

Abstract
Ectopic expression of C/EBPα in p210BCR/ABL-expressing cells induces granulocytic differentiation, inhibits proliferation and suppresses leukemogenesis.
To dissect the molecular mechanisms underlying these biological effects, C/EBPα-regulated genes were identified by microarray analysis in 32D-p210BCR/ABL cells. One of the genes whose expression was activated by C/EBPα in a DNA binding-dependent manner in BCR/ABL-expressing cells is the transcriptional repressor Gfi-1. We show here that C/EBPα interacts with a functional C/EBP binding site in the Gfi-1 5′ flanking region and enhances the promoter activity of Gfi-1. Moreover, in K562 cells, RNAi-mediated downregulation of Gfi-1 expression partially rescued the proliferation inhibitory but not the differentiation inducing effect of C/EBPα. Ectopic expression of wild type Gfi-1 but not of a transcriptional repressor mutant (Gfi-1P2A) inhibited proliferation and markedly suppressed colony formation but did not induce granulocytic differentiation of BCR/ABL-expressing cells. By contrast, Gfi-1 shRNA-tranduced CD34+ CML cells were markedly more clonogenic than the scramble-transduced counterpart.
Together, these studies indicate that Gfi-1 is a direct target of C/EBPα required for its proliferation and survival inhibitory effects in BCR/ABL-expressing cells.
INTRODUCTION
The transcription factor C/EBPα plays an essential role in regulating the balance between differentiation and proliferation during the early stages of myelopoiesis (1,2).
In many types of myeloid leukemia, C/EBPα is mutated with reduced activity or its expression is lowered (3,4), suggesting that decreased C/EBPα expression/activity is important for leukemogenesis.
Several mechanisms have been implicated in the inactivation of C/EBPα in myeloid leukemia. Mutations in the N- and C-terminus reduce the functional levels of C/EBPα and have the potential to generate mutant proteins with dominant-negative activity (5,6); these mutant proteins promote the development of acute leukemia when expressed from the C/EBPα gene locus (7,8).
C/EBPα expression/activity is also inhibited by transcriptional, post-transcriptional and post-translational mechanisms (9–12). In myeloid cells transformed by the p210BCR/ABL oncoprotein, expression of C/EBPα is repressed at the translational level by MAP kinase-dependent phosphorylation and stabilization of the RNA binding protein hnRNPE2 which binds the 5′ UTR of c/ebp′ mRNA and inhibits its translation (13, 14). Regardless of the mechanisms responsible for C/EBP′ loss of function, ectopic expression of C/EBP′ in myeloid leukemia lines and in primary blast cells induce differentiation and inhibits proliferation (13,15–17), further emphasizing the importance of genetic and/or functional inactivation of C/EBP′ for leukemogenesis and the therapeutic potential of restoring expression of functional C/EBP′ in leukemic cells. Mechanistically, it is unclear how ectopic expression/activation of C/EBP′ exerts its anti-leukemic effects in p210BCR/ABL-expressing cells; although the anti-proliferative effects of C/EBP′ depend, in part, on interaction with cell cycle regulatory and chromatin remodeling proteins (18–21), granulocytic differentiation is only induced by DNA binding and transcription activation-competent proteins in vitro and in leukemic mice (16,22). Consistent with this, leukemogenesis is suppressed more potently by transcription-activation competent C/EBP′ than by a DNA binding-deficient mutant (16). However, it is unclear whether transcription-regulated C/EBP′ targets may be, in part, responsible for its effects on cell proliferation and survival.
We searched for transcriptionally regulated biologically relevant targets of C/EBP′ by probing oligonucleotide microarrays with RNA isolated at early time points after activation of wild type or DNA binding deficient C/EBP′ in 32D-p210BCR/ABL cells. One of the genes whose expression was activated by C/EBP′ in a DNA binding-dependent manner is the transcriptional repressor Gfi-1 which is important for maintenance of hematopoietic stem cells and for differentiation of late granulocytic progenitors (23–27). Moreover, transformation of hematopoietic stem cells engineered to express a DNA binding deficient C/EBP′ mutant is associated with reduced expression of Gfi-1(8), raising the possibility that low levels of Gfi-1 are also expressed in AML with C/EBPα mutations potentially reducing the “quiescence” of these AML stem cells. We show here that Gfi-1 is a direct C/EBPα target and that its expression is required for C/EBPα-dependent inhibition of proliferation but not induction of differentiation in K562 cells. Consistent with these findings, expression of wild type Gfi-1 inhibited proliferation and colony formation of BCR/ABL-expressing cell lines and primary CML cells whereas Gfi-1 shRNA-transduced CD34+ CML cells were markedly more clonogenic of the scramble-transduced counterpart.
MATERIALS AND METHODS
Plasmids
p42C/EBPα-ER and K298E C/EBPα-ER, cloned in the XhoI-EcoRI- digested MigRI vector, were previously described (16).
MigRI-Gfi-1 and MigRI-Gfi-1 P2A were the kind gift of Dr Zhu (Laboratory of Immunology, NIH).
MigRI-Gfi-1-ER was generated bypolymerase chain reaction (PCR) as follows: the ligand-bindingdomain of the murine estrogen receptor (ER) was amplified using p42C/EBPα-ER as template with an upstream oligomer containing a5′-flapping BamHI site and a downstream oligomer containinga 3′-flapping EcoRI site. Gfi-1 was amplified by PCR from the MigRI-Gfi-1 plasmid withan upstream primer containing a 5′-flapping XhoI site and adownstream primer containing a 3′-flapping BamHI site without stop codon. Gfi-1 and ER PCR products were directionally clonedinto the XhoI-EcoRI-digested MigRI vector.
pRRLSIN.cPPT.PGK-GFP.WPRE was purchased by Addgene Plasmid Repository. pRRLSIN.cPPT.Gfi-1PGK-GFP.WPRE and pRRLSIN.cPPT.Gfi-1P2APGK-GFP.WPRE were generated by cloning Gfi-1 (PCR-amplified from the wild type or mutant MigRI-Gfi-1 plasmids) at the XhoI-EcoRV restriction sites upstream of the PGK-GFP cassette.. The proximal promoter of the mouse Gfi-1 gene (−500 to +150) cloned into the pGL3-basic vector was the kindgift of Dr P. Laslo (University of Chicago, Chicago, IL). pGL3 Gfi-1 (−500 to +150) with two substitutions (TCTGAAGCAA into TCTGAAACAG) of the three more conserved nucleotides of the putative C/EBPα binding site (C/EBP-MUT pGL3 Gfi-1) was generated by site-directed mutagenesis from wild type Gfi-1 (−500 to +150) pGL3 plasmid.
