

Abstract
In experimental studies, nonsteroidal anti-inflammatory drugs (NSAIDs) display promising antineoplastic activity, but toxicity resulting from cyclooxygenase (COX) inhibition limits their clinical use for chemoprevention. Studies suggest that the mechanism may be COX independent, although alternative targets have not been well defined. Here we show that the NSAID, sulindac sulfide (SS) inhibits cGMP phosphodiesterase (PDE) activity in colon tumor cell lysates at concentrations that inhibit colon tumor cell growth in vitro and in vivo. A series of chemically diverse NSAIDs inhibited cGMP hydrolysis at concentrations that correlate with their potency to inhibit colon tumor cell growth, while no correlation was observed with COX-2 inhibition. Consistent with its selectivity for inhibiting cGMP hydrolysis compared with cAMP hydrolysis, SS inhibited the cGMP specific PDE5 isozyme and increased cGMP levels in colon tumor cells. Of numerous PDE isozyme specific inhibitors evaluated, only the PDE5 selective inhibitor MY5445 inhibited colon tumor cell growth. The effects of SS and MY5445 on cell growth were associated with inhibition of β-catenin mediated transcriptional activity to suppress the synthesis of cyclin D and survivin, which regulate tumor cell proliferation and apoptosis, respectively. SS had minimal effects on cGMP PDE activity in normal colonocytes, which displayed reduced sensitivity to SS and did not express PDE5. PDE5 was found to be overexpressed in colon tumor cell lines as well as in colon adenomas and adenocarcinomas compared to normal colonic mucosa. These results suggest that PDE5 inhibition, cGMP elevation, and inhibition of β-catenin transcriptional activity may contribute to the chemopreventive properties of certain NSAIDs.
Keywords: colon cancer, NSAIDs, COX inhibitors, sulindac, PDE5
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase-2 (COX-2) selective inhibitors display promising chemopreventive efficacy, especially for colorectal cancer. Epidemiological studies, for example, have shown that long term use of NSAIDs is associated with reduced incidence of colorectal cancer in the general population by 30–50% (1, 2). Commonly prescribed for the treatment of inflammatory conditions, traditional NSAIDs inhibit both COX-1 and COX-2 enzymes, which catalyze the production of prostaglandins from arachidonic acid. Unfortunately, the depletion of prostaglandins by NSAIDs is associated with significant side effects including gastrointestinal ulcers, bleeding, and intestinal perforation. COX-2 selective inhibitors display reduced gastrointestinal toxicity but are associated with increased risk of myocardial infarction. These potentially fatal adverse effects outweigh the benefits of NSAIDs or COX-2 inhibitors for preventing colorectal cancer, which tend to require high dosages administered over a long period of time.
The mechanism of action that is responsible for the chemopreventive activity of NSAIDs is widely attributed to COX-2 inhibition, although a number of reports have suggested that a COX-independent mechanism may contribute to or be fully responsible for their anti-neoplastic activity (3–8). In support of this possibility, studies have shown that the non-COX inhibitory sulfone metabolite of the NSAID sulindac can inhibit human colon tumor cell growth and induce apoptosis in vitro (7, 8). Sulindac sulfone also inhibits tumorigenesis in the azoxymethane-induced rat model of colon carcinogenesis without suppressing prostaglandin levels in colonic mucosa (7–9). In clinical trials, sulindac sulfone (exisulind) caused regression of adenomas in patients with either familial (10) or sporadic polyposis (11) but did not receive FDA approval due to hepatotoxicity. Mechanistic studies have shown that sulindac sulfone and several structurally related analogs can inhibit cGMP phosphodiesterase (PDE) activity at concentrations that are comparable to those required to inhibit colon tumor cell growth and induce apoptosis (12–14). However, the specific PDE isozyme(s) responsible for its tumor cell growth inhibitory activity has not been identified, nor has this effect been well studied with regard to the chemopreventive efficacy of conventional NSAIDs.
PDE is a metallophosphohydrolase that specifically hydrolyzes the 3′,5′-cyclic phosphate moiety on cyclic nucleotides to a 5′ monophosphate, thereby deactivating cAMP or cGMP. PDE terminates second messenger signaling by degrading cyclic nucleotides, whereas inhibition of PDE activity blocks cyclic nucleotide degradation to mimic or amplify cyclic nucleotide signaling. The PDE superfamily consists of 20 distinct genes divided into 11 protein families (15). Despite the heterogeneity within the PDE superfamily, the catalytic domain is highly conserved across family members, yet minor changes within this domain determine the specificity of each isozyme family member for cAMP (PDE4, PDE7, PDE8), cGMP (PDE5, PDE6, PDE9), or both cAMP and cGMP (PDE1, PDE2, PDE3, PDE10, PDE11) as substrates. Both cAMP and cGMP have been shown to have antiproliferative and pro-apoptotic effects in many cell types (15, 16) and numerous studies have described the growth inhibitory and apoptosis inducing effects of various PDE inhibitors on certain tumor cell types, including colorectal cancer cells (17–20).
Sulindac is perhaps the most effective among the NSAIDs with regard to colon cancer chemoprevention, and its efficacy in numerous preclinical and clinical studies has been well documented. Most notably, sulindac has been shown to cause 60–70% reduction of adenoma size and number in patients with familial adenomatous polyposis (21). As reviewed previously (22), sulindac is a prodrug that requires metabolic activation for its anti-inflammatory activity. The sulfoxide moiety is reversibly reduced by colonic bacteria to form the active COX-inhibitory sulfide metabolite. The non-COX-inhibitory sulfone metabolite is irreversibly generated by oxidation of either the sulfide or the sulfoxide form in the liver. While both metabolites have been shown to inhibit tumor cell growth and induce apoptosis (7, 8), only the sulfone metabolite has been studied with regard to cGMP PDE inhibition in colon tumor cells (12). Recently, we reported that sulindac sulfide (SS) can selectively inhibit PDE5 activity in human breast tumor cells and that this activity is closely associated with its ability to inhibit growth and induce apoptosis (23). Additionally, other studies using PDE5 antisense have shown the importance of PDE5 expression and activity for colon tumor cell growth and survival (20, 24). Here we characterize the cGMP PDE inhibitory activity of SS and other NSAIDs and provide evidence that the tumor cell growth inhibitory and apoptosis inducing activity of SS is the result of cGMP elevation caused by selective inhibition of the PDE5 isozyme, which is overexpressed in colon tumor cells compared with normal colonocytes.
