which a transparent bottomless box that allowed unrestrained movement of the mouse was placed. Two investigators sharing a stopwatch observed the animal's behavior, which was also a video recorded. The latency to the first hindpaw bite, vigorous lift and shake of a hindpaw, or jump before the cut-off time was identified. The video record was used to verify stopwatch readings.
Stress-induced analgesia (SIA)
Inescapable foot shock was used to produce SIA. After obtaining baseline hot-plate measurements, animals were put individually into a metal grid-floor box (ENV-414S-SR, Med Associates Inc., St. Albans, VT) and received continuous, 2-second interval, scrambled foot shock with intensity 0.6 mA for 1 min. Immediately after the shock the animals were retested on the 55 °C hot-plate. When drug effects were evaluated the baseline hot-plate response latency step was omitted. No animals were injured during the experiments.
Formalin test
The formalin test was conducted in clear Plexiglas boxes over a mirror. Mice were familiarized with the box over 2 sessions of 5 min each one and two days before the experiment. On the day of the experiment, after 2 h for acclimatization to the room, 20 μl of 2% formalin (0.74% formaldehyde) was injected subcutaneously into the dorsal part of the left hindpaw with a 30-gauge needle. Immediately after the injection the mice were released into a box and their behavior for the next hour video recorded. Each episode of hindpaw biting, licking or shaking was defined as a response. Responses were counted for each minute of the first five-minute period and for one minute out of every three minutes subsequently, to the end of 1 h. Mice were euthanized immediately after the formalin test.
Pharmacological treatment
Intracerebroventricular drug administration
A 26-gauge stainless steel guide cannula (Plastics One, Roanoke, VA) was implanted into the left lateral ventricle (anteroposterior: −0.6 mm, lateral: +1.2 mm and dorsoventral: −2.0 mm from bregma) of isoflurane anesthetized animals in a stereotaxic apparatus, and secured to the skull with three screws and acrylic dental cement. Stylets were placed in each cannula to maintain patency. Animals were handled daily and the stylets manipulated to acclimate the animals to the microinjection procedure and reduce tissue growth on the cannulae tips.
On the day of the experiment, after 1 h acclimation to the procedure room, a 33-gauge internal injector with a 0.5 mm projection was inserted into the cannula and 1.0 μl vehicle containing 0.1% bovine serum albumin (Equitech-Bio Inc., Kerrville, TX) in saline, 500 pmol of TIP39 or 5, 50 or 500 pmol HYWH infused into the lateral ventricle over 5 min. The injectors were removed 1 min after the completion of the infusion and the animals were immediately tested. In dose–response experiments animals received only one HYWH dose. TIP39 and HYWH were dissolved and diluted in 10 mM Tris HCl, 6%DMSO, at pH 7.0. TIP39 has the sequence (single letter amino acid codes) SLALADDAAFRERARLLAALERRHWLNSYMHKLLVLDAP. The underlined and bold amino acids are replaced by His, Tyr, Trp, His to create H3Y4W5H6-TIP39, referred to as HYWH. Both peptides were synthesized by Midwest Biomedicals (Mokena, IL). Mice were euthanized at the end of these experiments and the brains dissected to verify cannula placement.
Intrathecal injection
Mice were lightly anesthetized with isoflurane (until the corneal reflex disappeared), the lower backs shaved, and the skin swabbed with Betadine. The body was flexed abdominally and stretched by gently pulling the tail. A 30-gauge needle attached to a gas-tight Hamilton syringe was inserted at a 90° angle between the spinous process of the fifth lumbar vertebra and the sacral crest. Once the tip of the needle penetrated the skin and the underlying ligaments, the angle of the needle was changed to 45° and advanced into the epidural space. A successful lumbar puncture was marked by a reflexive tail-flick or a hind leg kick. Drug or vehicle in a 5 μl volume was injected over 30 s. The entire procedure from the induction of the anesthesia to the needle withdrawal took less than 5 min to complete. The animals were tested 10 min after that.
Intraperitoneal injection of rimonabant and naloxone
TIP39-KO or PTH2R-KO mice with their corresponding WT littermates were assigned to groups receiving vehicle and groups receiving drug. Rimonabant (SR141716A; provided by the NIMH Chemical Synthesis and Drug Supply Program) was dissolved in 4% Tween 20, 8% DMSO, 20% H2O and saline quantum satis with the concentration adjusted so that 0.2 ml per animal was intraperitoneally injected. Naloxone hydrochloride dihydrate (Sigma-Aldrich, St. Louis, MO) was dissolved in 2% DMSO/0.9% NaCl for a dose of 1 mg/kg in 0.2 ml of solution. Mice were tested 30 min after the rimonabant or naloxone injection.
“Cannabinoid tetrad” measurements
The effect of a cannabinoid agonist on the TIP39-KO line was assessed using the mouse “tetrad assay” adapted from Long (Long et al., 2009). First, 0.3 mg/kg of CP 55,940 (2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl) cyclohexyl]-5-(2-methyloctan-2-yl)phenol) (Tocris, Ellisville, MO) dissolved in 10% DMSO and brought to volume in normal saline (final concentration to provide 0.3 mg/kg in 0.2 ml solution) or vehicle were injected intraperitoneally. Thirty minutes after the injection, tests were performed in this order: catalepsy, locomotion, hot-plate and body temperature. Catalepsy was measured by placing the mouse's front paws on a 4.5 cm high 0.7 mm diameter metal bar, and measuring the amount of time that the mouse stayed absolutely still in 1 min. If the mouse moved, the timer was stopped, the animal put back in its original posture and the timer restarted. Locomotion was evaluated in a 42 × 21 cm Plexiglas box with the floor marked in 7 × 7 cm squares. Animals were familiarized with the box before the experiment. The number of hindpaw crossings of the floor lines was counted for 1 min from a video record. The hot-plate test was performed as described above, at 55 °C. A “Thermalert TH-5” apparatus (Clifton, NJ) with lubricated rectal probe was used to measure body temperature.
Anatomical techniques
In situ hybridization histochemistry for CB1 and FAAH mRNA
Tissue preparation and probe hybridization were performed as described previously (Bago et al., 2009). Briefly, 12 μm thick coronal sections were cut from unfixed rapidly frozen brains and thaw mounted onto positively charged slides that were stored at −80 °C until hybridization. 35S-labeled riboprobes were synthesized from polymerase chain reaction products with recognition sites for T7 RNA polymerase incorporated into the antisense strand that corresponded to bases 4324–4715 of mouse CB1 (GenBank accession # NM_007726) and bases 1636–2656 of mouse FAAH (GenBank accession # NM_010173). Following hybridization and washes, slides were dipped in Ilford nuclear emulsion (Harman Technology Ltd., Mobberley, UK) and stored at 4 °C for 15 to 18 days. Slides were developed and fixed with Kodak Dektol developer and Kodak fixer, counterstained with Giemsa, and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA). Dark field images of atlas-matched sections were acquired with identical lighting and exposure conditions using an Olympus IX70 microscope and Photometrics CoolSnap FX camera. The signal intensity was evaluated using NIH ImageJ. A region of interest was outlined and the integrated signal intensity of that region obtained. The signal from three nearby areas of similar size with only background labeling was then obtained. The mean intensity per area of the background regions was multiplied by the area of the region of interest and then subtracted from the integrated signal intensity of that region. This measure was obtained for two sections per animal for each brain area analyzed, and the mean of these two determinations used as the measure for that animal. Data from four separate experiments were calculated as percent WT and then pooled, with a final N of 13 to 14 animals per group.
Immunohistochemistry for PTH2R and VGlut2
The animals were perfused with 4% paraformaldehyde and 40 μm sections were cut from the brain areas of interest. The free floating sections were incubated with a rabbit antibody directed at the PTH2R (Faber et al., 2007) for 48 h at 4 °C followed by primary antibody detection using ABC reagents (Vectastain Elite, Vector Laboratories, Burlingame, CA) and DyLight 488 (Pierce, Rockford Il) conjugatedtyramine. Then, sections were incubated with a guinea pig antibody against VGlut2 (gift from Dr. Jeffery Erickson; 1:1000), which was visualized with an Alexa 594 labeled secondary antibody (InVitrogen, Carlsbad, CA). The labeling pattern obtained with both primary antibodies was the same as that previously described for the corresponding antigen (Faber et al., 2007; Varoqui et al., 2002). The PTH2R antibody labels cells transfected with a human PTH2-R cDNA but not the parent cells or cells transfected with a PTH1-R cDNA by immunohistochemistry and only labels extracts from PTH2-R expressing cells by western blot (Usdin et al., 1999a,b). The VGlut2 antibody labels extracts from VGlut2 expressing cells but not parent PC12 or VGlut1 or VGlut3 expressing cells (Schafer et al., 2002).
The anatomical levels of sections and specific regions were identified using the Franklin and Paxinos (Franklin and Paxinos, 2008)) atlas. Immunolabeling was visualized in representative sections using a Zeiss LSM 510 confocal microscope in the NINDS Intramural Light Imaging Facility. Z-series were analyzed using Volocity software (Improvision, Waltham MA). Parameters for the high magnification images shown in Fig. 16 were Z-series 1024X1024X15, scaling 0.14 μm×0.14 μm×0.2 μm, 63× oil, electronic zoom ×3, wavelengths 488 nm and 543 nm. Voxels with suprathreshold signal in both red and green channels were pseudocolored yellow and the images rendered by the command 3D opacity.
Fig. 16.
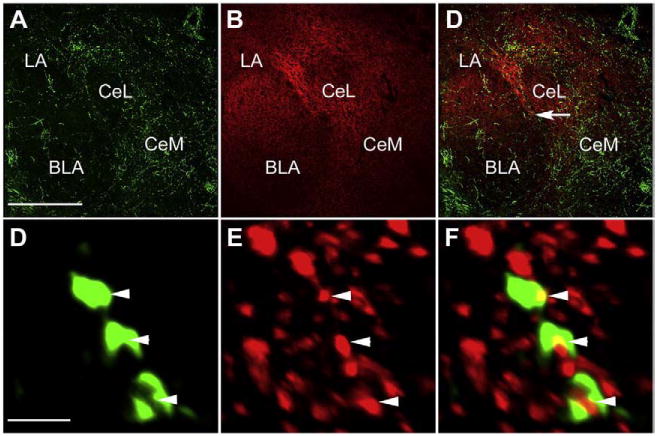
PTH2R and VGlut2 colocalization in the amygdala. Confocal images show PTH2R-ir fibers (A, green) and VGlut2-ir (B, red) in amygdaloid nuclei. In amygdaloid nuclei PTH2R-ir overlaps VGlut2-ir (merged images in C). High magnification image in panel F show colocalization of PTH2R-ir punctae (D) with VGlut2-ir punctae (E) from area indicated with arrow in panel C. Arrowheads indicate structures with PTH2R-ir/VGlut2-ir colocalization. The high magnification images are 3D reconstructions from a 3 μm Z-stack with colocalized voxels pseudocolored yellow (See Materials and methods). Abbreviations used: BLA – basolateral amygdaloid nucleus, CeL – centrolateral amygdaloid nucleus, CeM – centromedial amygdaloid nucleus, LA – lateral amygdaloid nucleus. Scale bars = 300 μm for the low and 2 μm for the high magnification images. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Statistical analysis
Data are presented as mean ± standard error of the mean (S.E.M.) unless stated otherwise. Student's t-test was used for two groups comparisons. One-way ANOVA with repeated measures followed by Newman–Keuls Multiple Comparison Test was used for drug dose responses and two-way ANOVA followed by Bonferroni Posttest for drug and genotype interaction. The accepted level of significance was P<0.05 in all tests.
