

Abstract
The clinical experimental agent, β-lapachone (Arq 501), can act as a potent radiosensitizer in vitro through an unknown mechanism. In this study, we analyzed the mechanism to determine whether β-lapachone may warrant clinical evaluation as a radiosensitizer. β-lapachone killed prostate cancer cells by NAD(P)H:quinone oxidoreductase 1 (NQO1) metabolic bioactivation, triggering a massive induction of reactive oxygen species (ROS), irreversible DNA single strand breaks (SSBs), PARP-1 hyperactivation, NAD+/ATP depletion, and μ-calpain-induced programmed necrosis. In combination with ionizing radiation (IR), β-lapachone radiosensitized NQO1+ prostate cancer cells, under conditions where nontoxic doses of either agent alone achieved threshold levels of SSBs required for hyperactivation of PARP-1. Combination therapy significantly elevated SSBs, γ-H2AX foci formation, and poly(ADP-ribosylation) of PARP-1, which were associated with ATP loss and induction of μ-calpain-induced programmed cell death. Radiosensitization by β-lapachone was blocked by the NQO1 inhibitor, dicoumarol, or the PARP-1 inhibitor, DPQ. In a mouse xenograft model of prostate cancer, β-lapachone synergized with IR to promote antitumor efficacy. NQO1 levels were elevated in ~60% of human prostate tumors evaluated relative to adjacent normal tissue, where β-lapachone might be efficacious alone or in combination with radiation. Our findings offer a rationale for clinical assessment of β-lapachone (Arq501) as a radiosensitizer in prostate cancers that overexpress NQO1, offering a potentially synergistic targeting strategy to exploit PARP-1 hyperactivation.
Keywords: β-Lapachone, PARP-1 Hyperactivation, Programmed Necrosis, Prostate Cancer, Radiosensitization, Synergy
Introduction
Prostate cancer is the most common non-cutaneous cancer in men in the United States. It occurs with the highest incidence (25%) of all cancers and is the second leading cause of cancer-related death in men (1). Radiation therapy (XRT) using fractionated low doses of ionizing radiation (IR) is the most heavily utilized therapeutic method for treating primary prostate cancers. However, traditional external beam fractionated XRT using total IR doses of <68 Gy have only limited curative potential for locally advanced stages of prostate cancer, with a high (~70%) five-year relapse rate (2). These lower doses are not efficacious for treating prostate cancer due to intrinsic radio-resistance (3, 4), while accumulated high doses of IR cause severe side-effects, such as urinary and bowel dysfunction, erectile dysfunction and infertility. These complications are caused by damage to surrounding normal tissue by surgery, XRT and/or chemotherapies, as a result of the lack of tumor-selectivity. Efficacious and tumor-selective synergistic strategies for treating human prostate cancers are in great demand.
β-Lapachone (β-lap, 3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran5-6-dione, aka., Arq 501), a novel antitumor quinone, has shown promise alone as a tumor-selective chemotherapeutic agent in cancers that over-express endogenous NAD(P)H:quinone oxidoreductase-1 (NQO1, E.C. 1.6.99.2), a two-electron oxidoreductase. We previously showed that NQO1 metabolized β-lap through a futile cycle with the parent quinone converted to a highly unstable hydroquinone form, utilizing dramatic levels of NAD(P)H. As a result, high levels of reactive oxygen species (ROS) are created, causing DNA lesions in NQO1+ cells (5–7). At LD90 and higher doses of β-lap (≥4 μMol/L), poly(ADP-ribose) polymerase-1 (PARP-1), a DNA damage sensor, is hyperactivated and extensive NAD+/ATP depletion ensues. PARP-1 hyperactivation required rapid Ca2+ release from endoplasmic reticulum (ER) stores, a result of ROS formation (8). Subsequently, loss of NAD+/ATP results in influx of Ca2+ leading to the activation and nuclear translocation of μ-calpain. Activation of μ-calpain causes a unique caspase-independent programmed necrotic cell death (8, 9). Importantly, β-lap killing of cancer cells is NQO1-specific and independent of cell cycle status, caspase activities or Rb and p53 statuses (6, 7, 10–12). Since NQO1 is highly expressed in many human cancers, including prostate, lung, pancreatic and breast, β-lap has become an attractive agent for selective cancer chemotherapy.
PARP-1 is an abundant nuclear enzyme essential for repair of DNA single strand breaks (SSBs) and an important damage sensor, for which numerous groups are developing inhibitors (13–15), particularly after its identification as a synthetic lethal target in BRCA1/2 breast cancers (16, 17). PARP-1 is essential for base excision (BER), SSB and possibly other DNA repair processes (18, 19). PARP-1 is activated after binding DNA strand breaks and uses NAD+ as a substrate to form long branched polymers of poly(ADP-ribose) (PAR). PARP-1-mediated poly ADP-ribosylation recruits various nuclear acceptor proteins, such as XRCC1, histones and PARP-1 itself to assemble other repair complexes to execute DNA repair [i.e., BER, SSB and double strand break (DSB) repair]. However, in response to excessive DNA damage, PARP-1 can be hyperactivated, converting its DNA repair capacity to initiation of programmed necrosis, due to dramatic NAD+ and ATP depletions. PARP-1 hyperactivation and programmed necrosis have been documented in several cellular responses, including ischemia-reperfusion, myocardial infarction and severe ROS-induced injury (20). To date, however, the only cytotoxic agent able to harness this cell death pathway in a tumor-selective manner, and at clinically relevant doses, is β-lap (6, 21).