Materials and Methods
Drugs and Reagents
MY5445 and NOR-3 were purchased from BioMol (Plymouth Meeting, PA). Recombinant PDE enzymes were purchased from BPS Biosciences (San Diego, CA). Anti-PDE antibodies were purchased from GeneTex (San Antonio, TX). Anti-rabbit and anti-goat horseradish peroxidase-conjugated secondary antibodies were obtained from Cell Signaling Technologies (Danvers, MA). DMSO was used as the vehicle for all compounds unless otherwise specified. All other drugs and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cells and Cell Culture
The human colon cancer cell lines HT-29, SW480, and HCT116 and the primary culture of normal fetal human colonocytes (FHC) were obtained from ATCC (Manassas, VA) and grown under standard cell culture conditions as recommended by ATCC. Cell counts and viability were determined by trypan blue exclusion using a hemacytometer. Only cultures displaying >95% viability were used for experiments.
Growth Assays
Tissue culture microtiter 96-well plates were seeded at a density of 5,000 cells per well. The inhibition of cell growth caused by treatment was determined after 72 hours of treatment using the luminescent Cell Titer Glo Assay (Promega), which measures viable cells based on ATP content. The assay was performed according to the manufacturer’s specifications.
Proliferation Assays
The antiproliferative activity of NSAIDs was determined by a reduction of DNA synthesis, which was measured by a radioactive thymidine incorporation assay. After treatment, cells were incubated at 37°C for 56 hours at which time a 1 μCi aliquot of 3H-thymidine (Amersham Biosciences) was added to each well. Cells were incubated another 16 hours for a total of 72 hours of treatment. Plates were harvested with a semiautomatic cell harvester onto filters that bind the DNA. Samples were counted in an automated scintillation counter to determine the amount of 3H-thymidine uptake.
Apoptosis Assay
Cells were seeded in 10 cm tissue culture dishes at a density of 1×106 cells per dish. After 48 hours of treatment, cells were collected and fixed with 4% formalin on ice for 15 minutes. Samples were stained for DNA strand breaks using the APO-BrdU TUNEL Assay Kit (Invitrogen). The assay was performed according to the manufacturer’s specifications. The percentage of TUNEL-positive cells was quantified using a Guava EasyCyte Plus flow cytometer. A minimum of 10,000 events were collected for each treatment group with use of minimal electronic compensation. Data was analyzed using CytoSoft 5.0 software (Guava Technologies).
Cell Lysis
Cells were harvested, vortexed in ice cold lysis buffer (20mM TrisAcetate, 5mM Magnesium Acetate, 1mM EGTA, 0.8% Triton X-100, 50mM NaF, and protease inhibitor cocktail at pH 7.4), and clarified by centrifugation at 10,000g for 10 minutes at 4°C. Protein content was determined using the BCA Protein Assay (Pierce) following the manufacturer’s specifications. The colon of normal rats was excised and scraped with a glass microscope slide to obtain normal colonocytes, which were then lysed as described.
Tissue Homogenization
HT-29 tumor cells were grown subcutaneously in athymic NCr-nu/nu mice. Tumors weighing 1–2 g each were homogenized in 100 mM Tris-HCl (pH 7.5), 10 mM NaCl, 1mM EGTA, 1mM DTT, 0.1% Triton X-100 and a protease inhibitor cocktail (Sigma). The extract was clarified by centrifugation at 20,000g for 1 h at 4°C. Protein content was determined using the BCA Protein Assay (Pierce) following the manufacturer’s specifications.
PDE Assays
cGMP PDE activity was measured using (3H) cGMP scintillation proximity (SP) enzyme assay kits (Amersham). The conditions of the assays were 50 mM Tris-HCl (pH 7.5), 8.3 mM MgCl2, 1.7 mM EGTA, 20 μM (3H) cGMP, 2% DMSO, and 0.2 mg/ml of HT-29 lysate in a volume of 100 μL. The reaction was allowed to proceed for 15 minutes at 30°C and terminated by adding SP beads, which bind the radiolabeled product. The beads were counted on a scintillation counter. PDE activity was also measured using the IMAP fluorescence polarization (FP) phosphodiesterase assay (Molecular Devices). The assay was modified, as described previously (23), to use fluorescein (Fl)-cAMP and tetramethylrhodamine (TAMRA)-cGMP as substrates, allowing for simultaneous measurement of cAMP and cGMP hydrolysis.
cGMP Assay
Cells were seeded at a density of 1×106 cells per 10 cm tissue culture dish. After 30 minutes of drug treatment, cells were lysed and assayed for cGMP content using the cGMP Direct Biotrak EIA kit (GE Biosciences) according to the manufacturer’s specifications.
Western Blots
Cell lysates (30 μg protein) were separated by SDS-PAGE in a 12% polyacrylamide gel followed by electrophoretic transfer to a nitrocellulose membrane. The membranes were blocked with 5% BSA in TBS containing 0.05% Tween-20. Incubation of membranes in primary and secondary antibodies was performed according to the manufacturers’ specifications.
Luciferase Reporter Assays
Cells were transiently transfected with the TOPFlash luciferase construct (MilliPROBE), β-galactosidase-expressing vector (Promega). After 24 hours of treatment, cells were lysed and both luciferase and β-galactosidase activities were measured. The luciferase activity was normalized to the β-galactosidase activity.
Tolerance Studies
The maximum tolerated dose (MTD) of sulindac was determined using 5–6 week old male NCr-nu/nu mice. Sulindac was administered by oral gavage at dosages of 50, 100, 200, or 400 mg/kg/day as a suspension in 0.5% CMC, 0.25% Tween 80 in water. Mice were observed daily and body weights measured twice weekly.
In Vivo Antitumor Efficacy
The antitumor efficacy of sulindac was evaluated using the human HT-29 colon tumor xenograft implanted subcutaneously in 5–6 week old male NCr-nu/nu athymic mice. Sulindac was administered orally once daily for 40 days at a dose of 50 mg/kg as a suspension in 0.5% CMC, 0.25% Tween 80 in water. The experiment consisted of a vehicle-treated control group and a sulindac treated group with each group containing ten mice. Treatment was initiated when the tumors reached a size of 75–200 mg. Mice were observed daily for mortality. Tumor dimensions and body weights were measured twice weekly starting on the first day of treatment. For all animal studies described above, mice were housed in micro-isolator cages, provided tap water and sterile pelleted diet ad libitum, and euthanized as per IACUC guidelines. All animal protocols were reviewed by Southern Research IACUC prior to experimentation.