Results
Nociception following acute PTH2R block
HYWH increases the response latency in tail-flick and hot-plate tests
ICV administration of the PTH2-R antagonist HYWH was antinociceptive in the tail-flick test (Fig. 1A, ANOVA, F3,53=16.19; P<0.001) and the hot-plate test (Fig. 1B, ANOVA, F3,44 =7.64; P<0.001). In the tail-flick test 500 pmol increased the response latency by 200%, from 1.4 ± 0.2 to 4.2 ± 1 s and in the hot-plate test it increased the response latency by 51%, from 14.1 ± 1.5 to 21.3 ± 4.1 s. Effects of lower doses were not statistically significant.
Fig. 1.
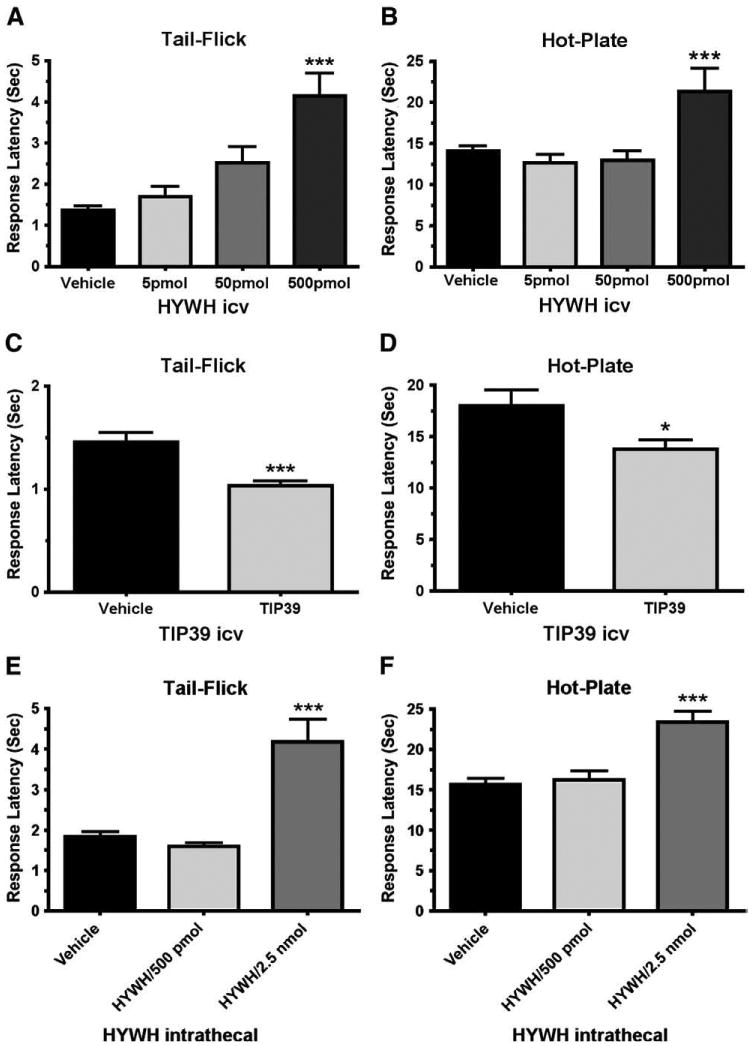
Antinociceptive effect of PTH2R block in the tail-flick and hot-plate tests. Tail-flick (A, C and E) or hot-plate (B, D and F) tests were performed following icv (A, B, C and D) or intrathecal (E and F) administration of the indicated amounts of the PTH2R antagonist HYWH or 500 pmol PTH2R agonist TIP39. The response latency significantly increased in both tests following icv administration of HYWH (500 pmol) and decreased following TIP39 administration. Intrathecally administered HYWH significantly increased the response latency only at a higher dose (2.5 nmol). *P<0.05, ***P<0.001.
The antagonist HYWH differs from TIP39, the PTH2-R's endogenous ligand, at only 4 out of 39 residues. As a test of the specificity of HYWH's effect on nocioception, we evaluated the effects of 500 pmol TIP39 (Figs. 1B and C), the amount of HYWH that significantly increased tail-flick and hot-plate response latency. The response latency of animals that received TIP39 decreased from a mean of 1.5 ± 0.2 s to 1.0 ± 0.1 s in the tail-flick test (Fig. 1C; Student's t-test, df=32, P<0.001) and from 18.0 ± 3.1 s to 13.7 ± 1.8 s in the hot-plate test (Fig. 1D; Student's t-test, df=32, P<0.05). These effects, decreases of 33% in the tail-flick and 24% in the hot-plate test, were in the opposite direction from the effect of HYWH.
Intrathecal administration of HYWH also affected nociceptive responses in the tail-flick (ANOVA, F2, 48=19.17; P<0.001) and hot-plate (ANOVA, F2, 51=15.36; P<0.001) tests, but required a greater dose. Intrathecally, 500 pmol HYWH did not affect the nociceptive response, but 2.5 nmol HYWH increased the responses latency by 133% in the tail-flick test, from 1.8 ± 0.3 to 4.2 ± 1.1 s and by 49% in the hot-plate test, from a baseline of 15.7 ± 2.1 to 23.4 ± 2.6 s (Figs. 1E and F).
There was no obvious effect of HYWH on the general behavior of mice. In some experiments there was a small increase in locomotion following icv infusion of 500 pmol, but there was no behavior that could be interpreted as sedation or deteriorated alertness.
ICV infusion of HYWH decreases nociceptive responses in the formalin test
Hindpaw formalin injection produced the expected biphasic nociceptive response. The response to formalin was decreased in animals administered 500 pmol of HYWH (icv) (Fig. 2). The effect of icv HYWH was much greater during the second phase (nociceptive responses number for vehicle, 90 ± 12.8; HYWH, 44 ± 5.4; Students t-test, df=29, P<0.01) than the first phase, in which the difference between the groups did not reach statistical significance. Intrathecal injection of 2.5 nmol HYWH did not have a significant effect in the formalin test (not shown).
Fig. 2.
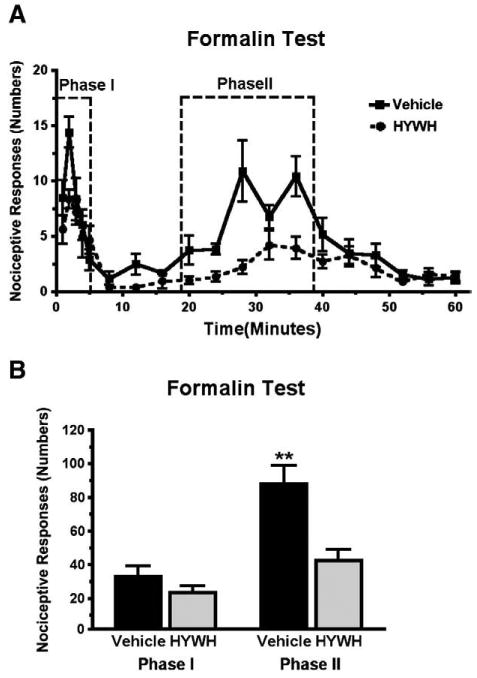
Antinociceptive effect of HYWH in the formalin test. Hindpaw nociceptive responses were counted over 60 min following icv infusion of 500 pmol HYWH and hindpaw formalin injection. A) The response is plotted in one-minute time bins, collected each minute for the first 5 min and then every third minute. B) Comparison of the total number of responses summed in each the two test phases as indicated in panel A. **P<0.01.
Nociception in TIP39-KO mice
TIP39-KO mice had decreased responses in the hot-plate and formalin tests
Nociception by the TIP39-KO mouse line was assessed in the tail-flick test, the tail-immersion test, the hot-plate test, and the hindpaw formalin test. The hot-plate test was performed at three temperatures, because the rate of heating may affect which nociceptors are activated and subsequent signaling pathways (Yeomans et al., 1996). TIP39-KO and WT mice did not differ in response latency in the tail-flick test (Fig. 3A), as reported previously (Fegley et al., 2008) or in the tail-immersion test at 55° (Fig. 3B). The KO animals also did not differ from WT at temperatures of 42 °C and 48 °C in the hot-plate test (Figs. 3C and D). However, the TIP39-KO mice had a decreased response at 55 °C, with a mean latency of 25.6 ± 2.9 a, while WT mice from the same breedings had an average latency of 16.7 ± 1 s, or a difference of 53% (Students t-test, P<0.05, df=27, Fig. 3E). We used the TIP39-KO mice to perform a second test of the specificity of HYWH. Icv infusion of HYWH into TIP39-KO mice was without effect on the response latency in the tail-flick and hot-plate tests, in an experiment in which the antagonist had the previously observed effect in WT mice, and in which the response of the TIP39-KO mice was also the same as previous experiments (Fig. 4).
Fig. 3.
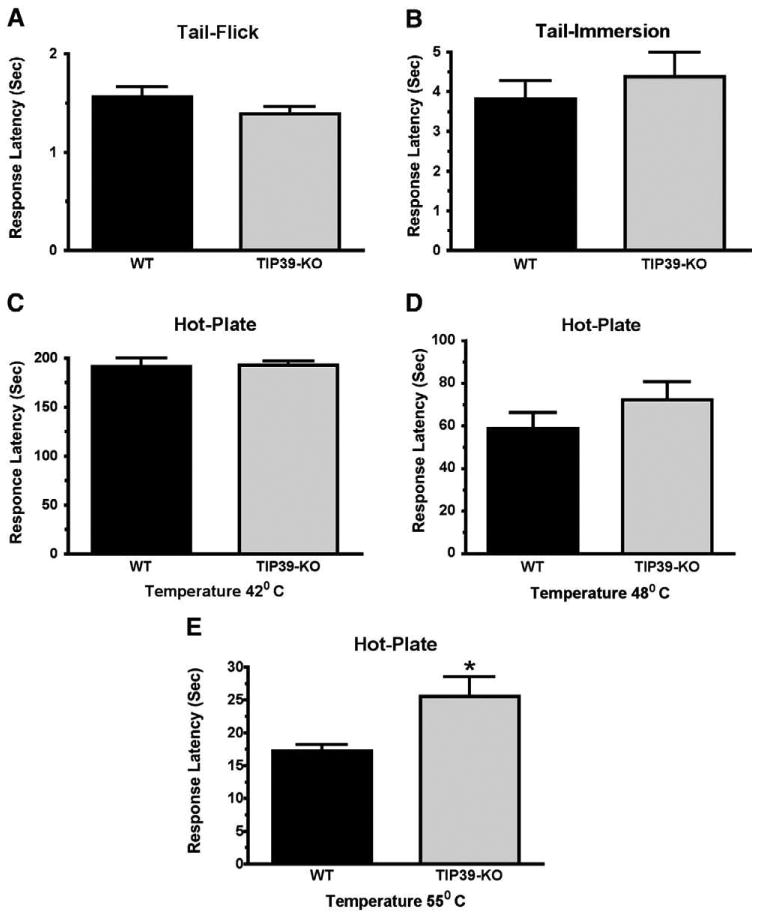
Comparison of the responses of TIP39-KO and WT mice in the tail-flick and hot-plate tests. The difference between TIP39-KO and WT mice was not significant in the tail-flick test (A), tail-immersion test at 55 °C (B) or in the hot-plate test performed at 42 °C (C) or 48 °C (D). The TIP39-KO had significantly greater response latency in the hot-plate test at 55 °C. *P<0.05 (E).