We previously showed that β-lap was an efficient radiosensitizer of specific cancer cells when given immediately after or during IR in vitro (22, 23). However, these studies stalled due to a lack of knowledge concerning the agent’s mechanism of action and an inability to efficaciously deliver the drug. Based on our recent elucidation of β-lap’s mechanism of action as a single agent (8, 9), we hypothesized that PARP-1 hyperactivation was a key factor mediating synergy between sub-lethal doses of IR and β-lap. Here, we demonstrate for the first time that PARP-1 hyperactivation is the principle determinant governing β-lap radiosensitizing effects in NQO1+ human prostate cancer cells, causing early and rapid PAR-modified PARP-1 accumulation and synergistic ATP loss after IR + β-lap treatments. Along with dicoumarol (a specific inhibitor of NQO1), DPQ (a specific PARP-1 inhibitor) blocked dramatic ATP depletion and apoptosis, confirming an essential role of NQO1-dependent PARP-1 hyperactivation in mediating synergy between these two agents. Antitumor studies using PC-3 xenografts that have endogenously elevated NQO1 levels in athymic mice showed significantly enhanced anti-tumor efficacy using various combined sub-lethal doses of β-lap + IR. Thus, β-lap treatment in combination with XRT represents the first effective, tumor-selective therapy that exploits PARP-1 hyperactivation for the treatment of cancers that have elevated NQO1 levels.
Materials and Methods
Chemicals and Reagents
β-Lap was synthesized by Dr. Bill Bornmann (M.D. Anderson, Houston, TX), dissolved in Dimethylsulfoxide (DMSO) at 47 mmol/L, and concentrations verified by spectrophotometry. Hoechst 33258, hydrogen peroxide (H2O2), staurosporine, cytochrome c, etoposide, DPQ (3,4-dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline) and dicoumarol (24) were purchased from Sigma-Aldrich (St. Louis, MO).
Cell culture
PC-3, DU145 and LNCaP human prostate cancer cells were originally obtained from Dr. George Wilding (U. Wisconsin-Madison). PC-3 and DU145 cells were grown in RPMI 1640 Medium (Invitrogen, Carlsbad, CA) with 5% fetal bovine serum (FBS) and LNCaP cells were grown in Dulbecco’s minimal essential medium (DMEM, Invitrogen) with 10% FBS. Cells were cultured at 37 °C in a 5% CO2-95% air humidified atmosphere and were free of mycoplasma.
Western immunoblotting
Whole-cell extracts were prepared, proteins separated by SDS-PAGE, and Western blots developed using SuperSignal® West Pico Chemiluminescent substrate (Thermo Scientific, Rockford, IL), and exposed using autoradiography film (Denville Scientific Inc., Metuchen, NJ). An anti-human NQO1 antibody was kindly provided to us by Dr. David Ross (University of Colorado Health Science Center, Denver, CO) and used at a 1:5000 dilution overnight at 4 °C. Both PAR (BD Pharmingen, San Jose, CA) and γH2AX (Upstate, Billerica, MA) antibodies were used at 1:2000 and 1:1000 dilutions, respectively. β-Actin or α-tubulin levels were used as loading controls.
Relative survival assays
Relative survival (DNA content and colony forming assays) were assessed as described (25). Briefly for DNA content, cells were seeded at 5 × 103 per well in 48-well plates and allowed to attach overnight. Cells were then mock-treated or treated with various doses of β-lap (for 2 h) in the presence or absence of dicoumoral as indicated. Drug-free medium was then added and cells allowed to grow for 5–7 days until control cells reached ~100% confluence. DNA content was then determined by Hoeschst 33258 staining and fluorescence detection using a plate reader (Perkin-Elmer, Boston, MA) (21). Relative survival assays after combined treatment were confirmed by colony-forming ability assays (23). Results were reported as means ± SE from at least three independent experiments.
Total and oxidized glutathione assays
Disulfide and total glutathione (GSH and GSSG, respectively) levels were determined using a spectrophotometric recycling assay (26). After treatments, whole cell homogenates were prepared (11). Data were expressed as %GSSG/total, normalized to protein content using Lowry et al. (27). Shown are means ± SE for experiments performed at least three times.
Alkaline and neutral comet assays
DNA lesions, including total base damage, DSBs and SSBs, versus DSBs were assessed using single-cell gel electrophoretic comet assays under alkaline or neutral conditions, respectively (TREVIGEN, Gaithersburg, MD). Slides were stained with SYBR Green and visualized using a Nikon Eclipse TE2000-E fluorescence microscope (Melville, NY). Digital photomicrographs were taken and comet tail lengths quantified using NIH Image J software. Each datum point represents an average of 100 cells ± SE, and data are representative of experiments performed in triplicate.
Nucleotide analyses
Changes in intracellular NAD+ levels were measured (6) and levels were expressed as percent treated divided by control (% T/C) ± SE from at least three individual experiments. ATP levels were analyzed from whole-cell extracts using CellTiter-Glo Luminescent Cell Viability Assays (Promega, Madison, WI) following the manufacture’s instructions. Data were graphed as means ± SE from at least three independent experiments in triplicate.
Apoptotic assays
Apoptosis was quantified using ApoDirect™ (TUNEL) assays from BD Pharmingen (21). Samples were analyzed by using FC-500 flow cytometer (Beckman Coulter Electronics, Brea, CA) and Elite acquisition software. Data were expressed as means ± SE from three independent experiments.
Antitumor efficacy
Athymic nu/nu mice were purchased from Charles River Laboratories International, Inc (Wilmington, MA). All animals were housed in a pathogen-free facility with 24-h access to food and water. Experimental protocols were approved by the institutional Animal Care and Use Committee at the University of Texas Southwestern. PC3 cells (5 × 106) were subcutaneously injected into the right thighs of athymic nude mice and tumor volumes allowed to reach ~350 mm3. Mice (5 mice/group) were then randomly grouped with no statistical differences in tumor sizes among the six groups. Mice were then mock-treated or exposed to various IR doses followed immediately by treatment with various doses (10–30 mg/kg) of HPβ-CD-β-lap or HPβ-CD. When used, various doses of IR were given locally first to tumor sites with whole-body shielding. Mice were exposed to one treatment regimen, consisting of mock or XRT, immediately followed by HPβ-CD alone or various HPβ-CD-β-lap doses administered via tail vein injections for five IR + β-lap exposures. Tumor volumes were measured by caliper (length × width × width/2) every other day. Mice were sacrificed when tumors reached 2 cm3 or 10% total body weight.