Immunohistochemistry
Formalin-fixed paraffin embedded clinical specimens were obtained from the Fox Chase Cancer Center by Dr. Andre Klein-Szanto and processed for immunohistochemistry by avidin-biotin peroxidase detection. PDE5 antiserum was generated in sheep against a 16 amino acid synthetic peptide sequence contained within the high affinity cGMP binding domain of the enzyme. Anti-PDE5 IgG was purified using an antigen affinity column. Peptide, antibody, and affinity column were obtained from Bethyl Laboratories (Montgomery, TX).
Experimental Design and Data Analysis
Drug effects on cell growth and PDE activity were measured and the potency expressed as an IC50 value, which is the concentration resulting in 50% inhibition when compared to the vehicle control. For growth assays, the IC50 value was determined by testing a range of 8 concentrations with a minimum of 4 replicates per dose. Each assay utilized a minimum of 2 replicates. Dose response curves were constructed using Prism5 software (Graphpad), which calculates IC50 values using a four parameter logistic equation. All experiments were repeated a minimum of twice in order to determine the reproducibility of the results. All values represent a comparison between drug treatment at the specified concentration and vehicle treated controls. All error bars represent standard error of the mean (SEM). Calculation of p values was done by comparing the specified treatment group to vehicle treated controls using a student’s t test.
Results
NSAID inhibition of colon tumor cell growth correlates with cGMP PDE inhibition
To determine whether cGMP PDE represents a potential COX-independent target responsible for the antineoplastic activity of sulindac, we compared the non-COX-inhibitory sulfone and sulfoxide forms of the drug with the COX-1 and COX-2 inhibitory sulfide metabolite, the highly selective COX-2 inhibitor, rofecoxib, and the COX-1 selective inhibitor, indomethacin. These drugs were evaluated for their potency to inhibit purified COX-1 and COX-2 enzymes, to inhibit cGMP hydrolysis in lysates from HT-29 human colon tumor cells, and to inhibit proliferation of HT-29 colon tumor cells. As summarized in Table 1, sulindac sulfone and sulfoxide inhibited the proliferation of HT-29 colon tumor cells with IC50 values of 89 and 224 μM, respectively, despite lacking COX-1 or COX-2 inhibitory activity at concentrations up to 300 μM. Sulindac sulfide more potently inhibited proliferation of the colon tumor cells than either the sulfoxide or sulfone forms with an IC50 value of 34 μM. However, this effect required appreciably higher concentrations compared to those needed to inhibit COX-1 or COX-2. In contrast with COX inhibition, all three forms of sulindac inhibited cGMP PDE activity with IC50 values that were comparable to their respective IC50 values for inhibiting proliferation. Indomethacin also inhibited colon tumor cell growth and cGMP PDE, but with reduced potency compared with sulindac sulfide despite its superior potency to inhibit COX-1 or COX-2. Moreover, the highly potent and specific COX-2 inhibitor, rofecoxib, was unable to inhibit colon tumor cell proliferation or cGMP PDE activity. These results demonstrate that the potency for cGMP PDE inhibition but not the potency for COX-1 or COX-2 inhibition is closely associated with colon tumor cell growth inhibitory potency among sulindac and its metabolites.
Table 1.
Potency of the sulindac metabolites, the COX-2 selective inhibitor rofecoxib, and the COX-1 selective inhibitor indomethacin to inhibit COX-1, COX-2, cGMP hydrolysis in HT-29 colon tumor cell lysates, and colon tumor cell proliferation. Purified ovine COX-1 and COX-2 were used for the COX assays. cGMP hydrolysis was measured by the scintillation proximity assay using cell lysates obtained from HT-29 human colon tumor cells were used as the source of cGMP PDE isozymes. Antiproliferative activity was measured after 72 hours of drug treatment of HT-29 cells using a tritiated thymidine assay.
| Compound | IC50 (μM) | |||
|---|---|---|---|---|
| COX-1 | COX-2 | cGMP PDE | Proliferation | |
| Sulindac sulfide | 1.8 | 6.3 | 19.7 | 33.9 |
| Sulindac sulfone | >300 | >300 | 95.9 | 89.4 |
| Sulindac sulfoxide | >300 | >300 | 119.7 | 224 |
| Rofecoxib | >300 | 2.7 | >300 | >500 |
| Indomethacin | 0.017 | 1.0 | 24 | 136 |
A series of chemically diverse NSAIDs representative of the acetic acid, fenamate, and proprionic acid families of NSAIDs, as well as another COX-2 selective inhibitor, celecoxib, were compared for potency to inhibit COX-2, cGMP PDE, and HT-29 colon tumor cell growth (Figure 1A). Among these anti-inflammatory drugs, a strong positive correlation was observed between potency for HT-29 cell growth inhibition and potency for inhibition of cGMP PDE activity with an r2 value of 0.933 (Figure 1B). However, no correlation was observed by comparing potencies of these NSAIDs for COX-2 inhibition as previously reported (25) with potencies for HT-29 cell growth inhibition (Figure 1C). The salicylate family, which contains aspirin as well as non-carboxylate NSAIDs from the pyrazole (e.g. phenylbutazone) and oxicam (e.g. piroxicam) families displayed low potency for HT-29 growth inhibition and did not affect cGMP PDE activity (data not shown). These data demonstrate that a variety of chemically diverse NSAIDs can inhibit cGMP PDE activity and that this effect is associated with their in vitro growth inhibitory potency.
Figure 1.
