

Abstract
Fibrates are activators of peroxisome proliferator-activated receptor (PPAR) α. Pretreatment with fibrates has been shown to protect brain against ischemia in mice. We hypothesized that fibrates elevate superoxide dismutase (SOD) levels in the brain microvessels (BMV). BMV were isolated from male C57BL/6 and PPARα null mice that had been treated with fenofibrate or gemfibrozil for 7 days. To examine the effect of discontinuation of fenofibrate, another animal group treated with fenofibrate was examined on post-discontinuation day 3 (D-3). To examine whether SOD elevations attenuate oxidative stress in the ischemic brain, separate animals treated with fenofibrate for 7 days were subjected to 60 minutes focal ischemia on post-discontinuation day 0 (D-0) or D-3. Fenofibrate (30 mg/kg) increased mRNA levels of all three isoforms of SOD and activity level in BMV on D-0 but these effects were not detected on D-3. The elevations were not detected in PPARα null mice. SOD levels were also elevated by gemfibrozil (30 mg/kg). Fenofibrate significantly reduced superoxide production and protein oxidation in the ischemic brain at 30 minutes after reperfusion. Fenofibrate reduced infarct size measured at 24 hours after reperfusion on D-0; however, the infarct reduction was not seen when ischemia was induced on D-3. These findings suggest that fibrates elevate SOD in BMV through PPARα, which contributes to the infarct reduction, at least in part. Further studies are needed to establish the link between the SOD elevations and the brain protection by fibrates against ischemia.
Keywords: cerebral ischemia; middle cerebral artery occlusion, neuroprotection; oxidative stress, nitric oxide
1. Introduction
Peroxisome proliferator-activated receptor (PPAR) is a ligand-dependent transcription factor (Chinetti et al., 2000). Currently, three subtypes α, γ, and β (also called δ and NUC1) have been identified. PPARs have been shown to be involved in lipid and glucose metabolism and adipocyte differentiation; therefore, PPARs have gained great attention as a potential therapeutic molecular target for metabolic syndrome which predisposes patients to be more susceptible to stroke and vascular dementia (Kersten et al., 2000).
Fibrates were initially developed as drugs for normalizing cholesterol or triglyceride levels (Thorp and Warning, 1962). Later, fibrates were identified as ligands for PPARα (Issemann and Green, 1990). In addition to their lipid lowering action, antiinflammatory and antioxidative actions have been reported (Staels et al., 1998; Escher and Wahli, 2000). Fibrates also protect the CNS against several disease conditions including ischemic stroke (Inoue et al., 2003; Deplanque et al., 2003). We recently reported that two types of fibrate, fenofibrate and gemfibrozil, attenuated infarct size partly by improving cerebral blood flow (CBF) in the ischemic brain in wild-type mice (Guo et al., 2009, 2010). Ouk and colleagues (2009) reported that fenofibrate pre-treatment partially restored ex vivo endothelial function of rat middle cerebral arteries that were dissected from animals subjected to middle cerebral artery occlusion (MCAO) and reperfusion. They also observed that superoxide production in the ischemic brain was attenuated by fenofibrate pretreatment (Ouk et al., 2009).
Superoxide dismutases (SODs) are key enzymes that metabolize superoxide (Fridovich, 1986). Three isoforms of SOD are identified: cytosolic Cu/Zn SOD (SOD1), mitochondrial Mn SOD (SOD2), and extracellular Cu/Zn SOD (SOD3). All three isoforms have been shown as neuro-protective enzymes against cerebral ischemia (Kinouchi et al., 1991; Murakami et al., 1998; Sheng et al., 1999). In addition, recent studies using molecular approaches revealed the importance of SODs in vascular function in normal states and under pathological conditions (Faraci and Didion, 2004). Since PPARs are transcription factors, and since the promoter region of SOD genes contain the consensus sequence for PPAR binding (Lemay and Hwang, 2006), SOD levels may be influenced by activated PPAR. SOD1 gene induction through the peroxisome proliferator-responsive element (PPRE) has been shown in human HepG2 hepatome cells (Yoo et al., 1999). Another type of fibrate clofibric acid has been shown to increase SOD2 mRNA levels in HepG2 cells (Bécuwe et al., 1999). On the contrary, docosahexaenoic acid has been shown to inhibit SOD1 gene transcription through PPARα in a human ovarian carcinoma cell line (Tuller et al., 2009). Regarding the cerebrovascular system, Mysiorek et al. (2009) reported by using a mouse in vitro blood-brain barrier model that fenofibric acid treatment for 24 hours did not influence SOD gene transcription. However, the influence of chronic treatment with fibrates on SOD levels in brain vessels remains unstudied. In addition, little is known about the effect of fibrates on SOD levels in brain vessels at the animal level.
In the current study, we measured in mice the impact of 7 days treatment with two types of fibrate, fenofibrate and gemfibrozil, on the level of SOD in brain microvessels (BMV). BMV are substantially responsible for the total resistance to CBF and are important for the neurovascular coupling (Joó, 1985). We also measured the impact of fenofibrate on oxidative stress in ischemic brain. To further associate the effects on SOD levels with brain protection, we measured the influence of fenofibrate discontinuation for 3 days on infarct size at 24 hours after reperfusion following 60 minutes of MCAO (Fig. 1). In our previous studies, the current treatment protocol was effective both in improving CBF in ischemic brain and in reducing brain infarct volume in mice (Guo et al., 2009, 2010).