Cell cultures and viral infection
32D-p210BCR/ABL and derivative cell lines were cultured in Iscove modified Dulbecco medium (IMDM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine and penicillin/streptomycin (100μg/ml each). K562 cells and derivative cell lines were cultured in IMDM supplemented with heat-inactivated FBS. The32D-p210BCR/ABL-C/EBPα-ER and the K562-C/EBPα-ER lines were established in our laboratory and used in previously published studies (16,28). The other derivative cell lines were established in the laboratory for this study. The identity of each cell line was authenticated by expression of p210BCR/ABL and C/EBPα-ER and by sequence analysis. Fresh leukapheresis or peripheral blood samples were obtained with written informed consent from patients with CML in chronic phase (CP) (n = 5) or blast crisis (BC) (n=1). Samples were enriched for CD34+ cells using CliniMACS (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions. Purity of CML CD34+ cells was more than 95% and more than 95% were Ph1 as assessed by dual-color dual-fusion fluorescence in situ hybridization.
Peripheralblood (granulocyte colony-stimulating factor [G-CSF] mobilized)normal CD34+ cells were purchased from StemCell Technologies(Vancouver, BC). Normal and CML CD34+ cells were cultured in StemSpan SFEM medium(StemCell Technologies) supplemented with StemSpan CC100 (20ng/mL IL-3 and IL-6, 100 ng/mL Kit Ligand (KL), 100 ng/mL Flt-3 ligand;StemCell Technologies) and 50 ng/mL thrombopoietin (StemCellTechnologies).
For retroviral infections, Phoenix cells (kind gift of G. P. Nolan, Stanford University School of Medicine, Stanford, CA) were transiently transfected with the indicated plasmids. The infectious supernatant was collected 48 h later and was used to infect (a 48-h procedure) p210BCR/ABL-expressing cells. Twenty-four h later, infected cells were sorted (EPICS Profile Analyzer; Coulter, Hialeah, FL) for green fluorescence protein (GFP) expression.
Lentiviral stocks were prepared by transient co-transfection of 293T cells with the transfer vector, the pCMV R8.74 encoding Gag, Pol, Tat and Rev, and the pMD.G plasmid encoding VSV-G), and ProFection® Mammalian Transfection according to Promega guidelines. The infectious supernatant was collected 48 h later and used to infect (a 48 h procedure) p210BCR/ABL-expressing cells. Infected cells were selected by GFP positivity or puromycin resistance. CD34+ CML cells were transduced with the scramble or the Gfi-1 shRNA lentivirus (TRCN0000020468, Open Biosystems) after testing the titer of the infectious supernatants by assaying puromycin-resistant colony formation of K562 cells. Lentivirally-transduced CD34+ CML cells were assayed for colony formation in puromycin (3μM)-supplemented methylcellulose plates.
Cell proliferation, colony formation and differentiation assays
For proliferation (counts of viable cells) and colony formation assays, GFP-positive Gfi-1-transduced p210BCR/ABL cells were washed with phosphate-buffered saline (PBS) and seeded at a cell density of 105 cells/ml or plated in methylcellulose (250 to 5,000 cells/plate). MigRI-Gfi-1-ER cells were cultured in the presence of 4-HT (250 ng/ml) or pre-treated (6 h) with 4-HT before plating in methylcellulose in the presence of 4-HT (250ng/ml). Viable cells were counted by trypan blue exclusion. Colonies were counted 5 (32D-p210BCR/ABL cells) or 7 (K562 cells) days later.
Differentiation was assessed by May-Grunwald/Giemsa staining and by detection of differentiation-related markers (Gr-1, CD11b, CD15) with specific phycoerythrin (PE)-conjugated mouse monoclonal antibodies (PharMingen, San Diego, CA).
Reverse transcription-PCR analysis
For RT-PCR analysisof pre-mRNA expression, nuclear RNA was extracted from untreatedor 4-HT–treated 32D-p210BCR/ABL-C/EBPα-ER cells using the PARISkit (Ambion, Austin, TX) and nuclear RNA was DNase-treated usingTurbo DNase treatment and Removal Reagents (Ambion), reversetranscribed, and PCR-amplified with a pair of Gfi-1 intron 1 primers(upstream primer: 5′-ACACCGACTCTGCTCCAAG-3′ and downstream primer: 5′CTGCGGAAAAGATTCAGGAG-3′). For real time Q-PCR, total RNA was isolated using the RNeasy Mini kit (Qiagen), 2μg was reverse transcribed and the resulting first-strand cDNA used as PCR template. All reactions were done in triplicates. Primer pairs designed using the ABI Primer Expression Software are: murine Gfi-1 FW(5′-AGGAGGCACCGAGAGACTCA-3′), RV(5′-GGGAGGCAGGGAAGACATC-3′); murine HPRT FW(5′-AGTCCCAGCGTCGTGATTAGC-3′), RV (5′-GAGCAAGTCTTTCAGTCCTGTCC-3′). Real time PCR was done using iQ SYBR Green supermix (Biorad) on a My IQ thermocycler (Biorad). HPRT, a housekeeping gene with constant expression, was used as an internal control to normalize input cDNA.
shRNA Transfection
For shRNAs transfection using the Amaxa cell optimizationkit V (Amaxa, Gaithersburg, MD), cells were resuspended in the nucleofector V solutionand 100 μL of cell suspension at a density of 106/mL wasmixed with 2 μg of lentivirus human Gfi-1 shRNAs ( TRCN0000020466, Open Biosystem). The solution was added to Amaxa electrode cuvettes and electroporated in an Amaxa NucleofectorII, using program T-16 for K562 cells.