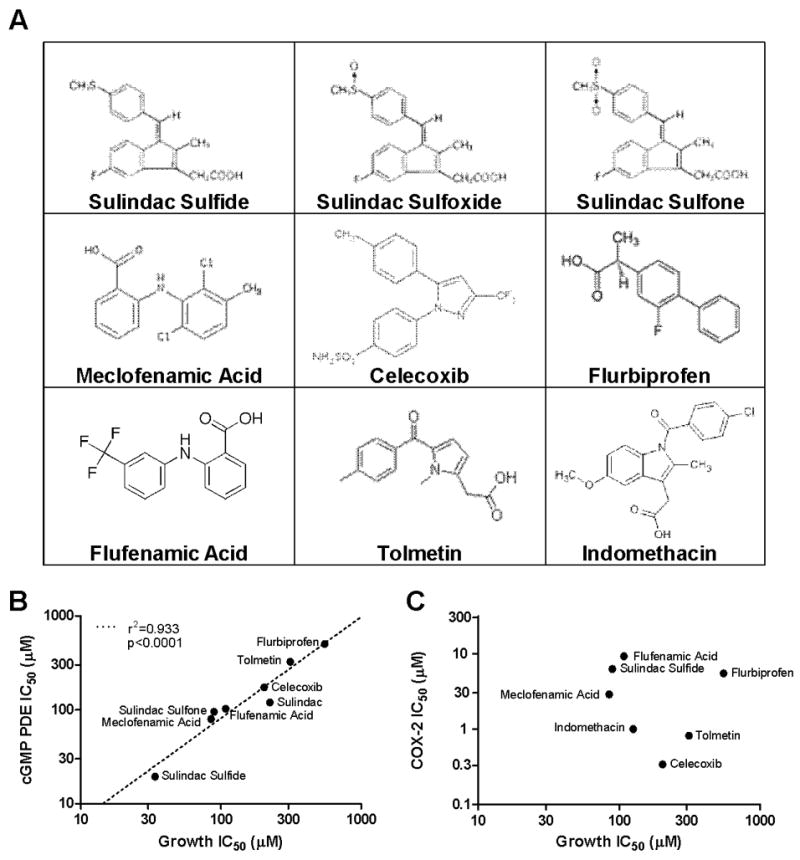
In vitro association between colon tumor cell growth inhibition and other molecular mechanisms for a panel of NSAIDs and COX-2 inhibitors. A, positive association between the potencies of for HT-29 growth inhibition and inhibition of cGMP hydrolysis in HT-29 cell lysates. B, no association between the potencies for HT-29 growth inhibition and inhibition of COX-2.
Sulindac sulfide selectively inhibits cGMP hydrolysis in colon tumor cells
Sulindac sulfide (SS) was further studied for its ability to inhibit cGMP hydrolysis and growth of the human colon cancer cell lines HT-29, SW480, and HCT116 compared with fetal human colonocytes (FHC), which are representative of a non-tumorigenic colon cell line. As shown in Figure 2A, SS selectively inhibited growth of the human colon tumor cell lines with IC50 values ranging from 73 to 85 μM compared to the IC50 value of 150 μM necessary for growth inhibition of normal colonocytes. In addition to inhibiting colon tumor cell proliferation, SS also induced apoptosis as measured by DNA fragmentation (Figure 2B). After 48 hours of treatment, 100 and 200 μM SS resulted in 6- and 15-fold increases, respectively, in the percentage of TUNEL positive HT-29 cells.
Figure 2.
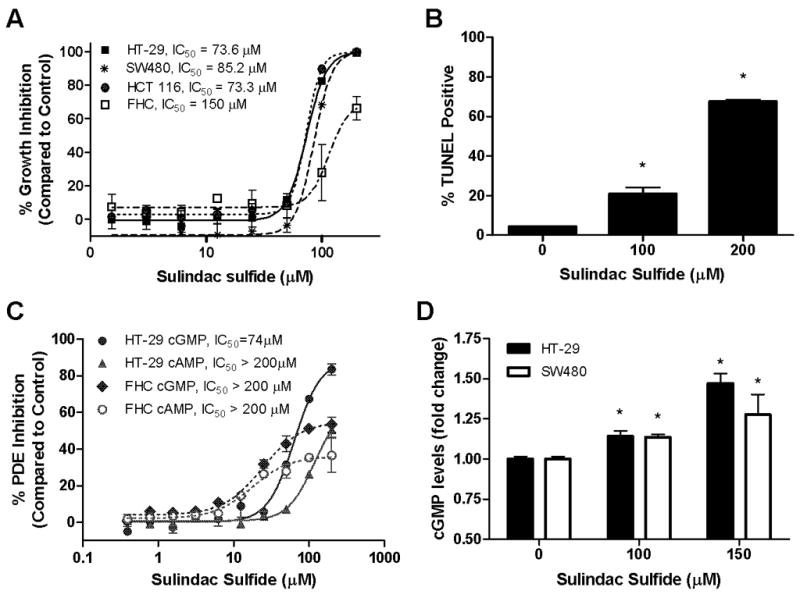
Colon tumor cell growth- and cGMP PDE-inhibitory activity of SS. A, dose-dependent growth inhibitory activity of SS after 72 hours of treatment. B, dose-dependent increase in apoptotic (TUNEL positive) HT-29 cells after 48 hours of SS treatment. C, dose-dependent cAMP (▲, ○) and cGMP (●, ◆) PDE inhibitory activity of SS in HT-29 cell lysates (solid lines) compared to FHC lysates (dashed lines). D, dose-dependent increase in intracellular cGMP levels in HT-29 and SW480 cells after 30 minutes of SS treatment. * represents that the treatment is significantly different than vehicle control with p<0.05.
Using a fluorescence polarization assay that we developed to simultaneously measure cAMP and cGMP hydrolysis, the effects of SS on PDE activity in lysates from HT-29 colon cancer cells and FHC were evaluated (Figure 2C). SS inhibited cGMP hydrolysis with an IC50 value of 74 μM in the HT-29 cell lysate, but appreciably higher concentrations were necessary to inhibit cAMP hydrolysis. Additionally, FHC displayed reduced sensitivity to the cGMP PDE inhibitory activity of SS, which paralleled its reduced sensitivity to growth suppression by SS.
To determine the relevance of cGMP PDE inhibition by SS in colon tumor cell lysates to the effects of the drug in intact cells, the intracellular levels of cGMP were measured after 30 minutes of SS treatment using a cGMP immunoassay (Figure 2D). SS treatment of HT-29 and SW480 colon tumor cells resulted in a significant increase in intracellular cGMP levels at concentrations of 100 and 150 μM, which is consistent with concentrations required to inhibit colon tumor cell growth and induce apoptosis.
cGMP elevation causes colon tumor cell growth inhibition
To determine if elevated levels of intracellular cGMP in colon tumor cells is sufficient to inhibit colon tumor cell growth, the effect of the nitric oxide donor and guanylyl cyclase activator NOR-3 on cell growth was evaluated in the HT-29, SW480, and HCT116 lines. NOR-3 inhibited growth in all three cell lines with IC50 values ranging from 59 to 74 μM (Figure 3A). Conversely, the adenylyl cyclase activator forskolin did not significantly affect growth of any of the cell lines at concentrations up to 100 μM (Figure 3B), suggesting that the growth inhibitory effect resulting from cGMP PDE inhibition is specific to cGMP and is not induced by cAMP.