Fig. 1.

Schematic diagram showing experimental designs. Experiment I measured SOD gene expression and enzymatic activity levels in nonischemic animals. Vehicle (Veh) or fenofibrate [30 mg/kg (F30) or 100 mg/kg (F100)] was administered for 7 days and brains were collected at 1 hour after the last administration. F30D-3, fenofibrate (30 mg/kg) was given for 7 days and brains were collected on post-discontinuation day 3 (D-3). Experiment II measured oxidative stress in ischemic brain at 30 minutes after reperfusion following 60 minutes of MCAO. Animals were treated with Veh, F30, or F100 for 7 days and subjected to MCAO at 1 hour after the last drug treatment. Sham, non-fenofibrate treated animals were subjected to sham surgery under anesthesia. Experiment III measured stroke outcome after 24 hours of reperfusion following 60 minutes of MCAO. NoTx, non-fenofibrate treated animals were subjected to stroke. D-0, animals were treated with fenofibrate (30 mg/kg) for 7 days and were subjected to stroke at 1 hour after the last fenofibrate administration. D-3, animals were treated with fenofibrate (30 mg/kg) for 7 days and were subjected to stroke 3 days after the last administration. Downward arrow and upward arrow indicate fenofibrate administration and MCAO, respectively. Scale bar, 1 day.
2. Results
2.1 Fenofibrate and gemfibrozil elevate SOD mRNA
Light microscopy observation showed that the isolated BMV contained primarily capillaries and venules, and, to a lesser extent, small arterioles. However, major cerebral arteries such as middle cerebral artery and basilar artery were not contained (Ospina et al., 2002). mRNA levels of all three isoforms of SOD were significantly elevated by fenofibrate in BMV when examined at 1 hour after the last administration (Fig. 2A). The elevations were more prominent at 30 mg/kg than 100 mg/kg on post-discontinuation day 0 (D-0). However, elevated SOD levels were not observed on post-discontinuation day 3 (D-3). We observed that fenofibrate also elevated PPARα mRNA levels on D-0 as we reported previously (data not shown) (Guo et al., 2010). There were positive correlations between PPARα and SODs on D-0 (Fig. 2B): SOD1, Pearson r = 0.84, P < 0.0001; SOD2, Pearson r = 0.69, P = 0.0031; and SOD3, Pearson r = 0.92, P < 0.0001 (n = 16 or 17 in each SOD isoform). Gemfibrozil also elevated mRNA levels of all three isoforms of SOD at 1 hour after the last administration (Fig. 2C). The elevations were more effective at 30 mg/kg than 120 mg/kg.
Fig. 2.

Effects of fibrates on SOD mRNA levels in brain microvessels in nonischemic wild-type mice. (A) mRNA levels of SOD after fenofibrate treatments for 7 days in wild-type mice. Brains were collected at 1 hour or 3 days after the last fenofibrate administration. mRNA levels were determined by quantitative PCR normalized by GAPDH mRNA levels in each sample. For each gene, the mean value in vehicle group was considered as 1 and relative level was calculated and expressed for fenofibrate groups. For Veh, F30, F100, and F30D-3, please refer to Figure 1, Experiment I. Error bars, SD. ***p < 0.001 by ANOVA followed by Scheffe (n = 5—7 in each group). (B) Relationship between SOD mRNA and PPARα mRNA level on D-0. (C) mRNA levels of SOD after gemfibrozil treatments for 7 days in wild-type mice. Gemfibrozil (30 or 120 mg/kg) or vehicle (Veh) was given for 7 days. Brains were collected at 1 hour after the last gemfibrozil administration. **p < 0.01 and ***p < 0.001 by ANOVA followed by Scheffe (n = 7 in each group).
2.2 Fenofibrate and gemfibrozil elevate SOD activity
We next measured SOD enzymatic activity level. Fenofibrate (30 mg/kg) elevated SOD activity on D-0; however, the activity returned to the basal level by D-3 (Fig. 3A). Gemfibrozil showed a similar increase in SOD activity level at 30 mg/kg (Fig. 3B).
Fig. 3.
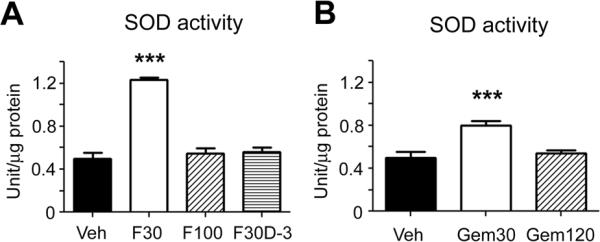
Effects of fenofibrate (A) and gemfibrozil (B) for 7 days on SOD enzymatic activity levels in wild-type mice. For Veh, F30, F100, and F30D-3 in (A), please refer to Figure 1, Experiment I. (B) Gemfibrozil (30 or 120 mg/kg) or vehicle (Veh) was given. Error bars, SD. ***p < 0.001 by ANOVA followed by Scheffe (n = 6 or 7 in each group).