Statistical analyses
For relative survival, different IR + β-lap combinations were fit with simple multi-target models in SigmaPlot for Windows Version 11.0. For synergy, a statistical definition of synergy (28) was used and calculations performed by fitting experimental data with the Machado and Robinson model using the R code (29). Equitoxic doses listed in Table 1 were calculated by parameters of the model of Machado and Robinson obtained in fitting. Regression analyses of tumor growth profiles in vivo in six tested groups were analyzed using a mixed model approach with AR (1) correlation structures. Log-rank tests were applied to survival analyses (Kaplan-Meier curves). In general, p values of ≤0.05 using two-sided student’s t-test were considered significant. All statistical analyses were performed using SAS 9.1 Service Pack 4.
Table 1.
Equitoxic doses comparing single to combined treatments in PC-3 cells
| IR (Gy) | β-Lap (μMol/L) | Equivalent dose of IR (Gy) | Equivalent dose of β-Lap (μMol/L) |
|---|---|---|---|
| 0 | 1 | 0.6 | 1 |
| 0 | 2 | 1.2 | 2 |
| 0 | 3 | 1.7 | 3 |
| 1 | 1 | 1.7 | 4.1 |
| 1 | 2 | 2.1 | 5.6 |
| 1 | 3 | 2.7 | 6.8 |
| 2 | 1 | 2.6 | 7.9 |
| 2 | 2 | 3.1 | 9.6 |
| 2 | 3 | 3.6 | 11.5 |
| 3 | 1 | 3.7 | 12.5 |
| 3 | 2 | 4.1 | 14.5 |
| 3 | 3 | 4.6 | 16 |
Assessment performed using Machado’s model. Values represent doses calculated from means for triplicate experiments from three independent experiments.
Results
β-Lap induces prostate cancer cell death via NQO1-induced ROS formation and SSBs
Our immunohistochemical (IHC) analyses of human prostate tumor and associated normal tissue revealed that ~60% of these cancers had elevated NQO1 levels (Supplemental Figure 1). Using human PC-3 prostate cancer cells that express high levels of endogenous NQO1, we showed that the cytotoxic effects of β-lap were NQO1-dependent (inhibited by dicoumarol Figure 1A). This was confirmed in DU145 and in NQO1-proficient (NQO1+) versus NQO1-deficient (NQO1−) LNCaP cells (Supplemental Figure 2). Importantly, only ~120 min exposure to 4 μMol/L β-lap was sufficient to achieve maximal cytotoxicity (Figure 1A), where significant levels of glutathione were oxidized (note rapid and elevated levels of %GSSG in 20–30 min, Figure 1B), suggesting dramatic ROS formation. Dramatic increases in SSBs were seen by alkaline comet assays, but DSBs, as assessed by neutral comet assays, were not noted (Figure 1C). Similar results were found using DU145, and NQO1+ LNCaP cells (Supplemental Figure 2). In contrast, NQO1- LNCaP cells were not responsive to β-lap as described (5).
Figure 1.
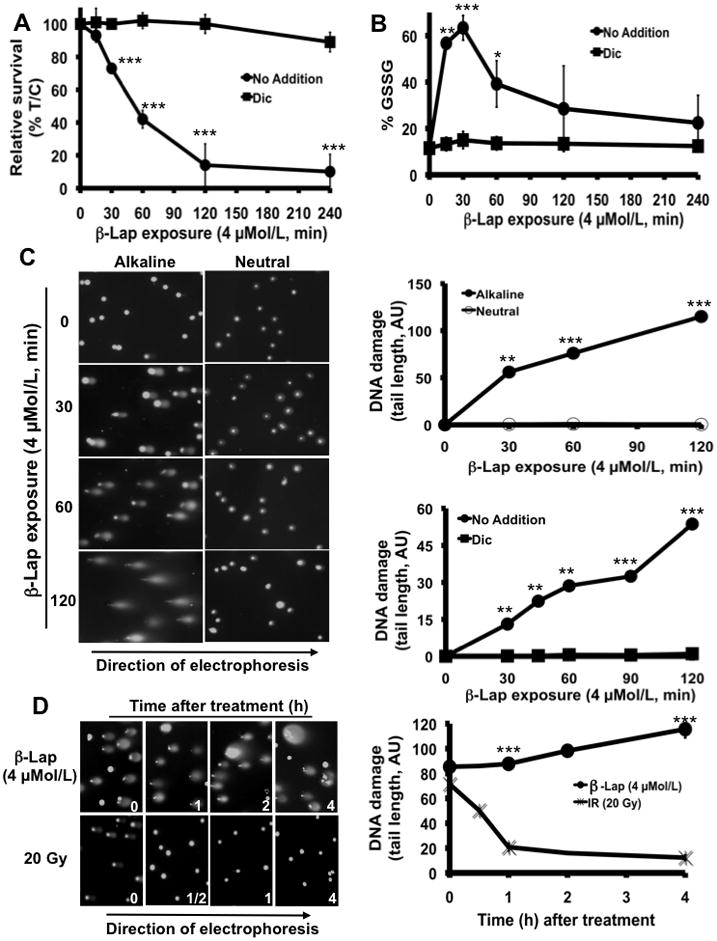
β-Lap-induced, NQO1-mediated ROS formation and SSBs are required for cell death in human prostate cancer cells. A, Relative survival of PC-3 cells after β-lap treatment in the presence or absence of dicoumarol (Dic, 40 μMol/L). Data are means ± SE for three independent experiments performed in sextuplicate. B, Reactive Oxygen Species (ROS) formation was indirectly monitored using the Oxidized Glutathione (GSSG) recycling assay in β-lap-exposed PC-3 cells in the presence or absence of 40 μMol/L Dic. Graphed results were means ± SE and represented three experiments performed in duplicate. C, Left panel, alkaline vs. neutral comet assays to assess total DNA damage or DNA double strand breaks (DSBs), respectively. H2O2 and Etoposide were used as positive controls for agents causing mostly SSBs or DSBs, respectively (not shown). Right panel, DNA damage assessment (arbitrary units (AU) of comet tail lengths) using NIH Image J software. Top graph as indicated, bottom graph (alkaline conditions), Data are means ± SE from 100 cells. D, DNA damage in PC-3 cells after IR (20 Gy) versus β-lap (4 μMol/L, 2h; left panel). Comet tail lengths (arbitrary units, AU) assessed using NIH Image J software. Data are means ± SE from 100 cells (right panel). Statistics (t-tests) were performed ***p < 0.001, **p < 0.01, *p < 0.05.