Figure 3.
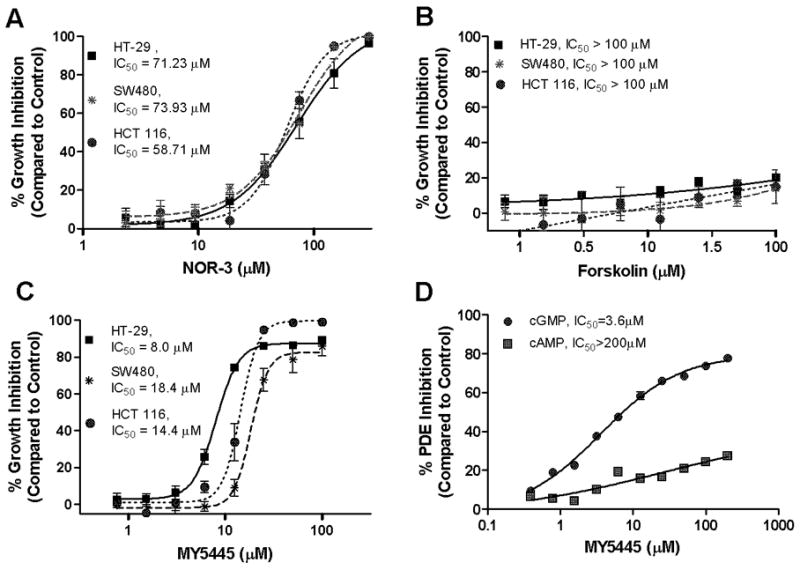
Effects of cyclic nucleotide elevating agents on growth of human colon cancer cells. A, dose-dependent growth inhibitory activity of the NO donor and guanylyl cyclase activator NOR-3 after 72 hours of treatment. B, non-growth inhibitory activity of the adenylyl cyclase activator forskolin after 72 hours of treatment. C, dose-dependent growth inhibitory activity of the PDE5 selective inhibitor MY5445. D, dose-dependent cAMP (■) and cGMP (●) PDE inhibitory activity of MY5445 in HT-29 cell lysates.
A panel of PDE isozyme specific inhibitors including vinpocetine (PDE1), EHNA (PDE2), milrinone (PDE3), rolipram (PDE4), MY5445 (PDE5), and others were evaluated for growth inhibitory activity in HT-29, SW480, and HCT116 colon tumor cell lines. Among these, only MY5445 was found to inhibit colon tumor cell growth with IC50 values ranging from 8–18 μM (Figure 3C). MY5445 also selectively inhibited cGMP PDE activity in HT-29 cell lysates with an IC50 value of 4 μM without substantially affecting cAMP hydrolysis (Figure 3D). Similar to SS, MY5445 inhibited proliferation and induced apoptosis of colon tumor cells (data not shown). However, other highly selective and potent PDE5 inhibitors including sildenafil and tadalafil did not affect colon tumor cell growth (data not shown).
Sulindac sulfide selectively inhibits PDE5
To determine which PDE isozyme is responsible for tumor cell growth inhibitory activity of SS, recombinant isozymes from all eleven isozyme family members were screened for sensitivity to SS using cGMP or cAMP as a substrate. As previously reported, the cGMP selective PDE5 isozyme was the most sensitive, although cGMP hydrolysis by PDE2 and PDE3 was also inhibited, albeit with reduced sensitivity (23). The difference in sensitivity is shown in Figure 4A with concentration-dependent inhibition of PDE5 by SS, resulting in an IC50 value of 38 μM. By comparison, SS inhibited cGMP hydrolysis by PDE2 and PDE3 with IC50 values of 97 and 84 μM, respectively. Consistent with its cGMP selectivity, SS did not significantly inhibit cAMP hydrolysis by PDE2 or PDE3 (data not shown). To assess the potential contribution of PDE2, 3, and 5 to cGMP degradation in whole cells, their activity and expression in colon cell lines was studied using PDE isozyme-specific antibodies and inhibitors. First, the levels of protein expression in HT-29 cells were determined by western blotting and compared to the levels of protein expression in FHC. As shown in Figure 4B, HT-29 cells expressed a single isoform of PDE3 and two splice variants of PDE5, but did not express PDE2. Conversely, FHC expressed PDE3 at levels higher than HT-29 cells, but lacked expression of PDE2 and PDE5. Other human colon cancer cell lines, HCT-116 and SW480, also expressed the two splice variants of PDE5 that were present in HT-29 (data not shown). To determine the importance of these isozymes to the overall cGMP hydrolytic capacity of the colon tumor cells, the cGMP PDE inhibitory activity of the PDE2 selective inhibitor EHNA, the PDE3 selective inhibitor milrinone, and the PDE5 selective inhibitor sildenafil were analyzed in HT-29 cell lysates. Consistent with the expression levels determined by Western blotting, milrinone and sildenafil inhibited cGMP PDE activity in the HT-29 lysate by 31 and 42%, respectively, while EHNA had only a modest effect of less than 12% inhibition (Figure 4C). The overexpression of PDE5 in colon tumor cells compared with FHC is consistent with the selectivity of SS for inhibition of PDE5 compared to other cGMP PDE isozymes and for inhibition of tumor cell growth and cGMP PDE compared to normal colonocytes.
Figure 4.

PDE isozyme expression and activity. A, inhibition of cGMP hydrolytic activity of recombinant PDE2, 3, and 5 by SS. B, relative levels of PDE2, 3, and 5 isozyme expression in HT-29 and FHC as measured by Western blotting with PDE family-specific antibodies. C, relative activities of PDE2, 3, and 5 families in HT-29 cell lysates as measured by overall inhibition of cGMP hydrolysis by the PDE2 selective inhibitor EHNA, the PDE3 selective inhibitor milrinone, and the PDE5 selective inhibitor sildenafil. D, PDE5 expression in normal colon mucosa, adenoma, and adenocarcinomas from human clinical specimens as determined by immunohistochemistry following avidin-biotin peroxidase labeling. E, effects of SS (top graph) or MY5445 (bottom graph) on Tcf/Lef transcriptional activity as measured by a luciferase reporter assay in HT-29 (hatched bars), HCT-116 (vertically stripped bars), and SW480 (horizontally stripped bars) colon cancer cells. F, time-dependent decrease in the expression of cyclin D1 and survivin in SW480 cells after treatment with SS or MY5445, with β-actin as a loading control.