2.3 Fenofibrate does not elevate SOD levels in PPARα null mice
The SOD increases induced by fenofibrate were not observed in PPARα null mice (Fig. 4), indicating that PPARα expression was required for the SOD elevations in both mRNA and activity levels.
Fig. 4.

Effects of fenofibrate (30 mg/kg) for 7 days on SOD mRNA (A) and SOD enzymatic activity levels (B) in brain microvessels in nonischemic PPARα null mice. mRNA levels were determined by quantitative PCR normalized by GAPDH mRNA levels in each sample. For each gene, the mean value in vehicle group was considered as 1 and relative level was calculated and expressed for fenofibrate groups. There was no significant difference compared with vehicle (Veh) treatment (n = 6 in each group).
2.4 Fenofibrate attenuates oxidative stress in the brain after ischemia/reperfusion
To examine whether the SOD elevation by fenofibrate attenuates oxidative stress in the ischemic brain after stroke, we measured the impact of fenofibrate on superoxide level and protein oxidation level in the ischemic hemisphere at 30 minutes of reperfusion after 60 minutes of MCAO. Compared to sham control, MCAO/reperfusion strongly increased both superoxide level and protein oxidation. Fenofibrate at 30 mg/kg significantly attenuated both oxidative stress indicators; however 100 mg/kg did not affect these indicators (Figs. 5 and 6).
Fig. 5.

Effects of fenofibrate on superoxide level in ischemic brain at 30 minutes after reperfusion following 60 minutes of MCAO. Mice were treated with vehicle (Veh) or fenofibrate [30 mg/kg (F30) or 100 mg/kg (F100)] for 7 days and subjected to MCAO at 1 hour after the last drug treatment. Non-drug treated animals that were subjected to sham surgery served as sham control. (A) fluorescent photomicrographs showing hydroxyethidium (Et) positive cells in the parietal cortex in the ischemic hemisphere. Scale bar, 100 μm. (B, C) bargraph showing Et positive cell number in the parietal cortex (B) and striatum (C) in the ischemic hemisphere. Error bars, SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared to Sham group and ##, p < 0.01 compared to Veh by ANOVA followed by Scheffe (n = 6 or 7 in each group).
Fig. 6.

Effect of fenofibrate on protein oxidation in the ischemic hemisphere at 30 minutes after reperfusion following 60 minutes of MCAO. Mice were treated with vehicle (Veh) or fenofibrate [30 mg/kg (F30) or 100 mg/kg (F100)] for 7 days and subjected to MCAO at 1 hour after the last drug treatment. Non-drug treated animals that were subjected to sham surgery served as sham control. (A) Western blot analysis showing derivitized protein carbonyls. Brain lysates (10 μg protein) were treated with (+) or without (−) 2,4-dinitrophenylhydrazine and separated by SDS-PAGE. Left end lane was loaded protein marker. (B) bargraph showing density of whole lane of each sample. Error bars, SD. **, p < 0.01 compared to Sham group and ##, p < 0.01 compared to Veh group by ANOVA followed by Scheffe (n = 6 or 7 in each group).
2.5 SOD elevation is associated with infarct size reduction by fenofibrate
To test whether increased SOD activity induced by fenofibrate is associated with neuroprotection, we measured the duration of infarct size reducing efficacy after cessation of fenofibrate treatment (Fig. 1, Experiment III). The D-0 group that was subjected to ischemia at 1 hour after the last fenofibrate treatment showed significantly smaller infarct volume compared with non-fenofibrate treated (NoTx) ischemic animals (Fig. 7A), confirming previously reported findings (Inoue et al., 2003; Deplanque et al., 2003; Guo et al., 2010). The attenuation was seen in the cortex but not in the subcortex (Figs. 7B and C). However, D-3 group that was subjected to ischemia at 3 days after the last fenofibrate treatment did not differ in infarct volume compared with the NoTx group (Fig. 7). There was no significant difference detected in neurological score (p = 0.56 by Kruskal-Wallis test), probably due to the insensitivity of the scoring method: NoTx (n = 13), [1 (0, 2, 0—3): median (first and third quartiles, range)]; D-0 (n = 10), [1.5 (1, 2, 0—3)]; D-3 (n = 11), [2 (0.5, 2, 0—3)]. CBF measured by laser Doppler flowmetry at the parietal skull over the ischemic core was significantly higher in the D-0 group when analyzed by repeated measures ANOVA (Table 1). During MCAO, all animals showed CBF less than 20% of the basal level and there was no difference among the groups. After reperfusion, CBF recovery was significantly better in the D-0 group (P < 0.05 by ANOVA followed by Scheffe).
Fig. 7.