Chromatin immunoprecipitation (ChIP) assays
Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP Assay Kit (USB Corporation, Cleveland, Ohio). 4 × 107 exponentially growing C/EBPα-ER-32Dp210-BCR/ABL cells (untreated and 4-HT-treated, 24 h) were cross-linked with 1% formaldehyde, incubated for 30 min at 37°C and treated with glycine (final concentration125 mM; 5 minutes at room temperature). Cells were then washed withice-cold PBS and resuspended in 1mL oflysis buffer with a protease inhibitor cocktail and sonicated at 28% power for 3 pulses of 10seconds each in a Branson Sonifier 450 (Branson Ultrasonics, Danbury, CT). Chromatin was pre-cleared with 50 μL protein A-agarose beads for 60 minat 4°C with rotation and pre-cleared lysates were immunoprecipitatedwith the anti C/EBPα antibody (8 μg; Santa Cruz Biotechnology, sc-61) at 4°C overnight withrotation. Immunoprecipitations without antibody (no antibody control) or an anti-rabbit IgG (8 μg;Thermo Scientific Pierce, Rockford, IL) were included with each experiment. Immune complexes were collected with 50 μL protein A-agarose beads for 60 minutesat 4°C with rotation (except for 10μL of supernatant of the no antibody control saved as input ) and washed once with 1 mL each of: low salt wash buffer (0.1% SDS; 1% Triton X-100; 2 mMEDTA; 20 mM Tris-HCl, pH 8.0; and 150 mM NaCl), high salt washbuffer (0.1% SDS; 1% Triton X-100; 2 mM EDTA; 20 mM Tris-HCl, pH 8.0; and 1.5 M NaCl), LiCl wash buffer (250 mM LiCl; 1% Nonidet P-40 [NP-40]; 1% sodium deoxycholate; 1 mM EDTA; and 10 mM Tris-HCl,pH 8.0); and twice with 10 mM Tris-HCl, pH 8.0; and 1 mM EDTA. Immune complexes were next eluted using freshly prepared elutionbuffer (1% SDS and 0.1 M NaHCO3). Cross-links were reversed byheating at 65°C overnight in the presence of 0.2 M NaCl. DNA was recovered using PrepEase DNA Clean-Up Kit (USB Corporation). ChIP DNA (2 μL) was used as a template for real time quantitative PCR using the Gfi-1 promoter (sense 5′GGGACTTAGTTTCTGAAGC3′, antisense 5′GTTTGCCTAGGGACTATGTG 3′ from −259 to −58) and G-CSFR promoter (sense 5′ TAGCTCACCAGTTTCCCTCA 3′ and antisense 5′GCAAGTTCCCGAAACTTCTA 3′, from −174 to + 8) primers.
Luciferase reporter assays
For luciferase assays, 293T cells were plated into 6-well plates at 5×104 cells per well and cultured overnight. Cells were transfected the following day with 1,5 μg of Gfi-1(−500+150) pGL3 or C/EBP-MUT Gfi-1(−500 to +150) pGL3, 1/50 of Renilla reporter, and ΔuORFC/EBPα-HA (p42 C/EBPα) or K298E C/EBPα-HA expression plasmid as required for a total of 6μg DNA/well using the Profection Mammalian Transfection System (Promega). After 48h, cells were washed, lysed and levels of firefly and Renilla luciferase activities measured using a dual luciferase assay kit (Promega). All transfections were performed in triplicates in three independent experiments.
Statistical Analyses
Means were compared using the unpaired, 2-tailed Student’s t test. A p value of less than 0.05 was considered statistically significant in all calculations.
RESULTS
Activation of C/EBPα induces Gfi-1 expression in p210BCR/ABL-expressing cells
Ectopic expression of C/EBPα promotes granulocytic differentiation, inhibits proliferation and suppresses leukemogenesis of p210BCR/ABL-expressing cells (13, 16). For optimal C/EBPα-dependent suppression of leukemogenesis, proliferation inhibition and differentiation induction are both required as the DNA binding-deficient K298E C/EBPα mutant which fails to induce granulocytic differentiation (16,22) is less effective of transcriptionally competent C/EBPα (16).
To identify potential mechanisms involved in the leukemia suppressive effects of C/EBPα, microarray hybridization studies were performed using RNA from 4-HT-treated 32D-p210BCR/ABL cells transduced with MigRI C/EBPα-ER or MigRI K298E C/EBPα-ER. Total RNA was extracted at 12 h post-4-HT treatment in three separate experiments and hybridized onto the Affymetrix MOE430_2 array, as described (28). We identified several genes whose expression was downregulated by wild-type C/EBPα but not by K298E C/EBPα, including the transcription factors c-Myb and GATA-2 (28). Instead, the transcription repressor Gfi-1 was among those whose mRNA levels were upregulated by C/EBPα in a DNA binding- dependent manner.
To confirm the microarray hybridization data, we assessed Gfi-1 mRNA levels in 4-HT-treated 32D-p210BCR/ABL cells expressing wild type or DNA binding deficient K298E C/EBPα-ER. Levels of Gfi-1 mRNA were upregulated as early as six hours post 4-HT treatment in cells expressing wild type C/EBPα but not in cells expressing the K298E mutant (Figure 1A).
Figure 1. Expression of Gfi-1 is induced by C/EBPα in BCR/ABL-expressing cells.