Colon adenomas and adenocarcinomas overexpress PDE5
The expression of PDE5 in normal colonic mucosa, adenomas, and adenocarcinomas from archival clinical specimens was determined by immunohistochemistry using a PDE5 specific antibody. Analysis of 11 specimens containing normal colonic mucosa demonstrated consistent negative labeling in the colonic epithelium, despite labeling of the muscularis mucosa. By contrast, 5 of 5 adenomas and 11 of 13 adenocarcinomas displayed positive labeling. Representative micrographs from normal colon mucosa, adenomas, and adenocarcinomas are shown in Figure 4D.
Tumor cell growth inhibitory activity of SS is associated with inhibition of β-catenin transcriptional activity
Previous studies reported that sulindac and its metabolites activate cGMP-dependent protein kinase (PKG), which can then phosphorylate β-catenin to promote its proteasomal degradation (12, 23). We evaluated the effects of SS and MY5445 on β-catenin signaling in colon cancer cells to determine if this mechanism is involved in the growth-inhibitory activity of these compounds. Both compounds resulted in a significant reduction of Tcf/Lef-mediated transcription as measured by a luciferase reporter assay in HT-29, HCT-116, and SW480 colon cancer cells (Figure 4E). While the length of treatment necessary to cause an effect differed between compounds and between proteins, SS and MY5445 treatment resulted in a significant reduction in expression of the proteins cyclinD1 and survivin, whose transcription is promoted by β-catenin signaling (Figure 4F). These data are consistent with cGMP-PKG mediated phosphorylation of β-catenin to induce degradation, thereby suppressing the transcription of key genes that regulate tumor cell proliferation and apoptosis.
Sulindac sulfide selectively inhibits cGMP hydrolysis in colon tumors
To further evaluate the tumor selectivity of SS, we measured the sensitivity of cGMP PDE activity in lysates from HT-29 tumors established in athymic mice to SS and MY5445 and compared the sensitivity to lysates obtained from normal rat colonic mucosa. Similar to the effects in cultured HT-29 tumor cells, both SS and MY5445 selectively inhibited cGMP PDE activity in lysates obtained from the HT-29 tumors. However, cGMP hydrolysis in normal colon mucosa displayed reduced sensitivity, comparable to cultured FHC (Figure 5A and B). SS displayed 4 fold greater potency for inhibition of the tumor cGMP PDE compared to that present in the normal tissue, whereas MY5445 displayed more than 10 fold greater potency. Both compounds inhibited cGMP PDE in the tumor lysate with IC50 values comparable to those necessary for cGMP PDE inhibition in cultured tumor cell lysates and for in vitro colon tumor cell growth inhibition. These results are consistent with increased expression and activity of PDE5 in colon tumor cells compared with normal colonocytes.
Figure 5.
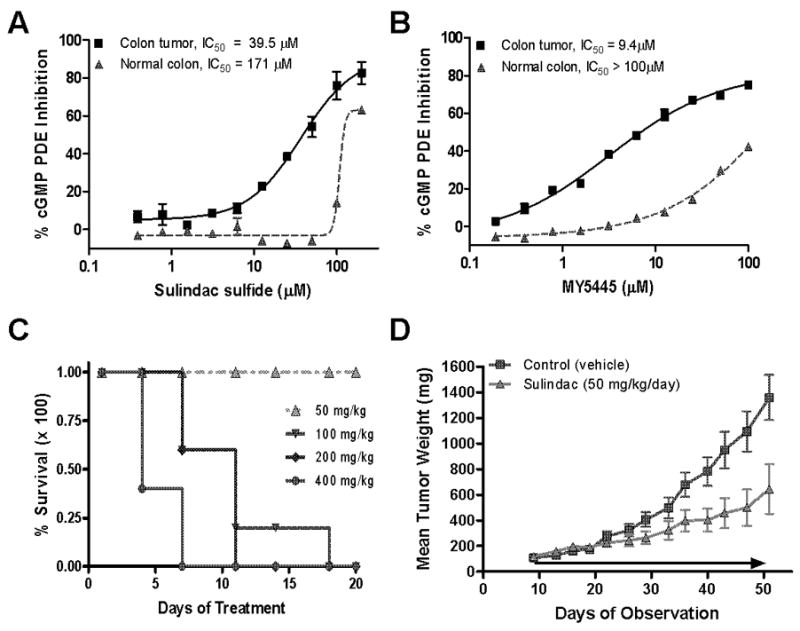
In vivo antitumor efficacy of sulindac. A, selectivity of SS for inhibition of cGMP hydrolysis in colon tumor homogenates (solid line) compared to homogenates of normal colonic mucosa (dashed line). B, selectivity of MY5445 for inhibition of cGMP hydrolysis in colon tumor homogenates (solid line) compared to homogenates of normal colonic mucosa (dashed line). C, MTD determination of sulindac in male athymic mice. D, dose-dependent inhibition of tumor growth in HT-29 xenograft mice treated with 50mg/kg/day compared to vehicle treated controls.
Sulindac inhibits colon tumor cell growth in vivo
To assess the in vivo pharmacological relevance of the above observations, the tumor growth inhibitory activity of sulindac was determined using the HT-29 colon tumor xenograft mouse model. Dosages of sulindac were selected based on a tolerance study following daily oral gavage for 20 days of treatment. The maximum tolerated dosage was determined to be approximately 50 mg/kg where no deaths (Figure 5A) or significant weight loss occurred. To determine antitumor efficacy of sulindac, two groups of ten mice each were inoculated subcutaneously with fragments of HT-29 tumors; one group was treated with vehicle (CMC) and the other was treated with sulindac at 50 mg/kg once daily by oral gavage. Sulindac treatment caused a significant reduction of tumor growth of approximately 55% by the end of treatment compared to mice treated with vehicle only (Figure 5D) without affecting body weight (not shown). In addition, levels of sulindac and its metabolites were measured in plasma collected 6 hours following a single oral dose of sulindac at 50 mg/kg. Sulindac generated plasma levels of 36.7 ± 10.2 μM for the sulfide, 8.9 ± 1.2 μM, for the sulfone, and 23.1 ± 5.5 μM, for the parent sulfoxide. These experiments show that SS is the predominant metabolite in plasma and that dosages of sulindac required for in vivo anti-tumor efficacy can generate plasma levels of SS that are comparable with the concentrations required to inhibit colon tumor cell proliferation and PDE5 activity in vitro.