Effects of fenofibrate on total (A), cortical (B), and subcortical (C) infarct size in C57BL/6 wild-type mice subjected to 60 minutes of MCAO and 24 hours of reperfusion. NoTx, non-fenofibrate treated group (n = 13). D-0, the group was treated with fenofibrate (30 mg/kg) for 7 days and was subjected to ischemia at 1 hour after the last fenofibrate administration (n = 10). D-3, the group was treated with fenofibrate (30 mg/kg) for 7 days and was subjected to ischemia 3 days after the last fenofibrate administration (n = 11). Horizontal bars, mean. Statistical analysis was done by one way ANOVA followed by Scheffe post hoc test.
Table 1.
%CBF measure by laser Doppler flowmetry at ischemic core
| NoTx (n=13) | D-0 (n=10) | D-3 (n=11) | |
|---|---|---|---|
| Before | 100 | 100 | 100 |
| During MCAO | 9.6 ± 2.6 | 11.7 ± 3.2 | 11.3 ± 4.3 |
| After reperfusion | 92.7 ± 14.1 | 108.0 ± 10.9* | 101.7 ± 7.6 |
Values are means ± SD. P < 0.01 by repeated measures ANOVA.
P < 0.05 compared with NoTx by ANOVA followed by Scheffe post hoc test.
3. Discussion
We showed at the animal level that fenofibrate significantly elevated mRNA levels of all three isoforms of SOD in BMV. We also confirmed our previous observation (Guo et al., 2010) that fenofibrate elevated PPARα mRNA (data not shown). There were strong positive correlations between PPARα and SODs. We also found that enzymatic activity levels of SOD in BMV were elevated by fenofibrate at 30 mg/kg but not 100 mg/kg. Fenofibrate attenuated oxidative stress indicators in the ischemic brain. The degree of SOD activity elevation and the degree of oxidative stress attenuation with different doses of fenofibrate were correlated with the previously reported CBF improvement in the brain after ischemia (Guo et al., 2010). Moreover, discontinuation of fenofibrate for 3 days returned both mRNA and activity levels of SOD in BMV to the basal levels by D-3. In parallel to the SOD levels in BMV, infarct size reduction was found on D-0 but not on D-3.
Another fibrate gemfibrozil similarly elevated SOD levels at 30 mg/kg but not 120 mg/kg. The effects of different doses of gemfibrozil on SOD levels were correlated with the previously reported protection against MCAO (Guo et al., 2009). In addition, fenofibrate did not influence the SOD levels in PPARα null mice. In our previous studies, fenofibrate did not provide protection against MCAO in PPARα null mice (Inoue et al., 2003; Guo et al., 2010). These findings agree with the possibility that fibrates elevate SOD levels through PPARα activation in BMV, which contributes to the cerebrovascular protection by scavenging oxygen radicals. Importantly, the current study showed that fenofibrate (30 mg/kg) profoundly attenuated superoxide increase in the cortex after MCAO/reperfusion. Such an attenuation was not significant in the striatum, which is consistent with the finding that fenofibrate did not reduce infarct volume in the subcortical region.
One limitation of this study is that we did not measure each isoform of SOD separately at the protein expression level. The currently used SOD activity assay does not differentiate the three SOD isoforms. Future studies with SOD isoform specific antibodies will help to address whether the elevations in the SOD genes are translated to the protein level, and if this is the case, which isoform of SOD is responsible for the activity increase by fenofibrate. It also should be noted that the elevations both at the mRNA and activity levels were observed at the intermediate dose (30 mg/kg) of fenofibrate. The higher dose (100 mg/kg) showed attenuated elevations at the mRNA level and no elevation at the activity level. Gemfibrozil showed similar dose-effect patterns. These findings suggest that SOD expression is regulated by a negative feedback loop through PPARα.
Fibrates are well known to elevate homocysteine levels in humans (de Lorgeril et al., 1999). Fenofibrate (100 mg/kg for 14 days) has been shown to double homocysteine level in wild-type mice but not in PPARα deficient mice (Legendre et al., 2002). Hyperhomocysteinemia has been suggested to contribute to vascular injury, including stroke (Saposnik et al., 2009). This adverse side effect on homocysteine level might counteract the beneficial effect of fibrates at 100 mg/kg. In addition, fibrates could also influence other molecules than PPARα, such as LXR, at higher concentrations (> 25 μM) (Thomas et al., 2003); thereby counteracting their beneficial effects. Since the brain protective efficacies by fibrates seem to be independent of their lipid normalizing properties (Deplanque et al., 2003; Inoue et al., 2003), a useful biomarker for monitoring appropriate dose of fibrates, specifically aiming at improving stroke outcome, needs to be identified.