Fig. 4.
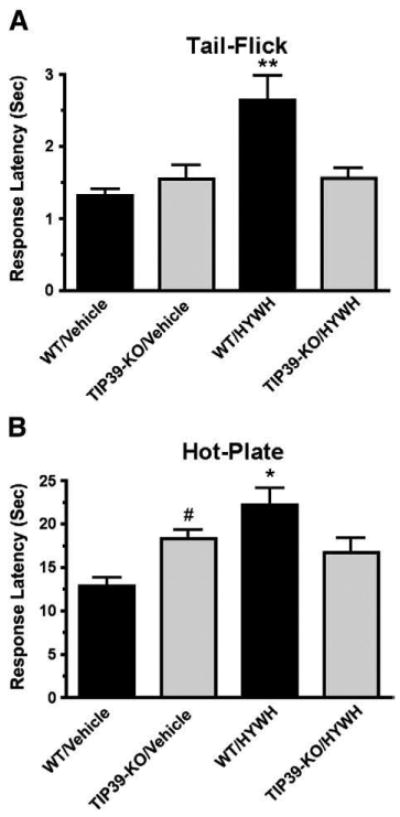
Comparison of the effect of HYWH on WT and TIP39-KO mice in the tail-flick and hot-plate tests. Tail-flick (A) or hot-plate (B) tests were performed following icv infusion of HYWH (500 pmol) or vehicle. HYWH significantly increased response latencies in WT but not TIP39-KO mice. **P<0.01, *P<0.05 for WT/HYWH vs. TIP39-KO/HYWH, #P<0.05 for WT/Vehicle vs. TIP39-KO/vehicle.
TIP39-KO mice responded less to hindpaw formalin injection than WT mice (Fig. 5A). The attenuated response to formalin was present in both phases of the test. In the first phase the mean number of nociceptive responses was 45 ± 6 for WT mice and 25 ± 4.8 for TIP39-KO mice (Student's t-test, P<0.05, df=12, Fig. 5B), while in the second phase the counts were 98 ± 17 in WT versus 50 ± 8.2 in TIP39 KO (Student's t-test, P<0.05, df=12, Fig. 5B).
Fig. 5.
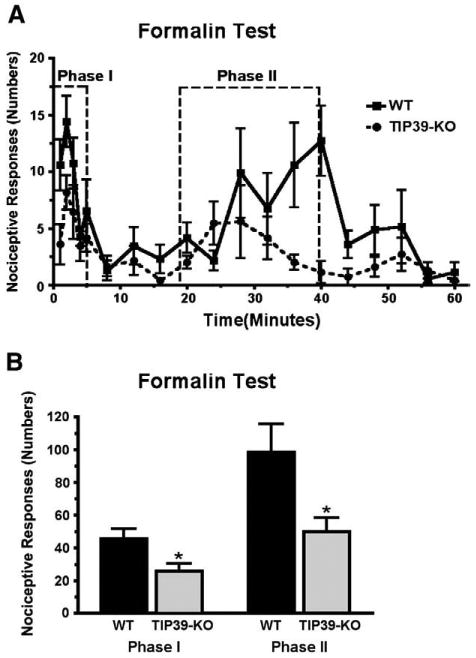
Comparison of TIP39-KO and WT mice in the formalin test. Hindpaw responses were counted over 60 min following formalin injection. A) The response is plotted in one-minute time bins, collected each minute for the first 5 min and then every third minute. B) Comparison of the total number of responses summed in each the two test phases as indicated in panel A. *P<0.05.
Rimonabant eliminates the difference in nociceptive response between TIP39-KO and WT mice
WT and TIP39-KO mice were tested on a 55 °C hot-plate 30 min after injection (intraperitoneal, ip) of the CB1 blocker rimonabant (Fig. 6A). Rimonabant did not affect the response of WT mice at any of the tested doses (0.1–5 mg/kg). However, it caused a dose-dependent increase in the nociceptive response of TIP39-KO mice that was significant at 1 mg/kg and 5 mg/kg, decreasing the latency by 48% and 59% respectively, from 17 ± 1.4 to 10.6 ± 1.5 and to 7 ± 0.7 s (ANOVA, F7,53=11.3, P<0.01 for TIP39-KO/Vehicle vs. TIP39-KO/1 mg/kg and P<0.001 for TIP39-KO/Vehicle vs. TIP39-KO/5 mg/kg, Fig. 6A). Naloxone (1 mg/kg, subcutaneous) did not affect the hot-plate response of the TIP39-KO mice, while the response latency of WT decreased (ANOVA, P<0.05 for WT/Vehicle vs. WT/naloxone, Fig. 6B).
Fig. 6.
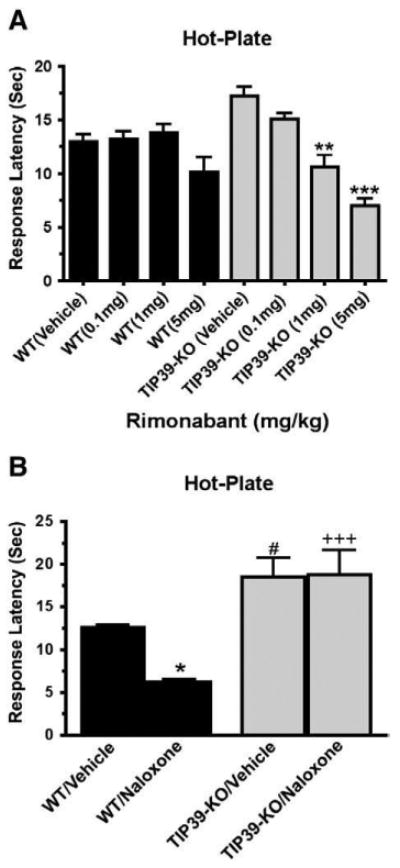
Effect of a CB1 or opioid receptor antagonist on the response of TIP39-KO and WT mice in the hot-plate test. The indicated doses of the CB1 antagonist rimonabant (A) or the opioid receptor antagonist naloxone (B; 1 mg/kg) were administered 30 min before the hot-plate test. The effect of rimonabant was significant only in TIP39-KO mice at 1 mg/kg (**ANOVA, F7,53=11.3, P<0.01) and 5 mg/kg (***ANOVA, F7,53 =11.3, P<0.001). There were significant differences between genotypes in the effect of naloxone and the post hoc test showed differences in WT/Vehicle vs. WT/Naloxone (*P<0.05), WT/Vehicle vs. TIP39-KO/Vehicle (#P<0.05) and WT/Naloxone vs. TIP39-KO/Naloxone (+++P<0.001).
Loss of TIP39 signaling increases stress-induced analgesia and decreases cannabinoid sensitivity
Augmented SIA after icv infusion of HYWH and in TIP39-KO mice
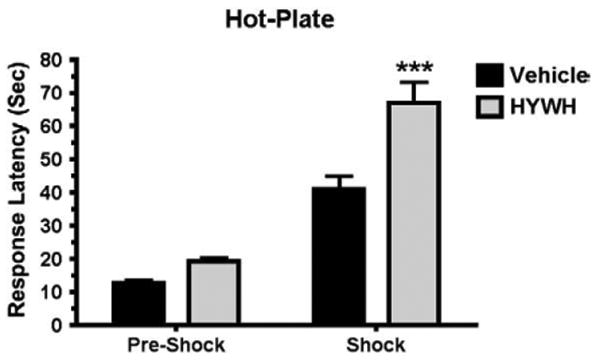
SIA has predominant opioid or non-opioid components, depending upon the type and intensity of the stressor (Mogil et al., 1996; Terman et al., 1986a,b). We adopted a test paradigmin which mice received an injection of vehicle or drug just after a baseline response was measured in the hot-plate test. Inescapable foot shock (0.6 mA for 1 min) was delivered 30 min after the injection and the animals were then retested on the hot-plate. As expected, this paradigm induced SIA in WT as well as the TIP39-KO mice (Fig. 7). The SIA in the KO group was much greater, with a mean response latency of 47.8 ± 6.9 s in comparison to the WT mean of 26 ± 2.6 s (ANOVA, followed by Bonferroni posttest, P<0.001 for WT/Vehicle/Shock vs. TIP39-KO/Vehicle/Shock), which is an increase over pre-shock response latency of 188.7% in WT and 288.9% in the TIP39-KO. Rimonabant (5 mg/kg ip) completely abolished the greater SIA in the knockouts, reducing their response latency to the latency of the WT group (ANOVA, by followed by Bonferroni posttest, P>0.05, for WT/Rimonabant/Shock vs. TIP39-KO/Rimonabant/Shock). Using one-way ANOVA to compare the differences between treatments within a genotype indicated that rimonabant significantly reduced SIA in the WT mice, from 26 ± 2.6 to 18.6 ± 2.4 s response latency (ANOVA, F3,67=21.8,P<0.05) and in the TIP39-KO, from 47.8 ± 6.9 s to 11.5 ± 1.6 s response latency (ANOVA, F3,47=16.1, P<0.01) (Fig. 7).
Fig. 7.
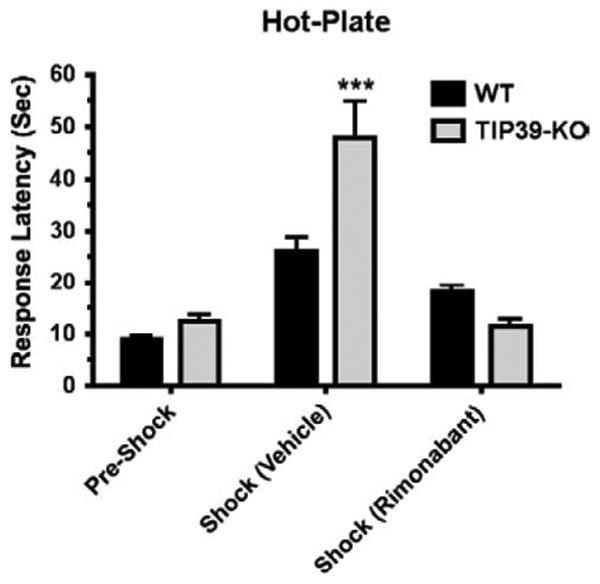
The effect of a CB1 antagonist on stress-induced analgesia in WT and TIP39-KO mice. SIA was much greater in TIP39-KO than WT mice (***Bonferroni posttest, P<0.001), and the SIA in TIP39-KO mice was completely blocked by 5 mg/kg rimonabant (TIP39-KO/shock/rimonabant vs. WT/Pre-Shock or WT/Shock/rimonabant, Bonferroni posttest, P>0.05).
The same experimental design was used to evaluate the effect of intracerebral infusion of HYWH on SIA (500 pmol icv) (Fig. 8). Two-way ANOVA indicated a significant effect of HYWH treatment 1 min before foot shock (P<0.001), a significant effect of foot shock (P<0.001) and significant interaction (P<0.05) between the two (ANOVA, Bonferroni Posttest, P<0.001, for Vehicle/Shock vs. HYWH/Shock). The antinociceptive effect of HYWH infusion was completely eliminated by pretreatment with rimonabant (5 mg/kg ip) (Fig. 9, ANOVA, P>0.05 for Vehicle/Rimonabant vs. HYWH/Rimonabant, P<0.05 for Vehicle/Vehicle vs. Vehicle/HYWH).