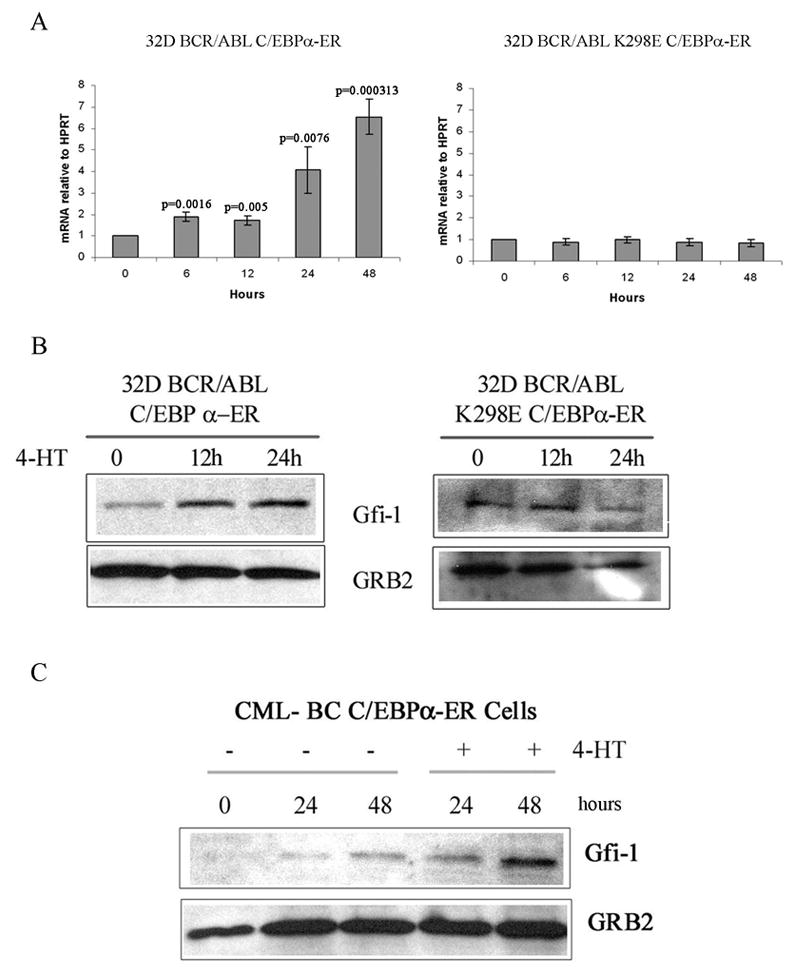
(A) Levels of Gfi-1 mRNA measured by real time PCR in 4-HT-treated C/EBPα-ER- or K298E C/EBPα-ER 32D-BCR/ABL cells; Western blot show expression of Gfi-1 in: (B) 4-HT-treated wild type C/EBPα-ER or K298E C/EBPα-ER 32D-BCR/ABL cells; (C) 4-HT-treated C/EBPα-ER-transduced CD34+ CML cells.
Consistent with the changes in mRNA expression, Gfi-1 protein levels increased upon activation of wild type C/EBPα but not of the K298E mutant (Figure 1B). We also tested whether activation of C/EBPα induced Gfi-1 expression in primary CML cells. To this end, CD34+ cells from a CML-BC patient (M351T mutation) were retrovirally transduced with MigRI C/EBPα-ER, GFP-sorted and treated with 4-HT to activate C/EBPα. After 24 and 48 h, lysates were prepared and used for Western blotting to assess Gfi-1 levels. Expression of Gfi-1 was induced upon C/EBPα activation (Figure 1C), consistent with the results in 32D-p210BCR/ABL cells.
The Gfi-1 promoter is activated by C/EBPα
To determine whether the effect of C/EBPα on Gfi-1 expression is due to a transcriptional mechanism, we first assessed expression of Gfi-1 pre-mRNA in nuclear RNA of untreated and 4-HT-treated C/EBPα-ER 32D-p210BCR/ABL cells. As shown in Figure 2A, levels of Gfi-1 pre-mRNA transcripts were increased approximately 3–5 fold upon 4-HT treatment. We then performed luciferase assays in 293T cells co-transfected with wild type or K298E C/EBPα expression plasmid and a Gfi-1 5′ flanking region (−500 to + 150) Luc plasmid (29) which contains a high- and a low-score putative C/EBP binding sites based on search of transcription factor binding site databases (30). In three separate experiments, wild type C/EBPα induced an approximately 3-fold increase (p=0.001) in Gfi-1-dependent luciferase activity (Figure 2B, upper panel), suggesting that Gfi-1 is a C/EBPα target.
Figure 2. Gfi-1 is a transcriptional target of C/EBPα.

(A) Steady-state Gfi-1 pre-mRNA levels in 4-HT-treated C/EBPα-ER-32D-BCR/ABL cells detected by RT-PCR (upper panel) and quantitated by gel densitometry (lower panel); (B) Effect of wild type or K298E C/EBPα on luciferase activity driven by the (−500 to +150) Gfi-1 5′ flanking region or by the corresponding promoter with mutated C/EBP binding site; (C), upper panel: nucleotide sequence of the murine Gfi-1 5′ flanking region shows (underlined) a putative C/EBP binding site; lower panels: ChIP assays (representative of two experiments) show the interaction of C/EBPα-ER to a segment of the Gfi-1 promoter containing the putative C/EBP binding site (−248 to −239) and to a segment of the murine G-CSFR promoter (−174 to + 8), containing a bona fide C/EBP binding site.