In the current study, fenofibrate showed better recovery of CBF at the ischemic core after reperfusion (Table 1). Our previous study using laser speckle imaging showed similar effects of fenofibrate in CBF recovery (Guo et al., 2010). The current study has not established the link between the SOD elevations and the improvement in CBF recovery. From the literature, several possibilities for the link may be speculated. One possibility may involve nitric oxide (NO) which is known to cause vascular relaxation (Iadecola, 1997). After ischemia/reperfusion, when superoxide is excessively generated, NO may rapidly react with superoxide (Beckman et al., 1990). Therefore, increased SOD activity, which enhances scavenging of superoxide, elevates the level of NO bioavailability. By this mechanism, the SOD elevation by fibrates may contribute to the better CBF maintenance in the ischemic brain. Consistent with this notion, faster recovery and improved maintenance of CBF after transient global ischemia/reperfusion have recently been reported in SOD1-overexpressing transgenic rats (Xu et al., 2009). Kirsch et al. (1993) reported that intravenous injection of polyethylene glycol (PEG)-conjugated SOD improved recovery of post-ischemic hypercapnic CBF response in piglets. Nelson et al. (1992) reported in cats that 15 minutes of complete global ischemia followed by reperfusion generated superoxide during the early reperfusion period but not during ischemia. The vasoconstrictor response to arterial hypocapnia was reduced by ischemia/reperfusion, which was prevented by pretreatment with SOD plus catalase. On the contrary, Rosenblum (1997) reported in mice that the impaired response to acetylcholine at 10 minutes after reperfusion following 10 minutes bilateral carotid artery occlusion was not recovered by SOD plus catalase. Fabian et al. (2004) reported that PEG-SOD did not influence CBF measured by laser Doppler during hyperoxygenation following hypoxia in rats although the treatment did inhibit superoxide production.
In addition, the reaction of superoxide with NO forms peroxynitrite, a potent oxidant (Beckman et al., 1990). Among multiple pro-oxidant actions of peroxynitrite, oxidation of the zinc-thiolate complex and tetrahydrobiopterin in endothelial NO synthase (eNOS) has been shown to divert electron flow through the enzyme to molecular oxygen rather than to L-arginine, shifting the enzyme function from NO production to superoxide formation, which is called eNOS uncoupling (Zou et al., 2002; Landmesser et al., 2003). The SOD elevations and subsequent inhibition of peroxynitrite formation may prevent this vicious cycle, maintaining the NO bioavailability within the brain tissue after ischemia/reperfusion. Consistent with this hypothesis, Nelson et al. (1992) reported in cats that pial vessel response to topical acetylcholine was converted to vasoconstriction after ischemia. But pretreatment with SOD plus catalase preserved normal vasodilator response to acetylcholine. In this scenario, fibrates may not affect CBF under normal conditions. In our previous study, fenofibrate did not influence CBF in the nonischemic brain (Guo et al., 2010).
Ouk et al. (2009) observed in rats a significant infarct reduction at 3 days after discontinuation of fenofibrate that was continued for 11 days. In our current study, discontinuation of fenofibrate treatment for 3 days abolished the beneficial effects (SOD elevation and infarct size reduction). Such different outcomes may be due to the different species of animals that were used. It is known that the rate of drug metabolism is different between rat and mouse. Our current study did not observe aggravated stroke outcomes after 3 days of fenofibrate discontinuation. Withdrawal of statins for 3 days, another type of lipid lowering drug, has been reported to adverse their effects on the cerebrovascular system, which is called “rebound phenomenon” (Endres and Laufs, 2006; Rosengarten et al., 2007). Our current study was primarily designed to detect beneficial effects of fenofibrate, but not intended to show its adverse side effects. Further studies with an appropriate design are needed to test whether fibrates show a rebound phenomenon after their withdrawal.
In summary, we showed at the animal level that continuous treatment with fibrates elevates SOD levels in BMV, which requires PPARα expression. These findings suggest that PPARα activation by fibrates elevates SOD levels. The elevated SOD levels were associated with brain protection against transient MCAO. It remains to be studied whether or not chronic fenofibrate treatment influences SOD levels also in larger resistance vessels including middle cerebral artery. Further studies are needed to investigate whether or not the SOD elevations by fibrates mediate the cerebrovascular protection and CBF improvement after ischemic stroke.
4. Materials and methods
4.1 Animals and drug treatment
Male C57BL/6J mice (6–8 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Fenofibrate and gemfibrozil were purchased from Sigma Aldrich (St. Louise, MO, USA). Male homozygous PPARα null mice (Lee et al., 1995) were obtained from our local mouse colony which was maintained by breeding homozygous PPARα-null males and females. Animals were housed with a 12 hour daily light/dark cycle. Food and water were provided ad libitum. All animal experiments were performed in accordance with the procedures approved by the Morehouse School of Medicine Institutional Animal Care and Use Committee. Fenofibrate (30 or 100 mg/kg), gemfibrozil (30 or 120 mg/kg), or vehicle (10 mL/kg of 0.5% carboxymethyl cellulose) was given through a feeding needle once per day for 7 consecutive days. The dosages were selected from our previous findings that fenofibrate and gemfibrozil produced cerebrovascular protection against ischemia (Guo et al., 2009, 2010).
4.2 Experimental design
Experiment I measured the impact of fenofibrate on brain SOD levels in nonischemic animals. Experiment II measured the impact of fenofibrate on oxidative stress in the ischemic brain at 30 minutes of reperfusion after60 minutes of middle cerebral artery occlusion (MCAO). Experiment III measured the influence of fenofibrate on infarct size after 60 minutes MCAO (Fig. 1). In Experiment III, one investigator treated all animals with fenofibrate or vehicle; then, treatment types were concealed until all data collection was completed. A surgeon, who was given the animals with identification codes (IDs) but without information on the treatment, performed MCAO. Neurological score was measured using a modified Bederson score at 24 hours after reperfusion; 0, normal; 1, decreased forepaw extension; 2, circling; 3, loss of postural reflex. The surgeon performed brain sectioning and staining with 2% 2,3,5-triphenyltetrazolium chloride at 24 hours after reperfusion. Different IDs were given to the collected brains and the IDs were concealed until infarct size measurement was completed by the third investigator.