DNA damage and repair responses of β-lap-treated NQO1-expressing PC-3 cells were compared to responses after IR treatment (Figure 1D). Extensive DNA lesions were noted in PC-3 cells after exposure to 4 μMol/L β-lap, equivalent to 20 Gy by alkaline assays. However, neutral comet assays revealed DSBs after IR, but not after β-lap exposures (not shown). Exposure of PC-3 cells to IR (20 Gy) resulted in DNA damage that was quickly repaired within 1 h post-treatment, whereas DNA damage created by 4 μMol/L β-lap was not repaired, but escalated over the 4 h time-period assessed, suggesting repair inhibition.
PARP-1 hyperactivation mediates β-lap-induced programmed cell death
Exposure of PC-3 cells to lethal doses of β-lap (Figure 2A) caused extensive PARP-1 hyperactivation, with significant PAR accumulation within 10–20 min that was blocked by dicoumarol (Figure 2B). Loss of PAR formation in β-lap-treated PC-3 cells noted from 40–60 min was most likely due to NAD+ substrate depletion (Figure 2C), as well as functional PARG (30). PARP-1 hyperactivation was accompanied by dramatic NAD+ and ATP losses as a function of (i) time (Figure 2C), where metabolite levels were exhausted within 120 min of β-lap exposure; and (ii) dose, where loss of ATP corresponded well with cytotoxicity (Figures 2A, 2D). Loss of intracellular nucleotide levels (NAD+/ATP) and lethality of β-lap-treated PC-3 cells were blocked by dicoumarol (40 μMol/L). Dicoumarol also prevented PARP-1 hyperactivation, NAD+ and ATP losses and cytotoxicity in DU145 cells after β-lap exposure (Supplemental Figure 3).
Figure 2.

NQO1-dependent cytotoxicity correlates with PARP-1 hyper-activation, nucleotide depletion and PAR formation in prostate cancer cells exposed to β-lap. A, Relative survival (DNA content left panel, colony forming right panel) of PC-3 cells exposed for 120 min to varying doses of β-lap in the presence or absence of dicoumarol (Dic, 40 μMol/L). Data are means ± SE for three independent experiments. B, Western blot analyses of PAR formation in PC-3 cells treated with DM (DMSO) or 4 μMol/L β-lap in the presence or absence of Dic at the indicated times. C, PC-3 cells were treated as in ‘B’, and cells harvested at indicated times were analyzed for NAD+ and ATP levels. Results are means ± SE for experiments performed three times in triplicate. D, ATP in β-lap-exposed PC-3 cells treated with various β-lap doses in the presence or absence of Dic (40 μMol/L). Data are means ± SE for six replicates from three independent experiments. Statistics (t-tests) were performed ***p < 0.001, **p < 0.01, *p < 0.05.
Synergy between IR and β-lap is mediated by DNA damage, reaching a threshold for PARP-1 hyperactivation
We previously reported that the combination of IR and β-lap synergistically killed specific cancer cells (31), however, the mechanism of synergy was not elucidated. PC-3 cells were treated with single doses of IR (1–3 Gy) followed by exposure to low, non-toxic doses of β-lap (1–3 μMol/L). Synergy was noted with all IR and β-lap combinations tested (Figure 3A), corresponding to synergistic increases of PAR levels after combined treatments, but not after single agent exposures. For example, dramatic PAR formation in PC-3 cells treated with 1 Gy + 3 μMol/L β-lap was noted at 60 min, with no apparent levels in cells after each agent alone (Figure 3B). Similar responses were noted using NQO1+ LNCaP and DU145 cells, but not in genetically matched NQO1- LNCaP cells (Supplemental Figures 4A and 5B, respectively). Synergy was prevented by dicoumarol in NQO1+ prostate cancer cells, corresponding to the formation of DNA lesions (noted by alkaline comet and γH2AX foci formation) that presumably reached threshold levels required for PARP-1 hyperactivation (Supplemental Figure 5). Synergy between IR and β-lap in PC-3 cells was accompanied by dramatic losses of ATP (Figures 3C–D for synergy between 2 or 3 Gy and 3 μMol/L β-lap) and NAD+ (not shown). Importantly, synergistic losses of ATP in PC-3 cells following 2 or 3 Gy + 3 μMol/L β-lap were prevented by pre- and co-treating cells with DPQ, a specific PARP-1 inhibitor (Figures 3C and 3D, respectively) that prevented β-lap-induced cell death alone in various endogenously over-expressing NQO1 cancer cells (6, 21). Synergistic ATP loss was also observed in NQO1+ LNCaP cells (Supplemental Figure 4B).
Figure 3.