Discussion
Epidemiological, pre-clinical, and clinical studies have shown convincing evidence that NSAIDs and COX-2 inhibitors have cancer chemopreventive efficacy, especially with regard to colorectal cancer. Unfortunately, gastrointestinal, renal, and cardiovascular toxicities that result from COX-1 and/or COX-2 inhibition limit their use for cancer chemoprevention. Here we show that the in vitro colon tumor cell growth inhibitory activity of a group of NSAIDs is closely associated with cGMP PDE inhibition. For instance, the potency of SS to inhibit HT-29 colon tumor cell growth was nearly identical to the potency for inhibition of cGMP PDE in tumor cell lysates, yet was more than an order of magnitude greater than the potency for inhibition of COX-1 or COX-2. Sulindac sulfone and sulfoxide did not inhibit COX but did inhibit tumor cell growth and cGMP PDE. Additionally, a strong positive correlation existed between the potencies of other chemically diverse NSAIDs for cGMP PDE inhibition and colon tumor cell growth inhibition, yet no correlation was apparent for COX-2 inhibition. Interestingly, the COX-2 selective inhibitor, rofecoxib, which did not inhibit tumor cell growth, also did not inhibit cGMP PDE, while another COX-2 inhibitor, celecoxib, did inhibit tumor cell growth at concentrations that inhibited cGMP PDE. These data suggest that cGMP PDE inhibition represents a COX-independent mechanism responsible for the in vitro tumor cell growth inhibitory activity of certain NSAIDs and COX-2 inhibitors that potentially could be targeted for the discovery of safer and more efficacious drugs for chemoprevention.
Because of its high potency to inhibit colon tumor cell growth, SS was used to investigate the role of cGMP in regulating tumor cell growth and to identify the PDE isozyme(s) responsible for its growth inhibitory activity. As previously reported, SS-induced growth inhibition was associated with inhibition of proliferation and induction of apoptosis, which occurred at concentrations comparable to those necessary for inhibition of cGMP hydrolysis in HT-29 cell lysates. The biological relevance of cGMP PDE inhibition in HT-29 cell lysates was confirmed using a cell-based immunoassay, which showed the ability of SS to increase cGMP levels in colon tumor cells at concentrations that inhibited growth and induced apoptosis. The ability of other cGMP signaling activators, including the guanylyl cyclase activator NOR-3 and the PDE5 specific inhibitor MY5445, to inhibit growth suggests that activation of cGMP signaling is sufficient for inhibition of colon tumor cell growth. Consistent with previous findings (12), we demonstrated that activation of the cGMP signaling pathway by compounds such as SS and MY5445 is associated with inhibition of β-catenin transcriptional activity, a pathway that is well documented to be associated with tumorigenesis. Therefore, inhibition of cGMP PDE, activation of cGMP signaling, and inhibition of β-catenin-mediated transcription by certain NSAIDs may fully account for their in vitro tumor cell growth inhibitory and apoptosis inducing activity and contribute to their anticancer efficacy.
Although activation of cGMP signaling is sufficient to inhibit colon tumor cell growth, nonselective activation of this pathway is not a plausible pharmacological approach to develop new chemopreventive agents due to the myriad of roles that cGMP plays in normal physiology and the potential for adverse effects. For this reason, we hypothesized that selective inhibition of one or more of the PDE isozymes expressed in tumor cells may provide a more effective strategy to selectively induce cGMP levels and kill neoplastic cells. Non-tumorigenic colon cells were not found to express the PDE5 isozyme that was expressed in colon tumor cell lines. The clinical relevance of this observation was confirmed by immunohistochemical analysis of human colon specimens, which showed increased PDE5 expression in adenomas and adenocarcinomas compared to normal colonicmucosa. Consistent with these observations, SS displayed selectivity for inhibition of cGMP hydrolysis by PDE5 compared to all other PDE isozymes and improved potency for both inhibition of colon tumor cell growth and cGMP PDE activity when compared to the non-tumorigenic colon cells. These results are consistent with previous studies that utilized PDE5 antisense in HT-29 human colon tumor cells to demonstrate that PDE5 activity is necessary for growth and survival of colon tumor cells (24).
The contradictory data showing that certain PDE5 selective inhibitors such as MY5445 can potently inhibit colon tumor cell growth while others such as sildenafil can not affect tumor cell growth are perplexing. However, highly potent PDE5 inhibitors, such as E4021 have previously been reported to be unable to induce a significant increase in intracellular cGMP levels in colon tumor cells despite inhibiting cGMP hydrolysis with purified isozyme preparations (12). There are several potential explanations for this discrepancy including an inability of the drug to access PDE5 within cancer cells due to confined subcellular localization of the enzyme or an active efflux of the drug from the cell thereby preventing sufficient intracellular accumulation. Whatever the reason certain PDE5 inhibitors are unable to induce cGMP signaling within colon tumor cells, the data presented here strongly suggests that PDE5 inhibition and cGMP elevation is responsible for the tumor cell growth inhibitory and apoptosis inducing activity of SS and possibly other NSAIDs.
We also show that SS not only inhibited cGMP hydrolysis in colon tumor cell lysates but also inhibited cGMP hydrolysis in homogenates of colon tumors grown in vivo. Additionally, SS displayed selectivity for inhibition of tumor tissue cGMP PDE compared to normal colon tissue cGMP PDE, mirroring the observations in cell cultures. Furthermore, the PDE5 selective inhibitor MY5445 exhibited a greater degree of selectivity for inhibition of cGMP PDE activity in tumor tissue homogenates compared to that exhibited by SS, further suggesting that selective inhibition PDE5 may be advantageous over nonselective PDE inhibition. Consistent with these observations, a significant reduction in colon tumor size compared to control was observed in HT-29 xenograft mice treated with 50 mg/kg/day of sulindac, a dosage that generated plasma levels of SS sufficiently high to inhibit cGMP PDE and, more specifically, PDE5 in vivo. While more work is necessary to confirm the direct effects of such in vivo treatments on cGMP signaling, these data suggest that inhibition of cGMP PDE and activation of cGMP signaling may play an important role in the in vivo antineoplastic activity of sulindac and potentially other NSAIDs. In support of this possibility, pharmacokinetic studies in humans have shown that clinically used dosages of sulindac can achieve plasma concentrations of the sulfide that are comparable to those required for PDE5 inhibition and cGMP elevation as we report here (21, 22). We conclude that PDE5 inhibition and subsequent elevation of intracellular cGMP levels and activation of cGMP signaling is an important mechanism responsible for the chemopreventive efficacy of sulindac and potentially other NSAIDs. The differential expression of PDE5 isozymes between normal and tumorigenic colon cells provides a COX-independent strategy to identify new drug candidates for chemoprevention. As such, further studies are warranted to determine the role of cGMP signaling in tumorigenesis.