4.3 Brain microvessel isolation and SOD assays
Brain microvessels (BMV) were isolated by multistep fractionations combined with the mesh technique (40 μm pore size) according to Ospina et al. (2002), with a minor modification. This method has been shown to separate efficiently microvessels from neurons (Guo et al., 2009). The isolated BMV were subdivided into two sets and used for mRNA analysis and SOD activity assay, respectively. Total RNA was isolated with Trizol (Invitrogen, Carlsbad, CA, USA). Polymerase chain reaction (PCR) amplifications were done with iQ SYBR Green Supermix kit (Bio-Rad, Hercules, CA, USA). Primer sequences for the studied genes are listed in Table 2. The fold expression of each gene was calculated by the comparative cross-threshold method using GAPDH as the reference gene.
Table 2.
Primer sequence for PCR analysis
| Target gene | Forward | Reverse |
|---|---|---|
| PPARα | 5'-gcagctcgtacaggtcatca-3' | 5'-ctcttcatccccaagcgtag-3' |
| SOD1 | 5'-gagacctgggcaatgtgact-3' | 5'-gtttactgcgcaatcccaat-3' |
| SOD2 | 5'-ccgaggagaagtaccacgag-3' | 5'-gcttgatagcctccagcaac-3' |
| SOD3 | 5'-atcccacaagcccctagtct-3' | 5'-gtgctatggggacaggaaga-3' |
| GAPDH | 5'-aactttggcattgtggaagg-3' | 5'-acacattgggggtaggaaca-3' |
For SOD activity assay, the BMV were homogenized in 50 mmol/L Tris-HCl (pH 7.0) at 4°C. Homogenates were centrifuged at 16,000 × g for 15 minutes at 4°C and the supernatant was used for further analysis using a commercially available kit (Wako Chemicals USA, Richmond, VA, USA). This detection kit is based on nitroblue-tetrazolium reduction by superoxide into diformazan. Superoxide was generated by adding a constant amount of xanthine and xanthine oxidase to the reaction solutions. SOD activity was determined by measuring the inhibition rate of diformazan production.
4.4 Middle cerebral artery occlusion
MCAO (60 minutes) was induced on the left side with an 8-0 nylon monofilament coated with silicone resin and hardener mixture (Heraeus, Hanau, Germany) under 1.5% isoflurane as previously described (Steele et al., 2008). The common carotid artery (CCA) and external carotid artery (ECA) were ligated with a 5-0 silk suture. A miniclip (Ohwatsusho, Tokyo, Japan) was temporally applied to the left internal carotid artery (ICA). Then, the filament was introduced into the ICA through the ECA, and advanced until the filament felt resistance. For reperfusion, the filament was withdrawn and the CCA was re-opened; however, the ECA remained permanently ligated. To confirm successful MCAO and reperfusion, CBF at the parietal skull over the ischemic core was monitored by laser Doppler flowmetry using a flexible fiber glass probe (FLO-C1, Omegawave, Tokyo, Japan). Sham surgery was conducted with ECA exposure but without filament insertion under anesthesia for 90 minutes.
4.5 Superoxide level measurement
In situ superoxide detection using hydroethidine (HEt) was performed as reported by Murakami et al. (1998). HEt rapidly penetrates into cells and reacts selectively with superoxide to form 2-hydroxyethidium (Et), which is a stable fluorescent product. Two hundred micro liters of HEt (Invitrogen, 1 mg/ml in phosphate-buffered saline containing 1% dimethyl sulfoxide) was administered intravenously 15 minutes before MCAO induction. At 30 minutes after reperfusion following 60 minutes of MCAO, the brains were collected. Brains were immersed in 4% paraformaldehyde solution overnight and coronal brain sections (20 μm thick) were made using a cryostat. Et fluorescent signal was observed at emission > 600 nm with a fluorescent microscope (Axioskop 2, Carl Zeiss). Images were taken from both cortex and striatum in the ischemic hemispheres using a 10× objective lens and a CCD camera (Axiocam HRc, Carl Zeiss) under the same exposure condition. Et-positive cell number was automatically determined using a computerized image analysis system (MCID, Imaging Research, Cambridge, UK).
4.6 Protein oxidation assay
OxyBlot (Millipore, Billerica, MA) is a semi-quantitative measurement using Western blot assay detecting carbonyl groups as a protein oxidation marker. Brain tissues were homogenized with lysis buffer using a tightly fitting homogenizer. The homogenates were centrifuged at 13000g for 20 minutes, and the supernatants were used for further analysis. The samples were treated with 2,4-dinitrophenylhydrazine (DPH) to derivatize the carbonyl groups to 2,4-dinitrophenylhydrazone. After the samples are neutralized, the samples (10 μg protein) were separated by SDS-PAGE and transferred onto a PVDF membrane. Immunoblotting was performed using rabbit primary antibodies against dinitrophenyl (1:300 dilution). By densitometry analysis of the immunoblots, total signal of a whole lane was measured in each sample. Samples that were not reacted with DPH were used to determine non-specific background noise for each sample. The background noise was subtracted from the density obtained with DPH-treatment for each sample; then, the obtained number was used for statistical analysis.