To further investigate the role of C/EBPα in transcriptional activation of Gfi-1, we performed ChIP assays to determine whether C/EBPα binds to the Gfi-1 promoter. Since expression of Gfi-1 is rapidly induced upon C/EBPα activation in 32D-p210BCR/ABL cells, ChIP assays were performed in untreated 32D-p210BCR/ABL-C/EBPα-ER cells and in cells treated for 24 h with 4-HT to activate C/EBPα; thereafter, the binding of C/EBPα to the Gfi-1 promoter was assessed by real-time quantitative PCR amplification of a segment of the Gfi-1 5′ flanking region which includes the high-score putative C/EBP binding site (nucleotides – 247 to −238, underlined in Figure 2C, upper panel). As shown in Figure 2C, lower panel, activation of C/EBPα by 4-HT treatment led to its interaction with nucleotides −259 to −58 of the Gfi-1 promoter. As control, C/EBPα bound to a segment of the G-CSFR promoter (−174 to + 8) containing a canonical C/EBP binding site (31).
To further assess the functionality of this C/EBP binding site at nucleotides −247 to −238 (Figure 2C) of the Gfi-1 promoter, we generated a mutated (TCTGAAGCAA into TCTGAAACAG) Gfi-1 (−500 to+150)-Luc plasmid and performed luciferase assays upon co-transfection of wild type or K298E C/EBPα and the mutated Gfi-1-Luc reporter plasmid in 293 cells; as shown in Figure 2B (lower panel), the mutated promoter was no longer transactivated by wild type C/EBPα, confirming that the segment of the Gfi-1 5′ flanking region bound by C/EBPα in the ChIP assays contains a functional C/EBPα binding site.
C/EBPα was also unable to transactivate a shorter Gfi-1 promoter (−41 to + 150) lacking putative C/EBP binding sites (not shown).
Expression of Gfi-1 is required for C/EBPα-dependent inhibition of proliferation and colony formation of K562 cells
To determine whether expression of Gfi-1 is required for the biological effects of C/EBPα in BCR/ABL-expressing cells, C/EBPα-ER K562 cells (28) were transduced or transfected with Gfi-1 shRNA lentiviruses and levels of Gfi-1, numbers of viable cells, colony formation and differentiation assessed after 4-HT treatment (250nM) to activate C/EBPα.
In C/EBPα-ER K562 cells transduced with a scramble (a murine Gfi-1 shRNA with seven mismatches relative to the human sequence) or a Gfi-1 shRNA (TRCN0000020468), 4-HT treatment induced an increase in Gfi-1 expression in the scramble- but not in the Gfi-1 shRNA-transduced cells (Figure 3A).
Figure 3. Expression of Gfi-1 is required for C/EBPα-dependent proliferation inhibition in K562 cells.

(A and E) Western blot shows Gfi-1 expression in 4-HT-treated scramble or Gfi-1 shRNA-transduced or transfected C/EBPα-ER/K562 cells; histograms show: proliferation of 4-HT-treated scramble (B and F) or Gfi-1 shRNA (C and G)-transduced or transfected C/EBPα-ER-K562 cells and methylcellulose colony formation of 4-HT-treated scramble or Gfi-1 shRNA-transduced or transfected C/EBPα-ER-K562 cells (D and H). Expression of Gfi-1 was detected by anti-Gfi-1 goat polyclonal antibody (N-20; Santa Cruz Biotechnology). For cell count assays, cells were seeded at 105cells/ml; for colony formation assays, 1,000 cells/plate were seeded. Data (expressed as % inhibition of 4-HT-treated vs untreated cells) are represented as the mean ± SEM of 2 (B-D) or 3 (F-H) independent experiments. * denotes that the differences in cell counts between Gfi-1 and scramble shRNA K562 cells (compare panels C,G and B,F) are statistically significant (p= 0.001 and 0.002, respectively ).
Activation of C/EBPα markedly reduced the number of scramble-transduced K562 cells (Figure 3B), whereas there was only a modest effect in Gfi-1 shRNA-transduced K562 cells (Figure 3C); activation of C/EBPα in scramble-transduced K562 cells markedly suppressed colony formation (approximately 85% inhibition) whereas it was less effective (approximately 50% inhibition) in Gfi-1 shRNA-transduced cells (Figure 3D). Comparable, although less strong, effects (probably reflecting residual expression of Gfi-1) were also observed upon 4-HT treatment of C/EBPα-ER K562 cells transfected with a different Gfi-1 shRNA lentivirus (TRCN0000020466) (Figure 3D-H).
These cells were also used to assess the role of Gfi-1 expression in C/EBPα-dependent induction of granulocytic differentiation.
Upon 4-HT treatment, scramble shRNA-transfected K562 C/EBPα-ER cells showed morphological features of granulocytic differentiation and increased levels of CD11b and CD15 (Figure 4); in cells transfected with the Gfi-1 shRNA, treatment with 4-HT induced morphological differentiation and an increase in CD11b and CD15 levels comparable to those in the control cells (Figure 4), indicating that increased Gfi-1 expression is not required for C/EBP-dependent induction of differentiation in K562 cells.
Figure 4. Expression of Gfi-1 is not required for C/EBPα-induced differentiation of K562 cells.

(A) Morphology; (B) Cd11b; and (C) CD15 expression in 4-HT-treated scrambled (SO) or Gfi-1 shRNA-transfected C/EBPα-ER/K562 cells.
Effects of Gfi-1 in BCR/ABL-expressing cell lines
To assess the effects of Gfi-1 in p210BCR/ABL-expressing cell lines, 32D-p210BCR/ABL and K562 cells were transduced with MigRI-Gfi-1 or MigRI-Gfi-1 P2A (a mutant deficient in transcriptional repression) and GFP-positive cells were utilized for cell counts of viable cells, colony formation and differentiation assays.
Cells expressing wild type Gfi-1 proliferated less than those expressing the Gfi-1 P2A mutant (not shown); the effects of wild type Gfi-1 were even more striking in clonogenic assays: in both cell lines, Gfi-1-expressing cells formed fewer colonies (approximately 80% less) than cells expressing the Gfi-1 P2A mutant (Figure 5A and 5B). However, ectopic Gfi-1 expression did not induce granulocytic differentiation as 32D-p210BCR/ABL and K562 cells remained morphologically undifferentiated and did not express the differentiation marker Gr-1 (32D-p210BCR/ABL cells) or CD11b and CD15 (K562 cells) (not shown).