Fig. 8.
PTH2R block increases stress-induced analgesia. HYWH (500 pmol, icv) or vehicle was infused just prior to foot shock, followed by the hot-plate test. (Shock/HYWH vs. Shock/vehicle, ***Bonferroni posttest, P<0.001).
Fig. 9.
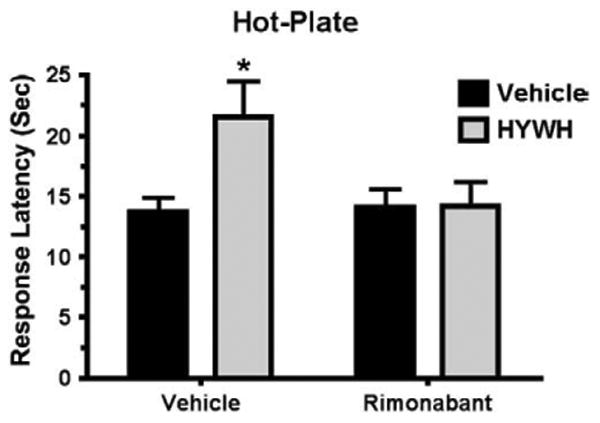
Rimonabant prevents HYWH potentiation of stress-induced analgesia. Mice were treated with rimonabant (5 mg/kg i.p.) or vehicle and 30 min later infused with HYWH (500 pmol, icv) or vehicle, then exposed to foot shock, followed immediately by the hot-plate test. Mice receiving HYWH had greater analgesia without (*Bonferroni Posttest, P<0.05), but not following rimonabant administration (P>0.5 for Vehicle/Rimonabant versus HYWH/Rimonabant).
TIP39-KO mice have decreased responses to a cannabinoid receptor agonist
Cannabinoid agonists produce a well-described tetrad of effects that consists of catalepsy, decreased locomotion, hypoalgesia and hypothermia. We observed the four effects following administration of CP55,940 (0.3 mg/kg i.p.) to WT mice (Fig. 10). In contrast, the only significant effect of CP55,940 observed in TIP39-KO mice was mild catalepsy. The agonist failed to decrease locomotion, increase hotplate response latency or cause hypothermia in TIP39-KO mice at the dose tested. Statistical analysis indicated a significant interaction of genotype with CP55,940 administration in all four tests (catalepsy P<0.001, locomotion P<0.05, nociception, P<0.001 and body temperature P<0.001).
Fig. 10.
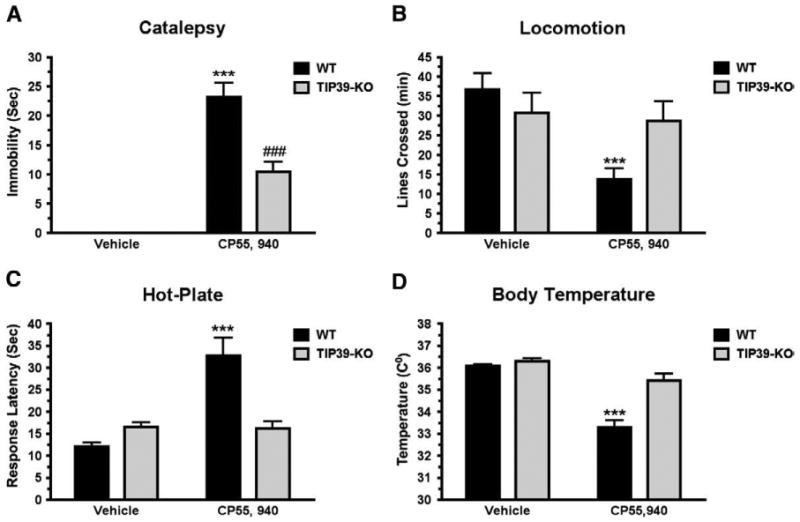
Cannabinoid “tetrad” response in TIP39-KO and WT mice. Tests were performed in the order catalepsy (A), locomotion (B), analgesia on hot-plate (C) and body temperature (D) following administration of the cannabinoid agonist CP55,940 (0.3 mg/kg, i.p.). The only test in which CP55,940 treated TIP39-KO mice differed from TIP39-KO mice receiving vehicle was catalepsy (vehicle treated animals displayed no catalepsy; Bonferroni Posttest, ###P<0.001. WT mice treated with CP55,940 differed from WT receiving vehicle in all four tests ***P<0.001.
CB1mRNA is decreased and FAAH mRNA is increased in the amygdala of TIP39-KO mice
Riboprobes directed at CB1 and FAAH produced labeling patterns similar to that described in the literature (Egertova et al., 2003; Mailleux and Vanderhaeghen, 1992) (Fig. 11). CB1 mRNA signal was present throughout most of the cerebral cortex, striatum, hippocampus (Figs. 11A and B) and amygdaloidal nuclei (Figs. 11C and D) with the typical high and low signal expressing cells. Hybridization to brain sections from a CB1 global knockout mouse (Zimmer et al., 1999) confirmed the specificity of the mRNA signal (not shown). Within the amygdala, the greatest level of CB1 mRNA signal was within the basolateral nucleus (BLA), followed by the basomedial nucleus. The central amygdala had a modest level of expression and all of the labeled cells were of the low signal type (Figs. 11C and D). The strongest FAAH mRNA signal was observed in the hippocampus (Figs. 11E and F) followed by the cerebral cortex and amygdala, where a uniform, dense, FAAH mRNA signal was present throughout the BLA, while a very low intensity signal was present in the central and basomedial nuclei (Figs. 11G and H). TIP39-KO mice had a significantly decreased CB1 mRNA signal in the BLA (by 63.2% ± 11.6; Student's t-test, df=34, P<0.001, Fig. 12I). The decrease included both low and high signal expressing cells and the latter were fewer in number (Student's t-test, df=34, P<0.01, Fig. 11J). The KO mice did not have any change in the CB1 mRNA signal intensity in the hippocampus or the striatum (Figs. 12K and L). In contrast, FAAH mRNA expression was increased by 182% ± 60.4 in the BLA of TIP39-KO animals (Student's t-test, df=17, P<0.01, Fig. 11M). The FAAH mRNA signal intensity was not significantly different in the dentate gyrus of the hippocampus (Fig. 11N). There was an inverse correlation between the changes in CB1 and FAAH mRNA expression level in the BLA, evaluated on adjacent sections (Linear Regression, r2=0.65, F1,16 =27, P<0.001, Fig. 11O).
Fig. 11.
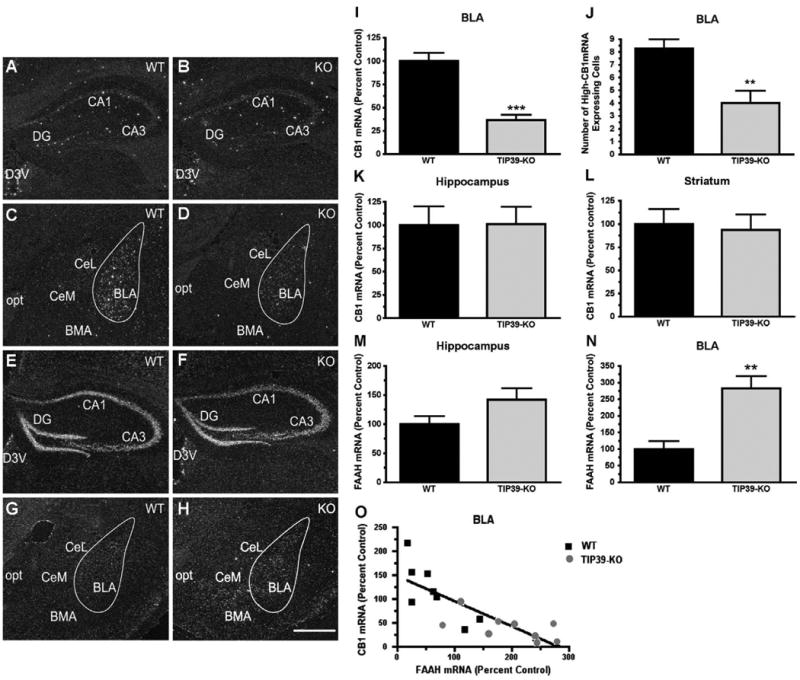
CB1 and FAAH mRNA expression in TIP39-KO and WT mice. Dark field images of emulsion dipped brain sections following in situ hybridization with 35S-riboprobes directed against CB1 or FAAH mRNA. Images from WT animals are in the left column and TIP39-KO mice in the right column. CB1 expression can be seen in the hippocampus (A, B) and in the amygdala (C, D). FAAH expression is shown in the hippocampus (E, F) and amygdala (G, H). Quantifications of the expression levels are shown at the right (I–N). O) Change in CB1 expression is plotted against change in FAAH expression, with each symbol indicating measurement of the two mRNA's on adjacent sections from one animal (Linear regression, Goodness of fit, F = 27, r2 = 0.65, P<0.001). **P<0.01, ***P<0.0001. Abbreviations: BLA – basolateral amygdaloid nucleus, anterior; BMA – basomedial amygdaloid nucleus, anterior; CA1 – field CA1 hippocampus; CA3 – field CA3 hippocampus; CeL – central amygdaloid nucleus, lateral; CeM – central amygdaloid nucleus, medial; D3V – dorsal 3 rd ventricle; DG – dentate gyrus; opt – optic tract. Scale bar – 100 μm.
Fig. 12.
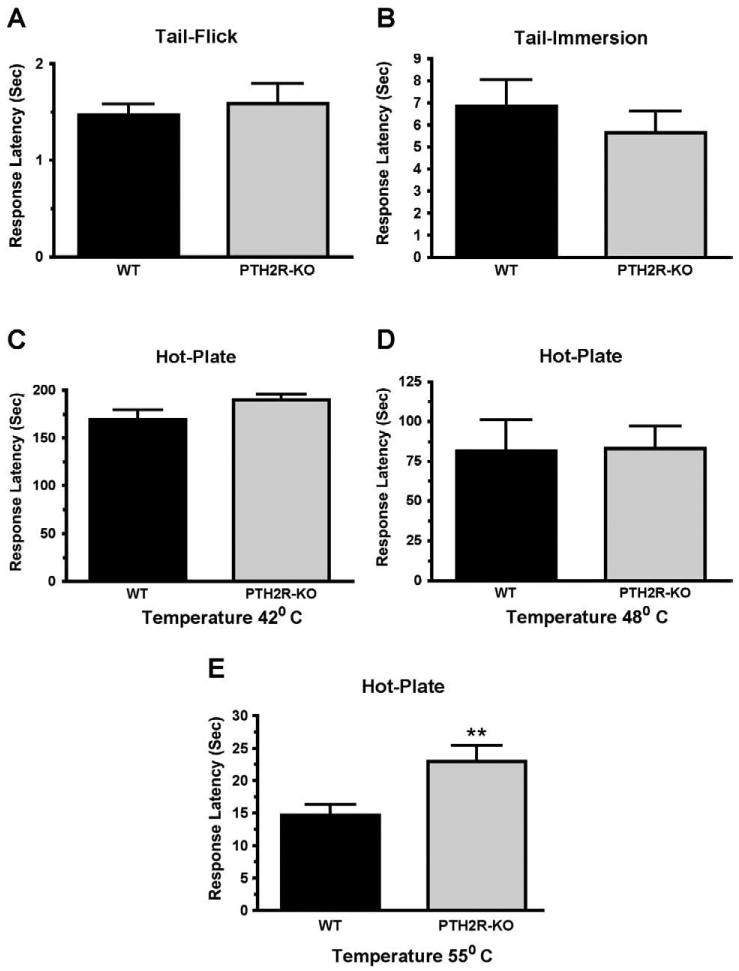
Comparison of the responses of PTH2R-KO and WT mice in the tail-flick, tail-immersion, and hot-plate tests. The difference between PTH2R-KO and WT mice was not significant in the tail-flick (A) or tail-immersion (B, 50 °C) tests, or in the hot-plate test at 42° (C) or 48 °C (D). The PTH2R-KO had significantly greater response latency in the hot-plate test at 55° (E). **P<0.01.