4.7 Infarct size measurement
Brains were collected at 24 hours after reperfusion and cut into 2 mm thick coronal sections. The brain sections were incubated with 2% 2,3,5-triphenyltetrazolium chloride and infarct area was determined using the MCID image analyzer.
4.8 Statistical analysis
Data for SOD, superoxide, and protein oxidation levels are presented as mean ± SD. Infarct volume data are shown by scatter plot. Difference among the groups in SOD, superoxide, and protein oxidation levels and infarct size were analyzed by one-way analysis of variance (ANOVA) followed by Scheffe post-hoc test. CBF data were analyzed by repeated measures ANOVA. Neurological score is expressed as [median (first and third quartiles, range)] and analyzed by Kruskal-Wallis test. P < 0.05 was considered statistically significant.
Acknowledgements
This work was supported in part by NS048532 and NS060659 from NIH/NINDS and S21MD000101 from NIH/NCMHD. The study was conducted in a facility constructed with support from Research Facilities Improvement Grant 1 C06 RR-07571 from NIH/NCRR. We thank Dr. Morris Benveniste of Department of Neurobiology at Morehouse School of Medicine for critical reading and comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest None
References
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bécuwe P, Bianchi A, Keller JM, Dauça M. Effects of the peroxisome proliferator clofibric acid on superoxide dismutase expression in the human HepG2 hepatoma cell line. Biochem. Pharmacol. 1999;58:1025–1033. doi: 10.1016/s0006-2952(99)00174-4. [DOI] [PubMed] [Google Scholar]
- Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000;49:497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- de Lorgeril M, Salen P, Paillard F, Lacan P, Richard G. Lipid-lowering drugs and homocysteine. Lancet. 1999;353:209–210. doi: 10.1016/S0140-6736(05)77220-2. [DOI] [PubMed] [Google Scholar]
- Deplanque D, Gele P, Petrault O, Six I, Furman C, Bouly M, Nion S, Dupuis B, Leys D, Fruchart JC, Cecchelli R, Staels B, Duriez P, Bordet R. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J. Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Laufs U. Discontinuation of statin treatment in stroke patients. Stroke. 2006;37:2640–2643. doi: 10.1161/01.STR.0000240690.69406.28. [DOI] [PubMed] [Google Scholar]
- Escher P, Wahli W. Peroxiome proliferator-activated receptors: insight into multiple cellular functions. Mutat. Res. 2000;448:121–138. doi: 10.1016/s0027-5107(99)00231-6. [DOI] [PubMed] [Google Scholar]
- Fabian RH, Perez-Polo JR, Kent TA. Extracellular superoxide concentration increases following cerebral hypoxia but does not affect cerebral blood flow. Int. J. Dev. Neurosci. 2004;22:225–230. doi: 10.1016/j.ijdevneu.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide dismutases. Adv. Enzymol. Relat. Areas. Mol. Biol. 1986;58:61–97. doi: 10.1002/9780470123041.ch2. [DOI] [PubMed] [Google Scholar]
- Guo Q, Wang G, Liu X, Namura S. Effects of gemfibrozil on outcome after permanent middle cerebral artery occlusion in mice. Brain Res. 2009;1279:121–130. doi: 10.1016/j.brainres.2009.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Wang G, Namura S. Fenofibrate improves cerebral blood flow after middle cerebral artery occlusion in mice. J. Cereb. Blood Flow Metab. 2010;30:70–78. doi: 10.1038/jcbfm.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- Inoue H, Jiang XF, Katayama T, Osada S, Umesono K, Namura S. Brain protection by resveratrol and fenofibrate against stroke requires peroxisome proliferator-activated receptor alpha in mice. Neurosci. Lett. 2003;352:203–206. doi: 10.1016/j.neulet.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Issemann I, Green S. Activation of a member of the steroid receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Joó F. The blood-brain barrier in vitro: Ten years of research on microvessels isolated from the brain. Neurochem. Int. 1985;7:1–25. doi: 10.1016/0197-0186(85)90002-6. [DOI] [PubMed] [Google Scholar]
- Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc. Natl. Acad. Sci. U S A. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch JR, Helfaer MA, Haun SE, Koehler RC, Traystman RJ. Polyethylene glycol-conjugated superoxide dismutase improves recovery of postischemic hypercapnic cerebral blood flow in piglets. Pediatr. Res. 1993;34:530–537. doi: 10.1203/00006450-199310000-00030. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre C, Causse E, Chaput E, Salvayre R, Pineau T, Edgar AD. Fenofibrate induces a selective increase of protein-bound homocysteine in rodents: a PPARalpha-mediated effect. Biochem. Biophys. Res. Commun. 2002;295:1052–1056. doi: 10.1016/s0006-291x(02)00814-8. [DOI] [PubMed] [Google Scholar]