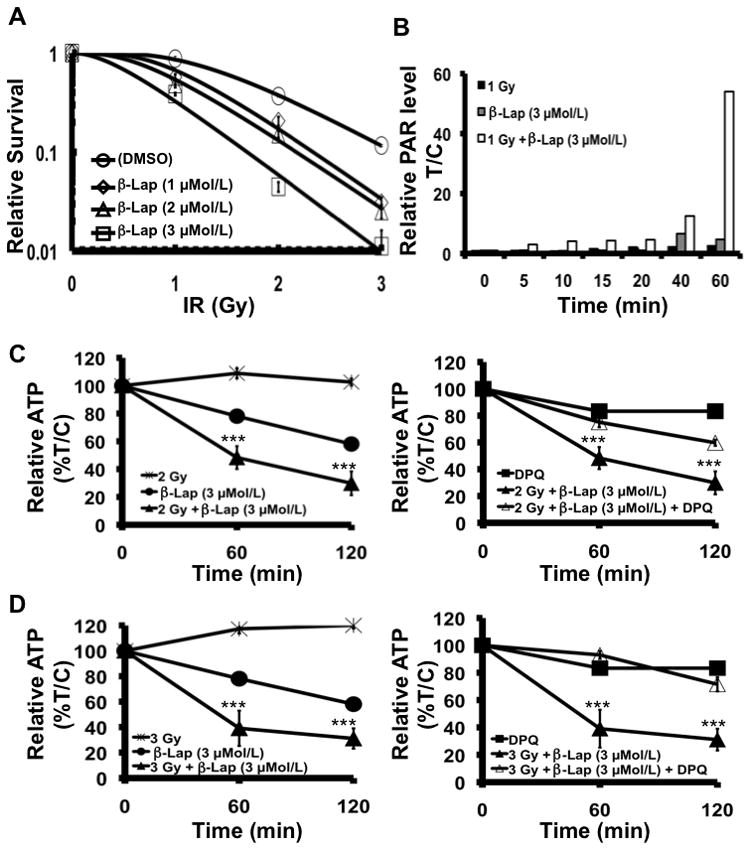
Synergy between IR and β-lap is mediated by PARP-1 hyper-activation. A, β-Lap exposure sensitizes PC-3 cells to IR. Results are means ± SE for three independent experiments repeated in triplicate. β12 > 1 (Machado’s model). B, Quantified PAR formation in PC-3 cells after various treatments. C-D, Synergistic ATP loss observed after IR + β-lap combinations (left panels). DPQ, a specific PARP1 inhibitor, blocked the synergistic ATP depletion effects of IR + β-lap (right panels). Results are means ± SE for experiments performed three times in octuplets. Statistics (t-tests) were performed comparing each datum point to those of single treatments. ***p < 0.001.
Synergy between IR and β-lap exposures involves atypical PARP-1 cleavage, and TUNEL+ programmed necrosis
Loss of survival as a result of β-lap treatment correlated well with TUNEL+ apoptotic responses (32). Synergistic cytotoxic responses of NQO1+ expressing PC-3 cells after IR + β-lap treatments were confirmed by analyzing apoptosis (Figure 4A). Treatment of PC-3 cells with 1–3 Gy, each in combination with 2 μMol/L β-lap, resulted in significant increases in apoptotic cells within 72 h (Figure 4A), corresponding directly to loss of colony forming ability (Figure 3A). Indeed, all combination therapies of IR with β-lap (Table 1) reveal synergy at these low doses of each agent. For example, treatment of PC-3 cells with nonlethal agents (alone) in combination with IR (i.e., 1 Gy + 1 μMol/L β-lap), was the same as treating cells with a lethal dose of 4 μMol/L β-lap. Similar responses were noted in NQO1+ LNCaP cells, in which synergistic levels of apoptosis and atypical PARP-1 cleavage at 72 h post-treatment were noted (Supplemental Figure 6). Synergy between IR and β-lap was prevented by dicoumarol, and not observed in NQO1-LNCaP cells. In contrast, different low doses of IR alone (i.e., 1–3 Gy) only led to 2 ± 2%, 5 ± 3% and 9 ± 3% apoptosis, respectively. Similarly, a low sublethal dose of 2 μMol/L β-lap in NQO1+ expressing PC-3 cells resulted in minimal apoptosis (i.e., 5 ± 2%, Figure 4A).
Figure 4.

Combined treatment with sublethal doses of IR and β-lap promotes apoptosis and atypical PARP-1 cleavage. A, PC-3 cells were exposed to the indicated treatments for 2 h, whole cell extracts prepared at 72 h, and apoptosis monitored by TUNEL reactions. Results are means ± SE from three independent experiments. Statistical differences between combined and single treatment regimen were indicated as *p<0.05. DPQ blocked apoptosis in all combinations (p < 0.01). B, PC-3 cells were treated as in A, above as indicated for 2 h and harvested at 48 h for Western blot analyses. A lethal β-lap dose of 4 μMol/L was used as positive control to indicate the ~60 kDa atypical cleaved PARP-1. TUNEL assay data were means ± SE from three independent experiments. UT: untreated.
Cell death caused by a lethal dose of β-lap in prostate cancer cells with endogenous elevation of NQO1 involves activation of μ-calpain and atypical cleavage of PARP-1 (9), as noted after 4 μMol/L β-lap treatment (Figure 4B, lane 7). Similarly, exposure of PC-3 cells with IR + β-lap involved synergistic apoptotic responses, above the additive levels of IR or β-lap alone. Atypical PARP-1 cleavage (i.e., formation of an ~60 kDa PARP-1 fragment) in combination-treated cells was noted (Figure 4B), resulting from activation and nuclear translocation of μ-calpain (9). Similar atypical PARP-1 cleavage events accompanied IR + β-lap synergy in NQO1+ LNCaP cells (Supplemental Figure 6B).
Efficacy in vivo of the combination of IR and β-lap
To date, efficacy of β-lap against human prostate cancer xenografts expressing elevated levels of endogenous NQO1 has not been demonstrated. Using the current clinical formulation of the drug (i.e., Arq 501), conjugated with HPβ-CD, we demonstrated significant efficacy of HPβ-CD-β-lap when administered at 10 or 20 mg/kg in combination with 2 Gy fractions of IR (Figure 5A). Mice (5/group) bearing PC-3 xenografts with an average tumor volume of ~350 mm3 were exposed to five doses of IR alone, HPβ-CD-β-lap alone, or IR + HPβ-CD-β-lap combinations every other day between days 1–9. All drug treatments were intravenous (iv) injection. Treatment of mice with HPβ-CD-β-lap at 10 or 20 mg/kg exhibited no antitumor efficacy, nor morbidity or mortality. Although mice treated with 2 Gy fractions (five treatments, every other day) resulted in significant tumor growth delay (ave: 26 days, Figure 5, Supplemental Figure 7A and Supplemental Table 1), combinations of IR (2 Gy) with 10 or 20 mg/kg HPβ-CD-β-lap resulted in significant tumor regression beyond additive from IR or HPβ-CD-β-lap treatments alone. Ninety percent (9/10) animals exposed to IR + 10 or 20 mg/kg HPβ-CD-β-lap were ‘apparently cured’, showing no further tumor growth for up to 150 days (Figure 5B, Supplemental Table 1). In contrast, 90% PC-3-bearing mice treated with HPβ-CD-β-lap alone (10 or 20 mg/kg) died within 40 days, similar to vehicle (HPβ-CD) alone control mice. Mice treated with IR (2 Gy) alone demonstrated an obvious delay in tumor growth (Figure 5A), however, all mice eventually died (sacrificed when tumor volumes were >10% of their weight). Finally, although mice treated with 2 Gy + 30 mg/kg HPβ-CD-β-lap resulted in statistically equivalent tumor growth delay and ‘apparent cures’ (Supplemental Table 1), we noted that the dose was close to the drug’s maximum tolerated dose (MTD, Supplemental Figure 7B). Collectively, we treated 15 mice (3X 5 mice/group) with 2 Gy + 10–30 mg/kg HPβ-CD-β-lap, noting dramatic synergistic responses, statistically superior (p < 0.001) to 2 Gy alone or HPβ-CD-β-lap alone (10–30 mg/kg) regimen (Supplemental Table 1). A representative experiment of these data is presented in Figure 5A.