The phenotype of PTH2R-KO mice matches TIP39-KO
PTH2R-KO mice have decreased responses in the hot-plate and formalin tests
Changes in the nociceptive behavior of PTH2R-KO mice were very similar to those of TIP39-KO mice. There was no difference between the response latency of PTH2R-KO and WT mice when tested using either a high intensity light beam in the tail-flick test or in the tail-immersion test at 50° (Figs. 12A and B). Like TIP39-KO mice, PTH2R-KO mice had increased response latency in the hot-plate test at 55 °C (Student's t-test, df=33, P<0.01, Fig. 12E) but not at lower temperatures (Fig. 12F). The results of the formalin test in the receptor KO mice also mirrored the results from the formalin test of the peptide KO mice (Fig. 13). PTH2R-KO had an average response number of 15 ± 3.8 versus 28 ± 3.6 in the WT (Student's t-test, df=12, P<0.01) in phase I and 69 ± 15.3 versus 169 ± 23.9 during phase II (Student's t-test, df=12, P<0.001, Fig. 13B). In PTH2R-KO mice CB1 mRNA was decreased by 60% ± 23.2 and FAAH mRNA was increased by 247% ± 82.6 (Student's t-test, df=10, P<0.05) in the BLA, changes similar to the TIP39-KO mice.
Fig. 13.
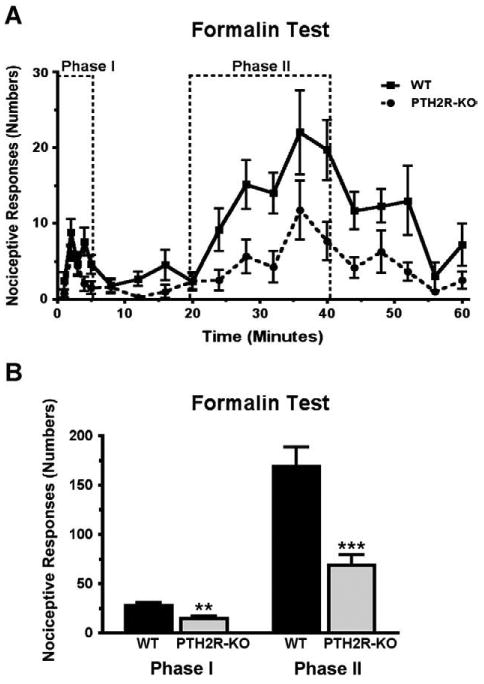
Comparison of WT and PTH2R-KO mice in the formalin test. Hindpaw responses were counted over 60 min following formalin injection. A) The response is plotted in one-minute time bins, collected each minute for the first 5 min and then every third minute. B) Comparison of the total number of responses summed in each the two test phases as indicated in panel A. There were significant differences between the genotypes in both phases **P<0.01, ***P<0.001.
Increased SIA in PTH2R-KO is blocked by rimonabant
Since, in experiments described above, 5 mg/kg (ip) rimonabant decreased the difference between TIP39-KO and WT animals in the hot-plate assay at baseline, and after stress, we evaluated this dose in PTH2R-KO mice (Fig. 14). The results were similar, with a significant main effect of treatment (P<0.001), genotype (P<0.05) and an interaction (P<0.05) in a two-way ANOVA. In WT mice the response latency increased from a baseline of 15.6 ± 2.1 s to 55.3 ± 13.9 s following shock. The increase in response latency following shock was significantly greater in the PTH2R-KO mice (increasing from 21.1 ± 4.5 s to 82.1 ± 11.1 s; 48% greater than the effect in WT), and prior rimonabant administration decreased the effect of shock in the KO animals, bringing the response latency following shock to that of WT mice (ANOVA, Bonferroni Posttest, P<0.01, for WT/Vehicle/Shock vs. PTH2R-KO/Vehicle/Shock, and P<0.05 for WT/Rimonabant/Shock vs. PTH2R-KO/Rimonabant/Shock).
Fig. 14.
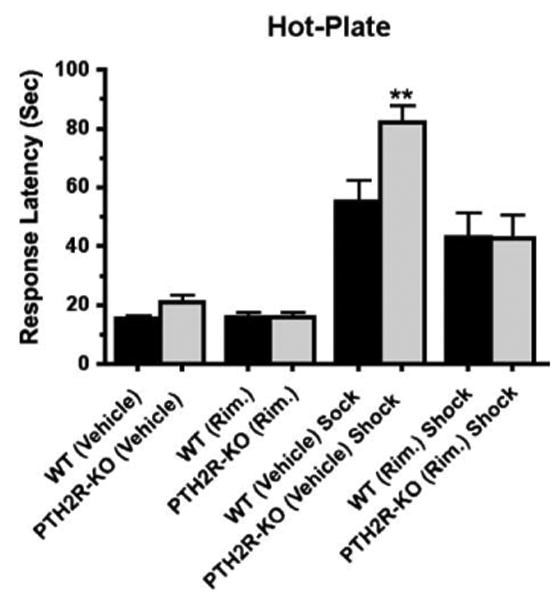
Evaluation of stress-induced analgesia in PTH2R-KO mice. WT or PTH2R-KO mice were given rimonabant (5 mg/kg i.p.) or vehicle and after 30 min subjected to foot shock (or not) and evaluated in the hot-plate test. SIA was significantly greater in PTH2R-KO injected with vehicle than the WT (**Bonferroni Posttest, P<0.01), and this increase was completely blocked by rimonabant (P>0.05).
Cannabinoid agonist administration has similar effects in PTH2R-KO and TIP39-KO mice
Intraperitoneal injection of 0.3 mg/kg CP 55,940 into PTH2R-KO and matched WT mice produced the typical CB1 agonist response tetrad in the WT animals (Fig. 15). This dose produced some catalepsy and a decrease of body temperature in PTH2R-KO but their response to CP 55,940 was very different than the WT, except in body temperature (ANOVA, followed by Bonferroni Posttest, catalepsy P<0.001, locomotion P<0.05, analgesia P<0.001 and body temperature P>0.05 for WT/CP 55,950 vs. PTH2R-KO/CP55 940, Figs. 15A and D). CP55,940 failed to produce analgesia in the PTH2R-KO mice at this dose (ANOVA, significant main effect on genotype (P<0.5), treatment (P<0.001) and interaction (P<0.01).
Fig. 15.
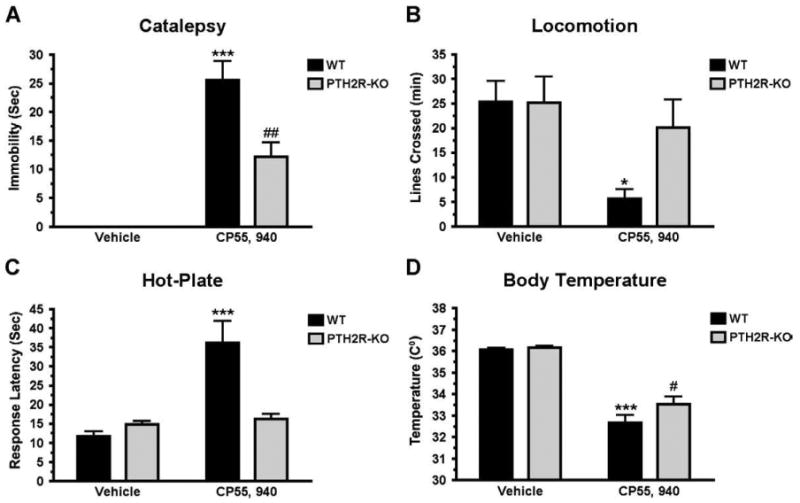
Cannabinoid “tetrad” response in PTH2R-KO and WT mice. Tests were performed in the order catalepsy (A), locomotion (B), analgesia on hot-plate (C) and body temperature (D) following administration of the cannabinoid agonist CP55,940 (0.3 mg/kg, i.p.). Catalepsy (##P<0.01) and body temperature decrease (#P<0.05) were significant in PTH2R-KO mice receiving CP55,940. WT mice treated with CP55,940 differed from WT receiving vehicle in all four tetrad tests – catalepsy (A) (***P<0.001), (B) locomotion (*P<0.05), analgesia (C) (***P<0.001) and body temperature (C) (***P<0.001).
PTH2R-ir colocalizes with VGlut2-ir in amygdaloid nuclei, PAG and spinal cord dorsal horn
PTH2R/VGlut2-ir colocalization was reported previously in the hypothalamus of rat (Dobolyi et al., 2003a,b) and monkey (Bago et al., 2009) but not investigated outside the hypothalamus. Nerve fibers labeled with a PTH2R directed antibody had a distribution like that previously described in rat (Wang et al., 2000), and consistent with the distribution of PTH2R synthesizing cells observed by in situ hybridization in rat and mouse (Faber et al., 2007). PTH2R-ir fibers were abundant in most amygdaloid nuclei, with exception of the centrolateral nucleus (Fig. 16A), and were present in the PAG and the spinal cord dorsal horn (not shown). VGlut2-ir was present in each of these brain regions, as previously described (Kaneko and Fujiyama, 2002; Todd et al., 2003; Varoqui et al., 2002) (Fig. 16B). VGlut2-ir was more widespread than PTH2R-ir, and the VGlut2-ir puncta were more numerous than PTH2R-ir structures, but most of the PTH2-ir colocalized with VGlut2-ir (Figs. 16D, E and F).
Discussion
We found hypoalgesia in mice with decreased TIP39 signaling. Genetic deletion of the peptide's coding sequence, null mutation of its receptor, the PTH2R, and acute icv infusion of a PTH2R antagonist each decreased nociceptive responses. Administration of the CB1 blocker rimonabant dose-dependently reversed the effects of the loss of TIP39 signaling. A dose of the agonist CP55,940 that was an effective analgesic in WT mice was ineffective in KO mice. As discussed below, these results are consistent with the suggestion that TIP39 may normally modulate nociception through effects on endocannabinoid signaling. A predominantly supraspinal site for TIP39's function is suggested by a lower effective dose for the PTH2R antagonist following intracranial than intrathecal administration, and significant decreases in nociceptive responses in the hot-plate and formalin tests, but not the in the tail-flick test, in KO mice. Changes in CB1 and FAAH expression in the amygdala of KO mice are consistent with supraspinal effects of TIP39 that influence endocannabinoid signaling. The data point to a previously unknown mechanism for modulation of CB1 mediated analgesia, as a consequence of TIP39/PTH2R regulation of endocannabinoid activity.