Figure 5. Effect of constitutive Gfi-1 expression on colony formation of BCR/ABL-expressing cells.

Colonies (triplicate plates) were counted 7 days after plating the indicated number of retrovirally-transduced (GFP-positive) 32D-p210BCR/ABL (A) or K562 (B) cells. Representative of three experiments.
We also tested the effects of Gfi-1 after conditional activation of the chimeric Gfi-1-ER protein in BCR/ABL-expressing cells.
To this end, 32D-p210BCR/ABL and K562 cells were transduced with the MigRI-Gfi-1-ER retrovirus, sorted for GFP expression and treated with 4-HT to activate Gfi-1. Compared to untreated cells, 4-HT-treated cells showed reduced proliferation (Supplementary Figure 1, panels A and C) and were markedly less clonogenic when plated in methylcellulose ( Supplementary Figure 1, panels B and D). Inhibition of colony formation upon Gfi-1 activation was approximately 66% in 32D-BCR/ABL cells and 80% in K562 cells. 4-HT treatment of parental or MigRI-transduced K562 cells had no effect on cell numbers or colony formation (Supplementary Figure 1, panels E and F).
Modulation of Gfi-1 expression affects colony formation of CD34+ CML cells
The effect of ectopic Gfi-1 expression was also tested in normal and CML CD34+ cells using methylcellulose colony formation assays. Compared to cells expressing the P2A Gfi-1 mutant, expression of wild type Gfi-1 in normal CD34+ cells induced a decrease in colony formation (Figure 6A); the inhibitory effect of Gfi-1 was also noted in CML-CP CD34+ cells; in three separate experiments, cells transduced with wild type Gfi-1 were markedly less clonogenic (approximately 65–75% inhibition) of those transduced with Gfi-1P2A (Figure 6B).
Figure 6. Effect of ectopic Gfi-1 expression on colony formation of CD34+ CML cells.

Colony formation and Gfi-1 expression of: (A) human CD34+ normal cells transduced with wild type or P2A Gfi-1 lentivirus; (B) CD34+ cells from CML-CP samples transduced with wild type or transcriptional repressor mutant Gfi-1 P2A lentivirus (experiments I and II) or retrovirus (experiment III). In experiment II, cells were also transduced with the empty vector lentivirus. Cells (5 × 104 normal CD34+; 2.5 × 104, 5 × 104 or 1 × 105 for CML CD34+, experiment I, II and III, respectively) were plated after GFP sorting; colonies were scored 12 days after plating.
Colony formation of lentivirus-transduced CD34+ cells from CML sample #2-221 was similar to that of cells transduced with the Gfi-1 P2A mutant, suggesting that the inhibitory effect of wild type Gfi-1 in CD34+ CML cells is not artificially enhanced as result of stimulatory effects of the Gfi-1 P2A mutant reported for murine hematopoietic progenitors (32).
Hematopoietic stem cells from Gfi-1 knockout mice show reduced quiescence and enhanced self-renewal (23,24), but it is unknown whether Gfi-1 expression is also required to restrict proliferation of normal or transformed human stem cells/early progenitors.
To address this issue, CD34+ cells from patients (n=3) with chronic phase CML were transduced with stocks of a scramble or a Gfi-1 shRNA lentivirus (TRCN0000020468, capable of inducing optimal Gfi-1 downregulation in K562 cells, see Figure 3A) pre-tested for comparable viral titers and plated in methylcellulose (5.0 × 103 to 2 × 105 cells/plate) in the presence of a cytokine cocktail and puromycin (3μM). Colonies were scored 7–10 days after plating.
Gfi-1 shRNA-transduced CD34+ CML cells were markedly more clonogenic than the scramble-transduced counterpart (Figure 7). Methylcellulose plates seeded with 2 × 105cells/plate were solubilized with PBS and cells were recovered and counted; viable cells from plates seeded with Gfi-1 shRNA- transduced CML cells were much more numerous of those from plates seeded with scramble-transduced cells (360,000 ± 60,000 vs 35,000 ± 25,000).
Figure 7. Effect of Gfi-1 RNAi on colony formation of CD34+ CML cells.

CD34+ cells (5 × 105) from CML-CP patients (n=3) were transduced with a Gfi-1 (TRCN0000020468) or scramble shRNA lentivirus and plated in methylcellulose (5 × 104, 1 × 105 or 2 × 105 cells/plate) in the presence of a cytokine cocktail and puromycin (3μM). Colonies were scored 7–10 days later. Histograms show number of colonies from Gfi-1 or scramble shRNA-transduced cells. Inset shows Gfi-1 expression (detected by Western blotting) in scramble- and Gfi-1 shRNA-transduced CD34+ CML cells. Cell lysates were obtained at the end of the lentiviral infection (before plating in puromycin-supplemented methylcellulose), possibly explaining the residual Gfi-1 expression in the Gfi-1 shRNA-transduced sample.
Together, these findings support the important role of Gfi-1 expression in restricting the proliferation of early CML progenitors.
Discussion
To identify mechanisms responsible for the anti-leukemia effects of wild type C/EBPα, we searched for biologically relevant transcription-regulated C/EBPα targets by probing oligonucleotide microarrays with RNA from 32D-p210BCR/ABL cells expressing the 4-HT-regulated wild type C/EBPα or the K298E DNA binding deficient mutant.
One of the genes whose expression was induced by C/EBPα in a DNA binding-dependent manner is the transcription repressor Gfi-1. This gene attracted our attention because of its reported involvement in maintenance of hematopoietic stem/progenitor cells and in terminal differentiation of myeloid precursors (23–27, 33).
Moreover, a functional relationship between C/EBPα and Gfi-1 is suggested by a recent study demonstrating that murine hematopoietic stem cells engineered to express a DNA binding deficient C/EBPα mutant exhibit reduced quiescence and, among several “quiescence” genes, express lower levels of Gfi-1 than the normal counterpart (8).