Figure 5.

IR +β-lap elicit synergistic antitumor activity in PC-3 tumor xenografts. A, Anti-tumor efficacy using different treatment regimens in athymic mice bearing PC-3 xenografts. Mice bearing 350 mm3 PC-3 xenografts were treated once every other day starting on day one, for five treatments (Methods and Materials). Results are means ± SE. Mixed Model analyses showed ***p<0.0001 for combined vs single treatments, including untreated controls. B, Kaplan-Meier survival curves reflect significantly enhanced antitumor efficacy using various IR + β-lap regimen. ***p<0.001, *p<0.05. Log-rank p-values represent significance indices of each single versus combined treatments.
Discussion
In general, there is great need for improved combination therapy, where XRT is combined with tumor-selective therapies to increase antitumor efficacy while simultaneously decreasing normal tissue toxicity. For XRT, the intrinsic radioresistance of many tumors pose significant clinical obstacles, limiting efficacy (33). Since the first stereotactic body radiotherapy (SBRT) was developed in 1991, the method has been refined to decrease high doses of IR used in local primary or focal metastatic lesions (34, 35). Recently, hypofractionated SBRT delivered by CyberKnife using 3–5 fractionated high dose XRT resulted in minimal toxicity, lowering short-term PSA and preserving normal tissue function in men with localized prostate cancer (36, 37). However, such SBRT regimens are not suited to treat metastatic disease and long-lasting normal tissue toxicity is still problematic. Our data showed that XRT combined with β-lap post-treatments selectively killed NQO1+ prostate cancer cells, offering a strategy to use lower efficacious doses of IR for targeted therapy, while simultaneously eliminating prostate cancer micrometastases. This therapy should be applicable to all cancers that have elevated levels of NQO1.
β-Lap is the only agent that selectively kills tumor cells by PARP-1-mediated, μ-calpain-directed programmed necrosis at clinically relevant doses. Detailed knowledge of its mechanism of action can be exploited for improving XRT of tumors with elevated NQO1 levels. The tumor-selective nature of the therapy, for those individuals with elevated NQO1 levels, should allow reduced IR exposures, while achieving improved antitumor efficacy and simultaneously avoiding normal tissue complications. Importantly, resistance to β-lap-induced, NQO1-directed antitumor cytotoxicity has not been noted to date (10,12), most likely due to the diverse downstream effects of this drug in NQO1-containing cells, including dramatic alterations in nucleotide metabolism (i.e., NAD+/ATP losses), DNA damage, and loss of Ca2+ homeostasis.
DNA damage response (DDR) pathways have become new and effective targets to potentiate XRT. Considerable interest has been focused on targeting PARP-1 to eliminate DNA repair and enhance the sensitivity of cancer cells to other DNA-damaging agents, including chemotherapeutic agents and IR (39, 40). None of these approaches currently exploit PARP-1 hyperactivation, which requires a ‘threshold’ of DNA lesions to activate a suicide pathway. Our data suggest that massive levels of ROS formed by β-lap lead to persistent base lesions that are ultimately converted to SSBs by BER that, in turn, hyperactivated PARP-1. Due to dramatic NAD+/ATP losses, other repair processes that depend on energy are blocked, preventing repair of lethal DSBs created by IR. The delayed increase in γH2AX formation after PAR formation (Supplemental Figure 2B) is consistent with PARP-1 break protection, followed by exposure of SSBs and eventually DSB formation detected by ATM, resulting in γH2AX foci formation.
Current Phase I/II clinical trials of β-lap (i.e., Arq 501) were limited by high doses of HPβ-CD vehicle that caused hemolytic anemia, limiting efficacy. We show that non-toxic doses of HPβ-CD-β-lap can be delivered in combination with XRT for efficacious antitumor therapy of prostate cancers expressing endogenously elevated NQO1 levels. Efficacy of combination therapy was much greater than single regimens alone (Table 1). Synergy between IR and β-lap in vitro was determined using several applied models (28, 29, 41, 42), but the Machado model gave the most consistent results. Overall, the ability of β-lap to sensitize IR-exposed NQO1+ tumor cells relates to the higher DNA lesions created by the drug/radiation combination that meets a threshold level required for PARP-1 hyperactivation and synergistic cell death, consistent with prior findings (31).