Anatomical localization of TIP39 effects
PTH2-R and TIP39 containing fibers are present at multiple sites that contribute to pain modulation. Evidence in favor of a predominantly supraspinal location for the altered nociceptive processing in the KO mice, and for involvement of endogenous TIP39 in nociception, is provided by a lower effective dose for intracranial than for intrathecal HYWH administration, and by decreased responses of KO mice in the hot-plate test, which is thought to have a greater supraspinal component than the tail-flick test (Jensen and Yaksh, 1986) in which neither KO line differed from WT littermates. A supraspinal location for TIP39 effects on nociception is also suggested by the greater SIA in KO than in WT mice. This points to effects on descending modulation, rather than the sensory limb of nociception.
Intrathecal administration of an antibody to TIP39 has previously been shown to increase the response latencies in the tail-flick test and in response to plantar pressure, and intrathecal administration of TIP39 to potentiate nociceptive responses in the same assays (Dobolyi et al., 2002). These hypoalgesic effects of an antibody to TIP39, and pro-nociceptive effects of the peptide, are consistent with the effects we observe following administration of HYWH in this study, and imply that endogenous TIP39 makes some contribution to nociception at the spinal level. PTH2-Rimmunoreactive fibers are abundant in outer lamina of the spinal cord dorsal horn and are probably the processes of dorsal root ganglion neurons and of dorsal horn neurons that express PTH2-R mRNA (Dobolyi et al., 2002). Recent evidence suggests that the PTH2R expressing dorsal root ganglia cells may correspond to A-delta fibers (Matsumoto et al., 2010), but little is known about the spinal cord neurons. Only a few fibers containing TIP39 immunoreactivity are observed in the spinal cord (Dobolyi et al., 2003a,b). In contrast, TIP39-, as well as PTH2-R-like, immunoreactivity is abundant in a number of areas that contribute to the supraspinal mediation of pain, including the parabrachial nuclei, midline thalamic nuclei, amygdala, hypothalamus, and periaqueductal gray (Bago et al., 2009; Faber et al., 2007; Wang et al., 2000). The TIP39 fibers that project to each of these PTH2-R rich regions have been mapped to neurons in the medial paralemniscal nucleus, a distinct brainstem structure, or to neurons at the caudal border of the thalamus in the periventricular gray or posterior intralaminar nucleus (Dobolyi et al., 2010; Wang et al., 2006). Thus TIP39 may act simultaneously to modulate nociception at several sites. A high degree of colocalization between PTH2R- and VGlut2-ir suggests that PTH2R expressing cells are glutamatergic, and thus TIP39 may act by modulation of glutamatergic transmission in areas that contribute to descending modulation of nociception.
PTH2Rs are present in a number of peripheral tissues, but except for primary afferent neurons, it is not likely that loss of TIP39 effects at any of these sites would explain the nociception-related phenotype of the KO mice. Effects of TIP39 on contractility have been described in an isolated heart preparation (Ross et al., 2005), but we found no difference between WT and TIP39-KO mice in a number of cardiovascular parameters and no effect of intravenous TIP39 on heart rate or blood pressure in urethane anesthetized cannulated WT mice (Fegley et al., 2008). Since HYWH was administered icv and intrathecally, its effects are most likely to be central, and because of the similarity between the phenotypes of TIP39-KO and PTH2R-KO mice and the HYWH effects it seems unlikely that effects on the peripheral terminals of primary afferent neurons, or other peripheral actions make a major contribution to the observed effects.
Involvement of endocannabinoid signaling in TIP39 effects
One of the unexpected findings of this study is the discovery of a functional link between the TIP39/PTH2R system and the endogenous cannabinoid system in pain modulation. Blocking CB1 receptors by rimonabant completely eliminated the hypoalgesic effects of HWYH infusion, and the hypoalgesia of TIP39-KO and PTH2R-KO mice present in the hot-plate test.
Increased signaling via endocannabinoid or endogenous opioid systems may contribute to SIA (Lewis et al., 1980; Connell et al., 2006; Hohmann et al., 2005; Terman et al., 1986a,b; Valverde et al., 2000). SIA induced by high intensity stressful stimuli (Mogil et al., 1996; Terman et al., 1986a,b), including inescapable foot shock, has been shown to have a predominant non-opioid component in rat (Lewis et al., 1980; Terman et al., 1986a,b, 1984). Rimonabant lessened the SIA in WT mice in the paradigm we used, while naloxone had no effect (data not shown). Most striking, however, was the observation that SIA was much greater after infusion of the PTH2-R antagonist, and in the KO mice in comparison to WT mice. The inhibitory effect of rimonabant on the SIA was much greater in the knockouts indicating greater sensitivity to CB1 block. Thus, the significant augmentation of rimonabant-sensitive SIA observed following infusion of HYWH, or present in the TIP39-KO and PTH2R-KO mice, suggests that endogenous TIP39, via the PTH2R, may negatively modulate an effect of endogenous cannabinoids on pain.
Further support for a functional link between endocannabinoids and TIP39 signaling comes from the difference between the responses of WT and KO mice to the cannabinoid agonist CP 55,940. The tetrad of analgesia, hypolocomotion, catalepsy and hypothermia, is well described as in vivo assay of CB1 agonist effects (De Vry et al., 2004; Long et al., 2009; Martin et al., 1991). TIP39-KO and PTH2R-KO mice responded very differently than WT mice to CP 55,940, consistent with down regulation of CB1. Systemic injection of CP 55,940 caused catalepsy in the KO lines that was qualitatively similar to the effect in WT mice, but occupied less of the test period, indicating decreased sensitivity of the KO mice. The drug did not decrease locomotion or cause analgesia in the KO at a dose that was effective in WT mice. Body temperature was not affected at all in the TIP39-KO and was decreased by much less in the PTH2R-KO mice than in WT. Maintained catalepsy in the KO mice could be explained by a major contribution to catalepsy of cannabinoid interaction with the striatal dopaminergic system (Gough and Olley, 1977; Sanudo-Pena et al., 1999), in combination with the absence of PTH2R expression in the basal ganglia (Faber et al., 2007; Wang et al., 2000). Brain areas with major roles in temperature regulation (medial preoptic area) and pain control (anterior cingulate cortex, amygdala, PAG, raphe nuclei and spinal cord dorsal horn) have abundant PTH2R expression (Faber et al., 2007; Wang et al., 2000). Thus, endocannabinoid function may be affected in the KO mice only in brain areas that are under the influence of TIP39/PTH2R signaling. The reason for the difference in the effect of CP 55,940 on temperature in TIP39-KO and PTH2R-KO mice is not clear. TIP39 mRNA encodes a signal peptide with a signal peptide cleavage site, followed by an uncharacterized amino acid sequence, two basic residues, and then the purified and characterized active TIP39 peptide (Dobolyi et al., 2002). Loss of the uncharacterized intercalated peptide, which based on its sequence and unpublished experiments is unlikely to act on the PTH2R, could potentially contribute to differences in the phenotype of TIP39-KO and PTH2R-KO mice. Taken together, the response of TIP39-KO and PTH2R-KO animals to the cannabinoid antagonist and agonist could be explained by fewer available CB1 receptors in nociception processing brain areas. This explanation for the KO's responses to the drugs is supported by changes in CB1 mRNA level in the BLA, but not in the striatum and hippocampus. CB1 binding and mRNA are down regulated in many brain areas, including the amygdala, following chronic administration of cannabinoid agonists (Martini et al., 2010; Oviedo et al., 1993; Sim-Selley and Martin, 2002). Thus, the decreased CB1 mRNA expression in the BLA of KO mice could potentially reflect down regulation, resulting from increased levels of endocannabinoids in this nucleus.
The suggestion that TIP39-KO mice have increased endocannabinoid release in the amygdala is supported by the correlation between decreased CB1 and increased FAAH mRNA in the KO mice. While we are not aware that a causative relationship between endocannabinoid levels and FAAH expression has been demonstrated, it is consistent with the common theme of increased levels of a substrate leading to increased synthesis of enzymes required for its degradation. In addition to anandamide, FAAH metabolizes other proanalgesic endocannabinoids including 2-arachidonoylglycerol and oleoyl ethanolamide (Jhaveri et al., 2006; Maione et al., 2006), so the present data do not implicate a particular endocannabinoid. We speculate that altered endocannabinoid function in the BLA, as a consequence of the absence of TIP39 signaling, could be the functional basis for the nociceptive phenotype of TIP39-KO and PTH2R-KO mice.
Rimonabant and CP55,940 have off-target effects that could potentially contribute to the effects observed. However, this seems unlikely because the effects observed are on nociception, and CB1 involvement in nociception is well established. In addition, because the cannabinoid drugs were administered systemically their site of action is not known. Cannabinoid effects on SIA are thought to be entirely supraspinal, and CB1 and FAAH expression were changed in the amygdala of TIP39 and PTH2R KO mice, so while a peripheral site cannot be excluded, a supraspinal site, potentially the amygdala, can explain the observed effects.
Comparison with previous studies of TIP39 signaling in nociception
In a previous study we did not find any difference between the nociceptive behavior of TIP39-KO and WT mice in tail-flick and hot-plate tests (Fegley et al., 2008). We also did not find any difference in the tail-flick test between the WT and TIP39-KO mice or between PTH2R-KO and WT in this study. However, the antagonist HYWH had an antinociceptive effect when 2.5 nmol was injected intrathecally, but not when 500 pmol was administered, consistent with the previous study in which intrathecal administration of an antibody to TIP39 attenuated responses and TIP39 potentiated responses in the tail-flick test (Dobolyi et al., 2002). Our interpretation is that TIP39/PTH2R signaling at the spinal level probably has a relatively small influence on this test, and observation of it is highly dependent on the experimental conditions. There are likely to be compensatory effects in the KO mice that obscure it.
In contrast to a previous study, we did find a significant effect of TIP39 deletion on the animal's nociceptive response in the hot-plate test. This discrepancy may be explained by several factors that differ between the studies. The mice were singly housed for this study and given ample time for acclimatization to the test room. In addition, the accuracy of the experiment was improved by conducting the experiment with two observers and with analysis of video recorded sessions, which confirmed the results of live monitoring. The average response latency increased for WT mice from 8.0 ± 0.6 s in the previous study to 16.7 ± 1 s in this study. Finally, an equivalent finding from TIP39-KO and PTH2R-KO mice gives us confidence in the observation (Fig. 17).
Fig. 17.
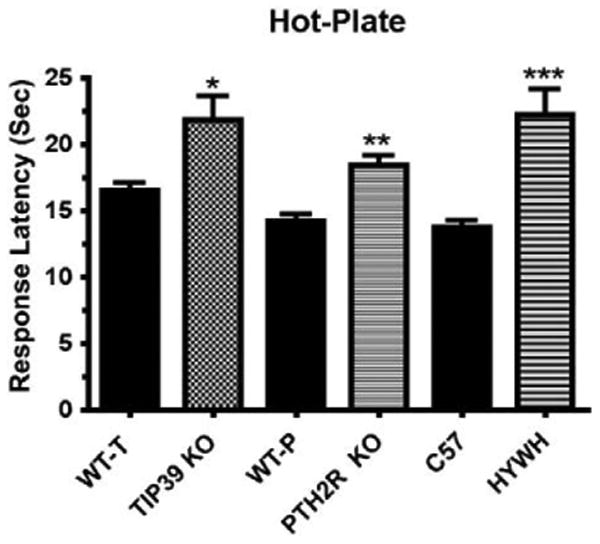
Responses of KO mice, littermate WT mice and C57Bl/6 J mice treated with HYWH (500 pmol, icv) or vehicle are compared in the 55 °C hot-plate test. Data are combined from hot-plate tests shown in Figs. 1B, 4D and 13E. (Statistical comparisons are fully explained in the original figures. The KO groups are compared with corresponding WT and the HYWH group with vehicle injected mice. *P<0.05, **P<0.01, ***P<0.001).