We show here that Gfi-1 is a direct transcriptional target of C/EBPα and its expression is required for the inhibitory effects of C/EBPα on proliferation and colony formation of p210BCR/ABL-expressing cells but not for induction of granulocytic differentiation. Consistent with these findings, ectopic expression of Gfi-1 also failed to induce granulocytic differentiation of 32Dp210BCR/ABL and K562 cells whereas it was highly effective in inhibiting proliferation and suppressing colony formation of p210BCR/ABL cells including CD34+ cells from CML patients.
These observations are apparently at odds with the notion that differentiation induction not proliferation inhibition depends on the activity of transcriptionally competent C/EBPα (16). Thus, the distinction in the biological effects of C/EBPα between transcription-dependent and independent mechanisms involved in induction of granulocytic differentiation and inhibition of proliferation, respectively, needs to be reassessed as this study shows that transcription mechanisms play also an important role in C/EBPα-dependent proliferation inhibition.
The importance of Gfi-1 expression for controlling the proliferation and survival of BCR/ABL-expressing cells was not limited to cells conditionally expressing a functional C/EBPα; Gfi-1 shRNA-transduced CD34+ CML cells were markedly more clonogenic than the scramble-transduced counterpart, supporting the essential role of Gfi-1 in restricting the proliferation of hematopoietic stem/progenitor cells as revealed by studies with murine Gfi-1 knockout hematopoietic cells (23, 24).
Although, at the moment, it is unclear whether normal and CML stem cells exhibit differential sensitivity to perturbation of Gfi-1 expression, understanding the mechanisms responsible for the biological effects of Gfi-1 could, conceivably, lead to the development of therapeutic strategies exploiting its effects.
The effects of Gfi-1 depend on its activity as transcriptional repressor as the P2A Gfi-1 mutant (which lacks a functional SNAG transcriptional repressor domain) had modest or no effects on proliferation and colony formation of p210BCR/ABL-expressing cells. Preliminary microarray analysis data upon Gfi-1 activation in K562 cells reveal that expression of several genes with established roles in hematopoietic stem cell proliferation and survival) (32, 34–38) was downregulated (c-Myb, STAT 5, Mcl-1) or upregulated (FOXO3a, PTEN).
Although it is unknown whether these are direct or indirect Gfi-1 targets, it seems likely that the effects of Gfi-1 in BCR/ABL-expressing cells could be recapitulated by strategies able to manipulate their expression. Indeed, co-expression of Mcl-1, STAT 5B and c-Myb almost completely reversed the anti-proliferative effect of Gfi-1 in K562 cells (data not shown).
In summary, we have identified Gfi-1 as a novel transcription target of C/EBPα which appears to play an important role as mediator of its inhibitory effects on proliferation and colony formation of p210BCR/ABL-expressing cells. These findings and those reported previously (28) are consistent with a model of C/EBPα function whereby it regulates, directly and indirectly, a network of transcription factors whose balanced expression might be necessary for controlling the proliferation versus the differentiation fate of early hematopoietic progenitors.
The fact that C/EBPα is structurally or functionally inactive in the majority of cases of acute myeloid leukemia (3, 4) further attests its essential role at the nodal points at which a balance in hematopoietic cell proliferation and differentiation needs to be maintained to prevent a potentially damaging disruption of tissue homeostasis.
Supplementary Material
Acknowledgments
This work was supported by National Cancer Institute grants CA 95111 and P01 78890 (B.C). M.R.L and A.R.S. were supported, in part, by a fellowship of the American-Italian Cancer Foundation (AICF). G.F-A. is supported by a fellowship of the Associazione Italiana Ricerca sul Cancro (AIRC).
References
- 1.Radomska HS, Huettner CS, Zhang P, Chang T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–4314. doi: 10.1128/mcb.18.7.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang PO, Iwasaki-Arai J, Iwasaki H, et al. Enhancement of hematopoietic self-renewal in the absence of the transcription factor C/EBPα. Immunity. 2004;21:853–863. doi: 10.1016/j.immuni.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- 4.Nerlov C. C/EBPα mutations in acute myeloid leukemias. Nat Rev Cancer. 2004;4:394–400. doi: 10.1038/nrc1363. [DOI] [PubMed] [Google Scholar]
- 5.Pabst T, Mueller BU, Zhang P, et al. Dominant-negative mutations of C/EBPα, encoding CCAAT/enhancer binding-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–270. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 6.Gombart AF, Hofmann WK, Kawano S. Mutations in the gene encoding the transcription factor CCAAT/enhancing binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood. 2002;99:1332–1230. doi: 10.1182/blood.v99.4.1332. [DOI] [PubMed] [Google Scholar]
- 7.Kirstetter P, Schuster MB, Bereshchenko O, et al. Modeling of C/EBP mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13:299–310. doi: 10.1016/j.ccr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Bereshcenko O, Mancini E, Moore S, et al. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPα mutant AML. Cancer Cell. 2009;16:390–400. doi: 10.1016/j.ccr.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 9.Pabst T, Mueller BU, Harakawa N, et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8; 21) myeloid leukemia. Nat Med. 2001;7:444–451. doi: 10.1038/86515. [DOI] [PubMed] [Google Scholar]
- 10.Westendorf JJ, et al. The t(8; 21) fusion product, AML-1-ETO, associates with C/EBPα, inhibits C/EBPα-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol. 1998;18:322–333. doi: 10.1128/mcb.18.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng R, Friedman A, Levis M, Li L, Weir EG, Small D. Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPα expression. Blood. 2004;103:1883–1890. doi: 10.1182/blood-2003-06-1978. [DOI] [PubMed] [Google Scholar]