Acknowledgments
We are grateful to Dr. Andre Klein-Szanto for providing clinical specimens and histopathological support for analysis of PDE5 expression in tissue sections and Dr. William Thompson for providing the PDE5 antibody.
Financial support: NIH grants R01 CA131378, R21 NS59509 and R03 CA128021
Abbreviations
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- FAP
familial adenomatous polyposis
- FP
fluorescence polarization
- GC
guanylyl cyclase
- MY5445
1-(3-chloroanilino)-4-phenylphthalazine
- NOR-3
(±)-(E)-Ethyl-2-((E)-hydroxyimino)-5-nitro-3-hexenamide
- NSAID
nonsteroidal anti-inflammatory drug
- PDE
phosphodiesterase
- PKG
cGMP dependent protein kinase
- SS
sulindac sulfide
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002 Feb 20;94(4):252–66. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 2.Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002 Mar;3(3):166–74. doi: 10.1016/s1470-2045(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 3.Alberts DS, Hixson L, Ahnen D, et al. Do NSAIDs exert their colon cancer chemoprevention activities through the inhibition of mucosal prostaglandin synthetase? J Cell Biochem Suppl. 1995;22:18–23. doi: 10.1002/jcb.240590804. [DOI] [PubMed] [Google Scholar]
- 4.Elder DJ, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res. 1997 Oct;3(10):1679–83. [PubMed] [Google Scholar]
- 5.Hanif R, Pittas A, Feng Y, et al. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol. 1996 Jul 26;52(2):237–45. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 6.Kashfi K, Rigas B. Is COX-2 a ‘collateral’ target in cancer prevention? Biochem Soc Trans. 2005 Aug;33(Pt 4):724–7. doi: 10.1042/BST0330724. [DOI] [PubMed] [Google Scholar]
- 7.Piazza GA, Rahm AK, Finn TS, et al. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction. Cancer Res. 1997 Jun 15;57(12):2452–9. [PubMed] [Google Scholar]
- 8.Piazza GA, Rahm AL, Krutzsch M, et al. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995 Jul 15;55(14):3110–6. [PubMed] [Google Scholar]
- 9.Piazza GA, Alberts DS, Hixson LJ, et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997 Jul 15;57(14):2909–15. [PubMed] [Google Scholar]
- 10.Stoner GD, Budd GT, Ganapathi R, et al. Sulindac sulfone induced regression of rectal polyps in patients with familial adenomatous polyposis. Adv Exp Med Biol. 1999;470:45–53. doi: 10.1007/978-1-4615-4149-3_5. [DOI] [PubMed] [Google Scholar]
- 11.Arber N, Kuwada S, Leshno M, Sjodahl R, Hultcrantz R, Rex D. Sporadic adenomatous polyp regression with exisulind is effective but toxic: a randomised, double blind, placebo controlled, dose-response study. Gut. 2006 Mar;55(3):367–73. doi: 10.1136/gut.2004.061432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson WJ, Piazza GA, Li H, et al. Exisulind induction of apoptosis involves guanosine 3′,5′-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. 2000 Jul 1;60(13):3338–42. [PubMed] [Google Scholar]
- 13.Piazza GA, Thompson WJ, Pamukcu R, et al. Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer Res. 2001 May 15;61(10):3961–8. [PubMed] [Google Scholar]
- 14.Whitehead CM, Earle KA, Fetter J, et al. Exisulind-induced Apoptosis in a Non-Small Cell Lung Cancer Orthotopic Lung Tumor Model Augments Docetaxel Treatment and Contributes to Increased Survival. Mol Cancer Ther. 2003 May;2(5):479–88. [PubMed] [Google Scholar]
- 15.Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995 Oct;75(4):725–48. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- 16.Sonnenburg WK, Beavo JA. Cyclic GMP and regulation of cyclic nucleotide hydrolysis. Adv Pharmacol. 1994;26:87–114. doi: 10.1016/s1054-3589(08)60052-6. [DOI] [PubMed] [Google Scholar]
- 17.Drees M, Zimmermann R, Eisenbrand G. 3′,5′-Cyclic nucleotide phosphodiesterase in tumor cells as potential target for tumor growth inhibition. Cancer Res. 1993 Jul 1;53(13):3058–61. [PubMed] [Google Scholar]
- 18.Wharton J, Strange JW, Moller GM, et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med. 2005 Jul 1;172(1):105–13. doi: 10.1164/rccm.200411-1587OC. [DOI] [PubMed] [Google Scholar]
- 19.Zhu B, Strada S, Stevens T. Cyclic GMP-specific phosphodiesterase 5 regulates growth and apoptosis in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005 Aug;289(2):L196–206. doi: 10.1152/ajplung.00433.2004. [DOI] [PubMed] [Google Scholar]
- 20.Zhu B, Strada SJ. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr Top Med Chem. 2007;7(4):437–54. doi: 10.2174/156802607779941198. [DOI] [PubMed] [Google Scholar]
- 21.Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993 May 6;328(18):1313–6. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 22.Davies NM, Watson MS. Clinical pharmacokinetics of sulindac. A dynamic old drug. Clin Pharmacokinet. 1997 Jun;32(6):437–59. doi: 10.2165/00003088-199732060-00002. [DOI] [PubMed] [Google Scholar]
- 23.Tinsley HN, Gary BD, Keeton AB, et al. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol Cancer Ther. 2009 Dec 8; doi: 10.1158/1535-7163.MCT-09-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu B, Vemavarapu L, Thompson WJ, Strada SJ. Suppression of cyclic GMP-specific phosphodiesterase 5 promotes apoptosis and inhibits growth in HT29 cells. J Cell Biochem. 2005 Feb 1;94(2):336–50. doi: 10.1002/jcb.20286. [DOI] [PubMed] [Google Scholar]
- 25.Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A. 1999 Jun 22;96(13):7563–8. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]