Comparison of acute block of TIP39 signaling and global peptide or receptor knockout
The results from HYWH infusion in WT mice were consistent with the differences observed between TIP39-KO or PTH2R-KO mice and WT mice, with only a few apparent exceptions. The first exception is the tail-flick test, in which the KO mice did not show any difference from the WT, but icv infusion of HYWH was hypoalgesic. While responses in tail-flick, hot-plate, and formalin tests are all subject to descending modulation, the circuits involved and/or thresholds for engagement must be somewhat different. We suggest that compensatory effects, in combination with less TIP39 influence on the tail-flick response, which has less supraspinal contribution at baseline, are responsible for the lack of change in the tail-flick test in the KO mice. The second difference between the results of HYWH infusion and the phenotype of the TIP39-KO and PTH2R-KO mice is that the HYWH infusion did not significantly decrease the pain response in the first phase of the formalin test, but both knockouts showed somewhat decreased responses in the first 5 min. Based on the distribution of TIP39 and the PTH2R, the endogenous peptide and the infused antagonist can potentially act to influence the response to formalin injection at forebrain, brainstem and spinal levels. The relatively slow diffusion rate of this 39-residue polypeptide could prevent HYWH from achieving a high enough concentration at the spinal level immediately after the formalin injection. HYWH-infused, TIP39-KO, and PTH2R-KO mice had identical attenuated pain responses in the second phase of the formalin test, the phase of inflammation (Hunskaar and Hole, 1986) and central sensitization (Dickenson and Sullivan, 1987). Since there was a decrease in phase I following HYWH infusion, which did not reach statistical significance, it is also possible that this apparent difference simply reflects a limitation in the statistical power of the icv infusion experiments.
Speculations on mechanisms underlying effects of modified TIP39 signaling
The changes in CB1 and FAAH mRNA expression patterns in the BLA provide an anatomical focal point for a plausible explanation of the nociceptive and stress-related behavior of TIP39-KO mice. TIP39-KO were hypo-responsive to high intensity thermal and chemical nociceptive stimuli, had exaggerated SIA, and in a previous study had increased fear and stress-related anxiety-like behavior (Fegley et al., 2008). A major role of the BLA in regulation of nociception, SIA, and fear and anxiety is well documented (Connell et al., 2006; Fox and Sorenson, 1994; Goncalves et al., 2008; Rogan et al., 1997). The BLA is also necessary for peripherally administered cannabinoids to have an effect on descending modulation of nociception (Manning et al., 2001). Intracerebral infusion of cannabinoid agonists into the BLA results in analgesia (Martin et al., 1999) and increases SIA (Connell et al., 2006). In our experiments, rimonabant, which does not inhibit SIA at the spinal level (Suplita et al., 2006), suppressed the difference between the TIP39-KO and the WT nociceptive behavior. Our speculation for the mechanism underlying the observed changes in BLA CB1 and FAAH mRNA is based on the presence of presynaptic, inhibitory, CB1 receptors on both GABAergic and glutamatergic neurons (Domenici et al., 2006; Katona et al., 2001, 2006; Marsicano and Lutz, 1999). Amygdaloid nuclei with major input to the BLA, including the lateral, basal and medial nuclei, and the central amygdaloid nucleus a major output of the BLA, all contain a dense network of PTH2R fibers and cells (Faber et al., 2007). Double labeling shows colocalization between the PTH2-R and VGlut2 in several brain areas (Dobolyi et al., 2006). In cells, in which it has been investigated, activation of the PTH2R increases cAMP accumulation and intracellular Ca2+ (Behar et al., 1996; Della Penna et al., 2003; Goold et al., 2001), so we infer that it is likely to facilitate neurotransmitter release. Thus, if TIP39 signaling contributes to the fine-tuning of glutamate release, its loss could lead to compensatory increased retrograde endocannabinoid signaling at glutamatergic and GABAergic synapses. Direct measurement of endocannabinoid and glutamate function in response to TIP39, HYWH, in KO mice, and following nociceptive stimuli will be required to test these hypotheses.
Conclusion
In summary, this study provides evidence that the endogenous peptide TIP39 modulates nociception. It may act in multiple brain regions. Current evidence suggests that its major effects are supraspinal. This study also provides evidence for a previously unknown pathway for functional cross talk between components of glutamatergic and endocannabinoid circuitry, via TIP39 signaling. We hypothesize that TIP39 signaling at a supraspinal level contributes to nociceptive sensitivity by modulating endocannabinoid activity. The data also suggest that TIP39 modulates stressor-related function in the amygdala as previous reports implied (Fegley et al., 2008; LaBuda et al., 2004). Thus, TIP39 may contribute to amygdalar processing of anxiety and stress generated by nociceptive signaling.
Acknowledgments
This work was supported by the Intramural Program of the National Institute of Mental Health, NIH. Jonathan Kuo, Milan Rusnak, and Dean Choi made valuable contributions to the work. We thank Laurence Coutellier for comments on the manuscript.
Abbreviations
- BLA
basolateral amygdaloid nucleus
- CB1
cannabinoid receptor 1
- FAAH
fatty acid amide hydrolase
- HYWH
Histidine3-Tyrosine4-Tryptophan5-Histidine6-TIP39
- icv
intracerebroventricular
- ip
intraperitoneal
- MPL
medial paralemniscal nucleus
- PAG
periaqueductal gray
- PTH2R
parathyroid hormone receptor 2
- SIA
Stress-induced analgesia
- SPF
subparafascicular area
- TIP39
tuberoinfundibular peptide of 39 residues
- VGlut2
vesicular glutamate transporter 2
Contributor Information
Eugene L. Dimitrov, Email: dimitrove@mail.nih.gov.
Emily Petrus, Email: petruse@umd.edu.
Ted B. Usdin, Email: usdint@mail.nih.gov.
References
- Almeida TF, Roizenblatt S, Tufik S. Afferent pain pathways: a neuroanatomical review. Brain Res. 2004;1000:40–56. doi: 10.1016/j.brainres.2003.10.073. [DOI] [PubMed] [Google Scholar]
- Bago AG, Dimitrov E, Saunders R, Seress L, Palkovits M, Usdin TB, Dobolyi A. Parathyroid hormone 2 receptor and its endogenous ligand tuberoinfundibular peptide of 39 residues are concentrated in endocrine, viscerosensory and auditory brain regions in macaque and human. Neuroscience. 2009;162:128–147. doi: 10.1016/j.neuroscience.2009.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon AW, Malmberg AB. Models of Nociception: Hot-Plate, Tail-Flick, and Formalin Tests in Rodents. Current Protocols in Neuroscience. 2007;41:8.9.1–8.9.16. doi: 10.1002/0471142301.ns0809s41. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar V, Pines M, Nakamoto C, Greenberg Z, Bisello A, Stueckle SM, Bessalle R, Usdin TB, Chorev M, Rosenblatt M, Suva LJ. The human PTH2 receptor: binding and signal transduction properties of the stably expressed recombinant receptor. Endocrinology. 1996;137:2748–2757. doi: 10.1210/endo.137.7.8770894. [DOI] [PubMed] [Google Scholar]
- Cliffer KD, Burstein R, Giesler GJ., Jr Distributions of spinothalamic, spinohypothalamic, and spinotelencephalic fibers revealed by anterograde transport of PHA-L in rats. J Neurosci. 1991;11:852–868. doi: 10.1523/JNEUROSCI.11-03-00852.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell K, Bolton N, Olsen D, Piomelli D, Hohmann AG. Role of the basolateral nucleus of the amygdala in endocannabinoid-mediated stress-induced analgesia. Neurosci Lett. 2006;397:180–184. doi: 10.1016/j.neulet.2005.12.008. [DOI] [PubMed] [Google Scholar]
- De Vry J, Jentzsch KR, Kuhl E, Eckel G. Behavioral effects of cannabinoids show differential sensitivity to cannabinoid receptor blockade and tolerance development. Behav Pharmacol. 2004;15:1–12. doi: 10.1097/00008877-200402000-00001. [DOI] [PubMed] [Google Scholar]
- Della Penna K, Kinose F, Sun H, Koblan KS, Wang H. Tuberoinfundibular peptide of 39 residues (TIP39): molecular structure and activity for parathyroid hormone 2 receptor. Neuropharmacology. 2003;44:141–153. doi: 10.1016/s0028-3908(02)00335-0. [DOI] [PubMed] [Google Scholar]
- Dickenson AH, Sullivan AF. Peripheral origins and central modulation of subcutaneous formalin-induced activity of rat dorsal horn neurones. Neurosci Lett. 1987;83:207–211. doi: 10.1016/0304-3940(87)90242-4. [DOI] [PubMed] [Google Scholar]
- Dimitrov E, Usdin TB. Tuberoinfundibular peptide Of 39 residues modulates the mouse hypothalamic-pituitary-adrenal axis via paraventricular glutamatergic neurons. J Comp Neurol. doi: 10.1002/cne.22462. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobolyi A, Ueda H, Uchida H, Palkovits M, Usdin TB. Anatomical and physiological evidence for involvement of tuberoinfundibular peptide of 39 residues in nociception. Proc Natl Acad Sci U S A. 2002;99:1651–1656. doi: 10.1073/pnas.042416199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobolyi A, Palkovits M, Bodnar I, Usdin TB. Neurons containing tuberoinfundibular peptide of 39 residues project to limbic, endocrine, auditory and spinal areas in rat. Neuroscience. 2003a;122:1093–1105. doi: 10.1016/j.neuroscience.2003.08.034. [DOI] [PubMed] [Google Scholar]
- Dobolyi A, Palkovits M, Usdin TB. Expression and distribution of tuberoinfundibular peptide of 39 residues in the rat central nervous system. J Comp Neurol. 2003b;455:547–566. doi: 10.1002/cne.10515. [DOI] [PubMed] [Google Scholar]
- Dobolyi A, Irwin S, Wang J, Usdin TB. The distribution and neurochemistry of the parathyroid hormone 2 receptor in the rat hypothalamus. Neurochem Res. 2006;31:227–236. doi: 10.1007/s11064-005-9011-9. [DOI] [PubMed] [Google Scholar]
- Dobolyi A, Palkovits M, Usdin TB. The TIP39–PTH2 receptor system: unique peptidergic cell groups in the brainstem and their interactions with central regulatory mechanisms. Prog Neurobiol. 2010;90:29–59. doi: 10.1016/j.pneurobio.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, Zieglgansberger W, Lutz B, Rammes G. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. 2006;26:5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egertova M, Cravatt BF, Elphick MR. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- Faber CA, Dobolyi A, Sleeman M, Usdin TB. Distribution of tuberoinfundibular peptide of 39 residues and its receptor, parathyroid hormone 2 receptor, in the mouse brain. J Comp Neurol. 2007;502:563–583. doi: 10.1002/cne.21330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley DB, Holmes A, Riordan T, Faber CA, Weiss JR, Ma S, Batkai S, Pacher P, Dobolyi A, Murphy A, Sleeman MW, Usdin TB. Increased fear- and stress-related anxiety-like behavior in mice lacking tuberoinfundibular peptide of 39 residues. Genes Brain Behav. 2008;7:933–942. doi: 10.1111/j.1601-183X.2008.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM. Anatomy and physiology of a nociceptive modulatory system. Philos Trans R Soc Lond B Biol Sci. 1985;308:361–374. doi: 10.1098/rstb.1985.0037. [DOI] [PubMed] [Google Scholar]