- 12.Radomska HS, Basseres DS, Zheng R, Zhang P, Dayaram T, Yamamoto Y, Sternberg, et al. Block of C/EBPα function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med. 2006;203:371–381. doi: 10.1084/jem.20052242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrotti D, Cesi V, Trotta R, et al. BCR-ABL suppresses C/EBPα expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30:48–58. doi: 10.1038/ng791. [DOI] [PubMed] [Google Scholar]
- 14.Chang JS, Santhanam R, Trotta R, Neviani P, et al. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2 dependent suppression of C/EBPα-driven myeloid differentiation. Blood. 2007;110:994–1003. doi: 10.1182/blood-2007-03-078303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tavor S, Park DJ, Gery S, Vuong PT, Gombart AF, Koeffler HP. Restoration of C/EBPα exression in BCR-ABL+ cell line induces terminal granulocytic differentiation. J Biol Chem. 2003;278:52651–52659. doi: 10.1074/jbc.M307077200. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari-Amorotti G, Keeshan K, Zattoni M, Guerzoni C, Iotti G, Donato N, Calabretta B. Leukemogenesis induced by wild-type and ST1571-resistant BCR/ABL is potently suppressed by C/EBPα. Blood. 2006;108:1353–1362. doi: 10.1182/blood-2006-01-011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pabst T, Mueller BU, Harakawa N, et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8 ;21) myeloid leukemia. Nat Med. 2001;7:444–451. doi: 10.1038/86515. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, Timchenko NA. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell. 2001;8:817–28. doi: 10.1016/s1097-2765(01)00366-5. [DOI] [PubMed] [Google Scholar]
- 19.Porse BT, Pedersen TA, Xu X, Lindberg B, Wewer UM, Friis-Hansen L, Nerlov C. E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell. 2001;107:247–58. doi: 10.1016/s0092-8674(01)00516-5. [DOI] [PubMed] [Google Scholar]
- 20.Müller C, Calkhoven CF, Sha X, Leutz A. The CCAAT enhancer-binding protein alpha (C/EBPalpha) requires a SWI/SNF complex for proliferation arrest. J Biol Chem. 2004;279:7353–8. doi: 10.1074/jbc.M312709200. [DOI] [PubMed] [Google Scholar]
- 21.D’Alo’ F, Johansen LM, Nelson EA, et al. The amino terminal of E2F interaction domains are critical for C/EBP alpha-mediated induction of granulopoietic development of hematopoietic cells. Blood. 2003;102:3163–3171. doi: 10.1182/blood-2003-02-0479. [DOI] [PubMed] [Google Scholar]
- 22.Keeshan K, Santilli G, Corradini F, Perrotti D, Calabretta B. Transcription activation function of C/EBPα is required for induction of granulocytic differentiation. Blood. 2003;102:1267–1275. doi: 10.1182/blood-2003-02-0477. [DOI] [PubMed] [Google Scholar]
- 23.Hock H, Haniblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, Orkin SH. Gfi-1 restricts proliferation and preserves functional integrity of hematopoietic stem cells. Nature. 2004;431:1002–1007. doi: 10.1038/nature02994. [DOI] [PubMed] [Google Scholar]
- 24.Zeng H, Yucel R, Kosan C, Klein-Hitpass L, Moroy T. Transcription factor Gfi-1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 2004;23:4116–4125. doi: 10.1038/sj.emboj.7600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menensez S, Antipin J, Reva B, Koff A, Nimer SD. P53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dahl R, Iyer SR, Owens KS, Cuylear DD, Simon MC. The transcriptional repressor GFI-1 antagonizes PU.1 activity through protein-protein interaction. J Biol Chem. 2007;282:6473–6483. doi: 10.1074/jbc.M607613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hock H, Hamblen MJ, Rooke HM, Trover D, Bronson RT, Cameron S, Orkin SH. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity. 2003;18:109–120. doi: 10.1016/s1074-7613(02)00501-0. [DOI] [PubMed] [Google Scholar]
- 28.Soliera AR, Lidonnici MR, Ferrari-Amorotti G, Prisco M, Zhang Y, Martinez RV, Donato NJ, Calabretta B. Transcriptional repression of c-Myb and GATA-2 is involved in the biologic effects of C/EBPalpha in p210BCR/ABL-expressing cells. Blood. 2008;112:1942–1950. doi: 10.1182/blood-2007-09-114975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR, Singh H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 30.www.gene-regulation.com.
- 31.Iida S, Watanabe-Fukunaga R, Nagata S, Fukunaga R. Essential role of C/EBPα in G-CSF-induced transcriptional activation and chromatin modification of myeloid specific genes. Genes to Cells. 2008;13:313–327. doi: 10.1111/j.1365-2443.2008.01173.x. [DOI] [PubMed] [Google Scholar]
- 32.Greig KT, Carotta S, Nutt SL. Critical roles for c-Myb in hematopoietic progenitor cells. Seminars in Immunology. 2008;20:247–256. doi: 10.1016/j.smim.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 33.Horman SR, Velu SC, Chaubey A, Bourdeau T, Zhu J, Paul WE, Gebelein B, Grimes HL. Gfi-1 integrates progenitor versus granulocytic transcriptional programming. Blood. 2009;113:5466–5475. doi: 10.1182/blood-2008-09-179747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuringa JJ, Chung KY, Morrone G, Moore MA. Constitutive activation of STAT5A promotes human hematopoietic stem cell self-renewal and erythroid differentiation. J EXP Med. 2004;200:623–635. doi: 10.1084/jem.20041024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wierenga ATJ, Vellenga E, Schuringa JJ. Maximal STAT5-induced proliferation and self-renewal at intermediate STAT5 activity levels. MolCell Biol. 2008;28:6668–6680. doi: 10.1128/MCB.01025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 37.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M, Chen C, Hosokawa K, Nakauchi H, Nakayama KI, Harada M, Motoyoma N, Suda T, Hirao A. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Grindlet JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfshl KS, Wiedemann LM, Wu H, Li L. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.