Here, we show that lethal doses of β-lap alone, or sublethal doses of IR + β-lap kill NQO1+ prostate cancer tumor cells as a result of extensive ROS formation, massive DNA damage, PARP-1 hyperactivation and dramatic energy depletion (Figures 1 and 2). Energy depletion, in turn, dramatically inhibits DNA repair. PAR formation was detected after combined IR + β-lap treatments, but not after single sublethal exposures of IR or β-lap alone in NQO1+ LNCaP cells. LNCaP NQO1- cells were not responsive (Supplemental Figure 4A). Downstream, dramatic metabolic changes culminated in catastrophic Ca2+ homeostasis. Atypical PARP-1 cleavage was noted after combination therapy, but not after sublethal IR or β-lap single treatments alone in NQO1+ human prostate cancer cells (Figure 4B, Supplemental Figure 6B), consistent with μ-calpain-mediated TUNEL+ programmed cell death responses after lethal doses of β-lap (6, 9). Since β-lap alone or XRT + β-lap combination therapy kills cells independent of cell cycle or p53 status, it represents a controllable synergistic therapy for slow-growing NQO1+ human prostate cancers, as well as for non-small cell lung, pancreatic, and breast cancer patients, whose elevated NQO1 levels are more frequent and even greater than those enzyme activities in associated normal tissue. Importantly, we demonstrate for the first time that XRT + HPβ-CD-β-lap caused significant long-term tumor regression with no tumor recurrence. The use of β-lap to synergize with IR (XRT) offers selectivity that can be rapidly individualized, such as for those prostate cancer patients whose NQO1 levels are elevated. Patients with easily detectable polymorphisms in NQO1 can be screened using blood-derived SNP analyses and excluded from treatment (43). While efficacious, we theorize that improved drug delivery using millirods for brachytherapy (44) or nanoparticle miceles (45) with XRT will further augment anti-tumor efficacy for the treatment of cancers with elevated NQO1 levels, such as for prostate cancer.
Supplementary Material
Acknowledgments
This work was funded by NCI/NIH grant # 2 R01 CA102792-08, and in part, by DoD grant #W81XWH-06-1-0198 to DAB and a DoD Post-Doctoral grant, #W81XWH-08-1-0 to YD.
We thank the Tissue Procurement and Biostatistics Shared Resources supported by the Harold C. Simmons Comprehensive Cancer Center at UT Southwestern Medical Center. This is CSCN #052 from the Cell Stress and Cancer Nanomedicine (CSCN) program.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Niciforovic A, Djordjevic J, Adzic M, Vucic V, Mitrasinovic PM, Radojcic MB. Experimental and systems biology studies of the molecular basis for the radioresistance of prostate carcinoma cells. Ann Biomed Eng. 2008;36:831–8. doi: 10.1007/s10439-008-9457-4. [DOI] [PubMed] [Google Scholar]
- 3.Buron C, Le Vu B, Cosset JM, et al. Brachytherapy versus prostatectomy in localized prostate cancer: results of a French multicenter prospective medico-economic study. Int J Radiat Oncol Biol Phys. 2007;67:812–22. doi: 10.1016/j.ijrobp.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 4.van Tol-Geerdink JJ, Stalmeier PF, Pasker-de Jong PC, et al. Systematic review of the effect of radiation dose on tumor control and morbidity in the treatment of prostate cancer by 3D-CRT. Int J Radiat Oncol Biol Phys. 2006;64:534–43. doi: 10.1016/j.ijrobp.2005.07.974. [DOI] [PubMed] [Google Scholar]
- 5.Planchon S, Pink J, Tagliarino C, Bornmann W, Varnes M, Boothman D. beta-Lapachone-induced apoptosis in human prostate cancer cells: involvement of NQO1/xip3. Exp Cell Res. 2001;267:95–106. doi: 10.1006/excr.2001.5234. [DOI] [PubMed] [Google Scholar]
- 6.Bentle MS, Reinicke KE, Bey EA, Spitz DR, Boothman DA. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J Biol Chem. 2006;281:33684–96. doi: 10.1074/jbc.M603678200. [DOI] [PubMed] [Google Scholar]
- 7.Bentle MS, Reinicke KE, Dong Y, Bey EA, Boothman DA. Nonhomologous end joining is essential for cellular resistance to the novel antitumor agent, beta-lapachone. Cancer Res. 2007;67:6936–45. doi: 10.1158/0008-5472.CAN-07-0935. [DOI] [PubMed] [Google Scholar]
- 8.Tagliarino C, Pink JJ, Dubyak GR, Nieminen AL, Boothman DA. Calcium is a key signaling molecule in beta-lapachone-mediated cell death. J Biol Chem. 2001;276:19150–9. doi: 10.1074/jbc.M100730200. [DOI] [PubMed] [Google Scholar]
- 9.Tagliarino C, Pink J, Reinicke K, Simmers S, Wuerzberger-Davis S, Boothman D. Mu-calpain activation in beta-lapachone-mediated apoptosis. Cancer Biol Ther. 2003;2:141–52. doi: 10.4161/cbt.2.2.237. [DOI] [PubMed] [Google Scholar]
- 10.Bentle M, Bey E, Dong Y, Reinicke K, Boothman D. New tricks for old drugs: the anticarcinogenic potential of DNA repair inhibitors. J Mol Histol. 2006;37:203–18. doi: 10.1007/s10735-006-9043-8. [DOI] [PubMed] [Google Scholar]
- 11.Reinicke K, Bey E, Bentle M, et al. Development of beta-lapachone prodrugs for therapy against human cancer cells with elevated NAD(P)H:quinone oxidoreductase 1 levels. Clin Cancer Res. 2005;11:3055–64. doi: 10.1158/1078-0432.CCR-04-2185. [DOI] [PubMed] [Google Scholar]
- 12.Bey EA, Wuerzberger-Davis SM, Pink JJ, et al. Mornings with Art, lessons learned: feedback regulation, restriction threshold biology, and redundancy govern molecular stress responses. J Cell Physiol. 2006;209:604–10. doi: 10.1002/jcp.20783. [DOI] [PubMed] [Google Scholar]
- 13.Drew Y, Plummer R. PARP inhibitors in cancer therapy: two modes of attack on the cancer cell widening the clinical applications. Drug Resist Updat. 2009;12:153–6. doi: 10.1016/j.drup.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Peralta-Leal A, Rodriguez MI, Oliver FJ. Poly(ADP-ribose)polymerase-1 (PARP-1) in carcinogenesis: potential role of PARP inhibitors in cancer treatment. Clin Transl Oncol. 2008;10:318–23. doi: 10.1007/s12094-008-0207-8. [DOI] [PubMed] [Google Scholar]