- Fox RJ, Sorenson CA. Bilateral lesions of the amygdala attenuate analgesia induced by diverse environmental challenges. Brain Res. 1994;648:215–221. doi: 10.1016/0006-8993(94)91120-7. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; New York, NY: 2008. [Google Scholar]
- Goncalves L, Silva R, Pinto-Rriberio F, Pego JM, Bessa JM, Petrovaara A, Sousa N, Almeida A. Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp Neurol. 2008;213:48–56. doi: 10.1016/j.expneurol.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Goold CP, Usdin TB, Hoare SR. Regions in rat and human parathyroid hormone (PTH) 2 receptors controlling receptor interaction with PTH and with antagonist ligands. J Pharmacol Exp Ther. 2001;299:678–690. [PubMed] [Google Scholar]
- Gough AL, Olley JE. delta9-Tetrahydrocannabinol and the extrapyramidal system. Psychopharmacology (Berl) 1977;54:87–99. doi: 10.1007/BF00426547. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Hunskaar S, Hole K. The formalin test in mice: dissociation between inflamatory and non-inflamatory pain. Pain. 1986;30:103–114. doi: 10.1016/0304-3959(87)90088-1. [DOI] [PubMed] [Google Scholar]
- Jensen TS, Yaksh TL. Comparison of antinociceptive action of morphine in the periaqueductal gray, medial and paramedial medulla in rat. Brain Res. 1986;363:99–113. doi: 10.1016/0006-8993(86)90662-1. [DOI] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J Neurosci. 2006;26:13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T, Fujiyama F. Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci Res. 2002;42:243–250. doi: 10.1016/s0168-0102(02)00009-3. [DOI] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo J, Usdin TB. Development of a rat parathyroid hormone 2 receptor antagonist. Peptides. 2007;28:887–892. doi: 10.1016/j.peptides.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBuda CJ, Usdin TB. Tuberoinfundibular peptide of 39 residues decreases pain-related affective behavior. Neuroreport. 2004;15:1779–1782. doi: 10.1097/01.wnr.0000134849.63755.15. [DOI] [PubMed] [Google Scholar]
- LaBuda CJ, Dobolyi A, Usdin TB. Tuberoinfundibular peptide of 39 residues produces anxiolytic and antidepressant actions. Neuroreport. 2004;15:881–885. doi: 10.1097/00001756-200404090-00030. [DOI] [PubMed] [Google Scholar]
- Lewis JW, Cannon JT, Liebeskind JC. Opioid and nonopioid mechanisms of stress analgesia. Science. 1980;208:623–625. doi: 10.1126/science.7367889. [DOI] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A. 2009;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailleux P, Vanderhaeghen JJ. Distribution of neuronal cannabinoid receptor in the adult rat brain: a comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience. 1992;48:655–668. doi: 10.1016/0306-4522(92)90409-u. [DOI] [PubMed] [Google Scholar]
- Maione S, Bisogno T, de Novellis V, Palazzo E, Cristino L, Valenti M, Petrosino S, Guglielmotti V, Rossi F, Di Marzo V. Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J Pharmacol Exp Ther. 2006;316:969–982. doi: 10.1124/jpet.105.093286. [DOI] [PubMed] [Google Scholar]
- Manning BH, Merin NM, Meng ID, Amaral DG. Reduction in opioid- and cannabinoid-induced antinociception in rhesus monkeys after bilateral lesions of the amygdaloid complex. J Neurosci. 2001;21:8238–8246. doi: 10.1523/JNEUROSCI.21-20-08238.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, Johnson MR, Melvin LS, Mechoulam R, Ward SJ. Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs. Pharmacol Biochem Behav. 1991;40:471–478. doi: 10.1016/0091-3057(91)90349-7. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Coffin PO, Attias E, Balinsky M, Tsou K, Walker JM. Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Res. 1999;822:237–242. doi: 10.1016/s0006-8993(98)01368-7. [DOI] [PubMed] [Google Scholar]
- Martini L, Thompson D, Kharazia V, Whistler JL. Differential regulation of behavioral tolerance to WIN55,212-2 by GASP1. Neuropsychopharmacology. 2010;35:1363–1373. doi: 10.1038/npp.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason P. Ventromedial medulla: pain modulation and beyond. J Comp Neurol. 2005;493:2–8. doi: 10.1002/cne.20751. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Kondo S, Usdin TB, Ueda H. Parathyroid hormone 2 receptor is a functional marker of nociceptive myelinated fibers responsible for neuropathic pain. J Neurochem. 2010;112:521–530. doi: 10.1111/j.1471-4159.2009.06473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Sternberg WF, Balian H, Liebeskind JC, Sadowski B. Opioid and nonopioid swim stress-induced analgesia: a parametric analysis in mice. Physiol Behav. 1996;59:123–132. doi: 10.1016/0031-9384(95)02073-x. [DOI] [PubMed] [Google Scholar]
- Oviedo A, Glowa J, Herkenham M. Chronic cannabinoid administration alters cannabinoid receptor binding in rat brain: a quantitative autoradiographic study. Brain Res. 1993;616:293–302. doi: 10.1016/0006-8993(93)90220-h. [DOI] [PubMed] [Google Scholar]
- Palkovits M, Helfferich F, Dobolyi A, Usdin TB. Acoustic stress activates tuberoinfundibular peptide of 39 residues neurons in the rat brain. Brain Struct Funct. 2009;214:15–23. doi: 10.1007/s00429-009-0233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Ross G, Engel P, Abdallah Y, Kummer W, Schluter KD. Tuberoinfundibular peptide of 39 residues: a new mediator of cardiac function via nitric oxide production in the rat heart. Endocrinology. 2005;146:2221–2228. doi: 10.1210/en.2004-1180. [DOI] [PubMed] [Google Scholar]
- Sanudo-Pena MC, Tsou K, Walker JM. Motor actions of cannabinoids in the basal ganglia output nuclei. Life Sci. 1999;65:703–713. doi: 10.1016/s0024-3205(99)00293-3. [DOI] [PubMed] [Google Scholar]
- Schafer MK, Varoqui H, Defamie N, Weihe E, Erickson JD. Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem. 2002;277:50734–50748. doi: 10.1074/jbc.M206738200. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Martin BR. Effect of chronic administration of R-(+)-[2, 3-Dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1, 2, 3-de]-1, 4-b enzoxazinyl]-(1-naphthalenyl)methanone mesylate (WIN55, 212–2) or delta(9)-tetrahydrocannabinol on cannabinoid receptor adaptation in mice. J Pharmacol Exp Ther. 2002;303:36–44. doi: 10.1124/jpet.102.035618. [DOI] [PubMed] [Google Scholar]
- Sugimura Y, Murase T, Ishizaki S, Tachikawa K, Arima H, Miura Y, Usdin TB, Oiso Y. Centrally administered tuberoinfundibular peptide of 39 residues inhibits arginine vasopressin release in conscious rats. Endocrinology. 2003;144:2791–2796. doi: 10.1210/en.2002-0017. [DOI] [PubMed] [Google Scholar]
- Suplita RL, II, Gutierrez T, Fegley D, Piomelli D, Hohmann AG. Endocannabinoids at the spinal level regulate, but do not mediate, nonopioid stress-induced analgesia. Neuropharmacology. 2006;50:372–379. doi: 10.1016/j.neuropharm.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Terman GW, Shavit Y, Lewis JW, Cannon JT, Liebeskind JC. Intrinsic mechanisms of pain inhibition: activation by stress. Science. 1984;226:1270–1277. doi: 10.1126/science.6505691. [DOI] [PubMed] [Google Scholar]
- Terman GW, Lewis JW, Liebeskind JC. Two opioid forms of stress analgesia: studies of tolerance and cross-tolerance. Brain Res. 1986a;368:101–106. doi: 10.1016/0006-8993(86)91046-2. [DOI] [PubMed] [Google Scholar]
- Terman GW, Morgan MJ, Liebeskind JC. Opioid and non-opioid stress analgesia from cold water swim: importance of stress severity. Brain Res. 1986b;372:167–171. doi: 10.1016/0006-8993(86)91472-1. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Hughes DI, Polgar E, Nagy GG, Mackie M, Ottersen OP, Maxwell DJ. The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. Eur J Neurosci. 2003;17:13–27. doi: 10.1046/j.1460-9568.2003.02406.x. [DOI] [PubMed] [Google Scholar]
- Usdin TB, Hilton J, Vertesi T, Harta G, Segre G, Mezey E. Distribution of the parathyroid hormone 2 receptor in rat: immunolocalization reveals expression by several endocrine cells. Endocrinology. 1999a;140:3363–3371. doi: 10.1210/endo.140.7.6855. [DOI] [PubMed] [Google Scholar]
- Usdin TB, Hoare SR, Wang T, Mezey E, Kowalak JA. TIP39:a newneuropeptide and PTH2-receptor agonist from hypothalamus. Nat Neurosci. 1999b;2:941–943. doi: 10.1038/14724. [DOI] [PubMed] [Google Scholar]
- Valverde O, Ledent C, Beslot F, Parmentier M, Roques BP. Reduction of stress-induced analgesia but not of exogenous opioid effects in mice lacking CB1 receptors. Eur J Neurosci. 2000;2:533–539. doi: 10.1046/j.1460-9568.2000.00929.x. [DOI] [PubMed] [Google Scholar]
- Varoqui H, Schafer MK, Zhu H, Weihe E, Erickson JD. Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci. 2002;22:142–155. doi: 10.1523/JNEUROSCI.22-01-00142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Palkovits M, Rusnak M, Mezey E, Usdin TB. Distribution of parathyroid hormone-2 receptor-like immunoreactivity and messenger RNA in the rat nervous system. Neuroscience. 2000;100:629–649. doi: 10.1016/s0306-4522(00)00282-7. [DOI] [PubMed] [Google Scholar]
- Wang J, Palkovits M, Usdin TB, Dobolyi A. Forebrain projections of tuberoinfundibular peptide of 39 residues (TIP39)-containing subparafascicular neurons. Neuroscience. 2006;138:1245–1263. doi: 10.1016/j.neuroscience.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Ward HL, Small CJ, Murphy KG, Kennedy AR, Ghatei MA, Bloom SR. The actions of tuberoinfundibular peptide on the hypothalamo-pituitary axes. Endocrinology. 2001;142:3451–3456. doi: 10.1210/endo.142.8.8308. [DOI] [PubMed] [Google Scholar]
- Willis WD, Westlund KN. Neuroanatomy of the pain system and of the pathways that modulate pain. J Clin Neurophysiol. 1997;14:2–31. doi: 10.1097/00004691-199701000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeomans DC, Pirec V, Proudfit HK. Nociceptive responses to high and low rates of noxious cutaneous heating are mediated by different nociceptors in the rat: behavioral evidence. Pain. 1996;68:133–140. doi: 10.1016/S0304-3959(96)03176-4. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]