- Lemay DG, Hwang DH. Genome-wide identification of peroxisome proliferator response elements using integrated computational genomics. J. Lipid Res. 2006;47:1583–1587. doi: 10.1194/jlr.M500504-JLR200. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J. Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysiorek C, Culot M, Dehouck L, Derudas B, Staels B, Bordet R, Cecchelli R, Fenart L, Berezowski V. Peroxisome-proliferator-activated receptor-alpha activation protects brain capillary endothelial cells from oxygen-glucose deprivation-induced hyperpermeability in the blood-brain barrier. Curr. Neurovasc. Res. 2009;6:181–193. doi: 10.2174/156720209788970081. [DOI] [PubMed] [Google Scholar]
- Nelson CW, Wei EP, Povlishock JT, Kontos HA, Moskowitz MA. Oxygen radicals in cerebral ischemia. Am. J. Physiol. 1992;263:H1356–H1362. doi: 10.1152/ajpheart.1992.263.5.H1356. [DOI] [PubMed] [Google Scholar]
- Ospina JA, Krause DN, Duckles SP. 17beta-estradiol increases rat cerebrovascular prostacyclin synthesis by elevating cyclooxygenase-1 and prostacyclin synthase. Stroke. 2002;33:600–605. doi: 10.1161/hs0202.102732. [DOI] [PubMed] [Google Scholar]
- Ouk T, Laprais M, Bastide M, Mostafa K, Gautier S, Bordet R. Withdrawal of fenofibrate treatment partially abrogates preventive neuroprotection in stroke via loss of vascular protection. Vascul. Pharmacol. 2009;51:323–330. doi: 10.1016/j.vph.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Rosenblum WI. Selective impairment of response to acetylcholine after ischemia/reperfusion in mice. Stroke. 1997;28:448–451. doi: 10.1161/01.str.28.2.448. [DOI] [PubMed] [Google Scholar]
- Rosengarten B, Auch D, Kaps M. Effects of initiation and acute withdrawal of statins on the neurovascular coupling mechanism in healthy, normocholesterolemic humans. Stroke. 2007;38:3193–3197. doi: 10.1161/STROKEAHA.107.491423. [DOI] [PubMed] [Google Scholar]
- Saposnik G, Ray JG, Sheridan P, McQueen M, Lonn E. Homocysteine-lowering therapy and stroke risk, severity, and disability: additional findings from the HOPE 2 trial. Stroke. 2009;40:1365–1372. doi: 10.1161/STROKEAHA.108.529503. [DOI] [PubMed] [Google Scholar]
- Sheng H, Bart RD, Oury TD, Pearlstein RD, Crapo JD, Warner DS. Mice overexpressing extracellular superoxide dismutase have increased resistance to focal cerebral ischemia. Neuroscience. 1999;88:185–191. doi: 10.1016/s0306-4522(98)00208-5. [DOI] [PubMed] [Google Scholar]
- Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, Najib J, Maclouf J, Tedgui A. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- Steele EC, Jr., Guo Q, Namura S. Filamentous middle cerebral artery occlusion causes ischemic damage to the retina in mice. Stroke. 2008;39:2099–2104. doi: 10.1161/STROKEAHA.107.504357. [DOI] [PubMed] [Google Scholar]
- Thomas J, Bramlett KS, Montrose C, Foxworthy P, Eacho PI, McCann D, Cao G, Kiefer A, McCowan J, Yu KL, Grese T, Chin WW, Burris TP, Michael LF. A chemical switch regulates fibrate specificity for peroxisome proliferator-activated receptor alpha (PPARalpha) versus liver X receptor. J. Biol. Chem. 2003;278:2403–2410. doi: 10.1074/jbc.M209629200. [DOI] [PubMed] [Google Scholar]
- Thorp JM, Warning WS. Modification of metabolism and disruption of lipids by ethyl chlorophenoxyisobutyrate. Nature. 1962;194:948–949. doi: 10.1038/194948a0. [DOI] [PubMed] [Google Scholar]
- Tuller ER, Beavers CT, Lou JR, Ihnat MA, Benbrook DM, Ding WQ. Docosahexaenoic acid inhibits superoxide dismutase 1 gene transcription in human cancer cells: the involvement of peroxisome proliferator-activated receptor alpha and hypoxia-inducible factor-2alpha signaling. Mol. Pharmacol. 2009;76:588–595. doi: 10.1124/mol.109.057430. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liachenko SM, Tang P, Chan PH. Faster recovery of cerebral perfusion in SOD1-overexpressed rats after cardiac arrest and resuscitation. Stroke. 2009;40:2512–2518. doi: 10.1161/STROKEAHA.109.548453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HY, Chang MS, Rho HM. Induction of the rat Cu/Zn superoxide dismutase gene through the peroxisome proliferator-responsive element by arachidonic acid. Gene. 1999;234:87–91. doi: 10.1016/s0378-1119(99)00176-6. [DOI] [PubMed] [Google Scholar]
- Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Invest. 2002;109:817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]