- 15.Drew Y, Plummer R. The emerging potential of poly(ADP-ribose) polymerase inhibitors in the treatment of breast cancer. Curr Opin Obstet Gynecol. 22:67–71. doi: 10.1097/GCO.0b013e328334ff57. [DOI] [PubMed] [Google Scholar]
- 16.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 17.Turner NC, Lord CJ, Iorns E, et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–77. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noel G, Giocanti N, Fernet M, Megnin-Chanet F, Favaudon V. Poly(ADP-ribose) polymerase (PARP-1) is not involved in DNA double-strand break recovery. BMC Cell Biol. 2003;4:7. doi: 10.1186/1471-2121-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–67. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 21.Bey EA, Bentle MS, Reinicke KE, et al. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci U S A. 2007;104:11832–7. doi: 10.1073/pnas.0702176104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boothman DA, Schlegel R, Pardee AB. Anticarcinogenic potential of DNA-repair modulators. Mutat Res. 1988;202:393–411. doi: 10.1016/0027-5107(88)90201-1. [DOI] [PubMed] [Google Scholar]
- 23.Boothman D, Pardee A. Inhibition of radiation-induced neoplastic transformation by beta-lapachone. Proc Natl Acad Sci U S A. 1989;86:4963–7. doi: 10.1073/pnas.86.13.4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermisson M, Wagenknecht B, Wolburg H, Glaser T, Dichgans J, Weller M. Sensitization to CD95 ligand-induced apoptosis in human glioma cells by hyperthermia involves enhanced cytochrome c release. Oncogene. 2000;19:2338–45. doi: 10.1038/sj.onc.1203554. [DOI] [PubMed] [Google Scholar]
- 25.Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem. 2000;275:5416–24. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- 26.Lee YJ, Galoforo SS, Berns CM, et al. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J Biol Chem. 1998;273:5294–9. doi: 10.1074/jbc.273.9.5294. [DOI] [PubMed] [Google Scholar]
- 27.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 28.Lee JJ, Kong M, Ayers GD, Lotan R. Interaction index and different methods for determining drug interaction in combination therapy. J Biopharm Stat. 2007;17:461–80. doi: 10.1080/10543400701199593. [DOI] [PubMed] [Google Scholar]
- 29.Machado SG, Robinson GA. A direct, general approach based on isobolograms for assessing the joint action of drugs in pre-clinical experiments. Stat Med. 1994;13:2289–309. doi: 10.1002/sim.4780132202. [DOI] [PubMed] [Google Scholar]
- 30.Min W, Wang ZQ. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front Biosci. 2009;14:1619–26. doi: 10.2741/3329. [DOI] [PubMed] [Google Scholar]
- 31.Boothman DA, Greer S, Pardee AB. Potentiation of halogenated pyrimidine radiosensitizers in human carcinoma cells by beta-lapachone (3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran- 5,6-dione), a novel DNA repair inhibitor. Cancer Res. 1987;47:5361–6. [PubMed] [Google Scholar]
- 32.Wuerzberger SM, Pink JJ, Planchon SM, Byers KL, Bornmann WG, Boothman DA. Induction of apoptosis in MCF-7:WS8 breast cancer cells by beta-lapachone. Cancer Res. 1998;58:1876–85. [PubMed] [Google Scholar]
- 33.Gerweck LE, Vijayappa S, Kurimasa A, Ogawa K, Chen DJ. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer Res. 2006;66:8352–5. doi: 10.1158/0008-5472.CAN-06-0533. [DOI] [PubMed] [Google Scholar]
- 34.Lo SS, Fakiris AJ, Chang EL, et al. Stereotactic body radiation therapy: a novel treatment modality. Nat Rev Clin Oncol. 2010;7:44–54. doi: 10.1038/nrclinonc.2009.188. [DOI] [PubMed] [Google Scholar]
- 35.Martin A, Gaya A. Stereotactic Body Radiotherapy: A Review. Clin Oncol (R Coll Radiol) 2010;22:157–72. doi: 10.1016/j.clon.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Friedland JL, Freeman DE, Masterson-McGary ME, Spellberg DM. Stereotactic body radiotherapy: an emerging treatment approach for localized prostate cancer. Technol Cancer Res Treat. 2009;8:387–92. doi: 10.1177/153303460900800509. [DOI] [PubMed] [Google Scholar]
- 37.King CR, Brooks JD, Gill H, Pawlicki T, Cotrutz C, Presti JC., Jr Stereotactic body radiotherapy for localized prostate cancer: interim results of a prospective phase II clinical trial. Int J Radiat Oncol Biol Phys. 2009;73:1043–8. doi: 10.1016/j.ijrobp.2008.05.059. [DOI] [PubMed] [Google Scholar]
- 38.Calabrese CR, Almassy R, Barton S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96:56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 39.Sakamoto-Hojo ET, Balajee AS. Targeting poly (ADP) ribose polymerase I (PARP-1) and PARP-1 interacting proteins for cancer treatment. Anticancer Agents Med Chem. 2008;8:402–16. doi: 10.2174/187152008784220302. [DOI] [PubMed] [Google Scholar]
- 40.Greco WR, Park HS, Rustum YM. Application of a new approach for the quantitation of drug synergism to the combination of cis-diamminedichloroplatinum and 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1990;50:5318–27. [PubMed] [Google Scholar]
- 41.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki M, Amano M, Choi J, et al. Synergistic effects of fradiation and beta-lapachone in DU-145 human prostate cancer cells in vitro. Radiat Res. 2006;165:525–31. doi: 10.1667/RR3554.1. [DOI] [PubMed] [Google Scholar]
- 43.Dong Y, Chin SF, Blanco E, et al. Intratumoral delivery of beta-lapachone via polymer implants for prostate cancer therapy. Clin Cancer Res. 2009;15:131–9. doi: 10.1158/1078-0432.CCR-08-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blanco E, Bey EA, Khemtong C, et al. Beta-lapachone micellar nanotherapeutics for non-small cell lung cancer therapy. Cancer Res. 2010;70:3896–904. doi: 10.1158/0008-5472.CAN-09-3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.