

Abstract
Processability remains a fundamental issue for the implementation of conducting polymer technology. A simple synthetic route towards processable precursors to conducting polymers (main chain and side chain) was developed using commercially available materials. These soluble precursor systems were converted to conjugated polymers electrochemically in aqueous media, offering a cheaper and greener method of processing. Oxidative conversion in aqueous and organic media each produced equivalent electrochromics. The precursor method enhances the yield of the electrochromic polymer obtained over that of electrodeposition, and it relies on a less corruptible electrolyte bath. However, electrochemical conversion of the precursor polymers often relies on organic salts and solvents. The ability to achieve oxidative conversion in brine offers a less costly and a more environmentally friendly processing step. It is also beneficial for biological applications. The electrochromics obtained herein were evaluated for electronic, spectral, and morphological properties.
Keywords: aqueous processing, conjugated polymers, electrochemistry, precursor, redox polymers
Introduction
Conducting polymers (CPs) have become increasingly studied in both the academic and industrial fields due to their exhibition of stability, high conductivities, and tunable optical properties.1 However, the preparation of CPs by electrodeposition suffers from the finite lifetime of the electrolyte solution due to the formation of byproducts which poison the bath. Additionally, the production of infusible CPs on the electrode surface hinders the use of electrochemical polymerization in industry, as it limits large-area processing and requires complicated patterned electrodes for more sophisticated designs. The use of any large-area substrate for optical applications, such as windows or displays, requires rigorous care against electrode defects. These concerns prompted the search for alternate routes to process CPs with greater efficacy. Precursor polymers have been used in the past; 2,3,4 we have developed a precursor polymer method wherein a soluble precursor may be cast, patterned, or otherwise processed and subsequently converted to its conjugated, electrochromic form. Conducting polymer networks have been formed from precursors by chemical5 and electrochemical6 methods of oxidation. Jang et al.7,8 developed a method to convert precursor polymers to their conductive forms in the solid swollen state. Advantages of solid state oxidative conversion using precursor polymers include higher yields, processability, control of morphology, and cheaper, cleaner processing.9,10 Precursor polymers are stable and exhibit high chain entanglements which provide a means to process and create conducting nanolines, thin films, and nanofibers.6,11,12 More recently, the Swager group has demonstrated the use of pendant norbornene moieties in block-copolymers in order to form nanostructures via microphase separation.9
Chahma et al. synthesized poly(3,4-ethylenedioxythiophene) (PEDOT) via electrochemical oxidative cleavage of silicon groups.13 Their work incorporated silated monomeric precursors of 3,4-ethylenedioxythiophene (EDOT); the silyl-carbon bonds were cleaved and polymerization was facilitated electrochemically at a lower oxidation potential. The optical and electronic properties remained intact, aside from subtle changes due to variations in the chain length and the morphology. That prompted Bokria et al. to produce an alternating copolymer of alkyl silanes and conjugated moieties 14 to create conjugated polymer upon silyl-carbon bond cleavage.7–13 Other precursor polymers have been used to coat gold nanoparticles in order to tune their surface plasmon resonance bands15 and in the facile assembly of electrochromic devices.16 Attention has also been gathering over the surface morphology and wettability of conjugated polymers films that are produced after polymerization. 17 We have taken advantage of this information and introduced a high molecular weight co-polymer of a silane and a conjugated moiety (main chain approach). Furthermore, we have also made an evaluation of the surface morphology with this procedure. This precursor polymer is soluble in the same common organic solvents as the corresponding monomer. The same chromophore can be attached as a pendant to a functionalized norbornene, which is then polymerized via ROMP (side chain approach). This soluble precursor, upon oxidation, couples into a crosslinked conjugated electrochromic network. These oxidations are carried out in traditional three-electrode cell configurations with organic solvents and organic salts. The expense and toxicity of the organic electrolyte baths make a green alternative particularly enticing. As a result, we have shown that both the main chain and side chain precursor systems were able to undergo conversion using brine (saturated sodium chloride in water) in place of the organic electrolyte. Brine conversion is not only less expensive but it is more environmentally friendly. Further, for biological applications, it allows the polymer to be converted in the medium in which it will eventually be switched. This eliminates the need to pre-cycle or “build in” electrochromic functionality, a procedure common for other processable electrochromics.18
The use of conjugated polymers for drug delivery has been reported in literature19 and more recently as a switchable interface with cells.20 Avoiding organic solvents is advantageous for biological applications, removing the possibility of trace compounds leeching from the polymer into a living system. Herein, we utilize the aforementioned processable precursor polymers to form a conjugated polymer under both organic and aqueous conditions. The synthesis of norbornene- and silane-based precursor polymers consisting of bis(3,4-ethylenedioxythienyl)thiophene (BEDOT-T) units is reported; these precursors produced equivalent electrochromic films via conversion in aqueous electrolyte as those formed using organic electrolyte.
Experimental
Materials
Tetrahydrofuran (THF) was purchased from Fisher Scientific and distilled over potassium metal, under nitrogen atmosphere. Acetonitrile (ACN) and dichloromethane (CH2Cl2) were purchased from Fisher Scientific and were distilled over calcium hydride (CaH2) under nitrogen atmosphere prior to use. 2.5 M n-butyllithium (n-BuLi) in hexanes, ferric chloride (FeCl3), anhydrous N,N-dimethylformamide (DMF), ethyl vinyl ether, ferrocene, silver nitrate, silver wire, 3-methyl thiophene, petroleum ether, n-pentane, sodium hydroxide, chloroform (CHCl3) were purchased from Fisher Scientific and used as received. 2,5-dibromothiophene was purchased from Oakwood Chemicals, Inc. and used as received. 3,4-Ethylenedioxythiophene (EDOT) and dichlorodi-n-octylsilane were purchased from Aldrich Chemical Co. and Gelest, Inc. respectively, and distilled under vacuum prior to use. Hydrazine hydrate solution in water (appx. 24%) was purchased from Riedel-de Haen and used as received. Magnesium bromide diethyletherate (MgBr2.(Et2O)2), bis(diphenyl phosphino)propane nickel(II) chloride (NiCl2.dppp), Grubbs Generation I alkylidene catalyst, deuterated chloroform (CDCl3) and tetra-n-butyl ammonium hexafluorophosphate (TBAPF6) were purchased from Aldrich Chemical Co. and used as received. Sodium hydride was purchased from Aldrich and stored in a desiccator when not in use. 5-Norbornene-2-methanol was purchased from Aldrich. N-Bromosuccinimide (NBS) was from Aldrich and was recrystallized from distilled water and vacuum dried for 24 hrs. 2,2′-Azobis(2-methylpropionitrile) (AIBN) from Aldrich was purified by recrystallization from methanol. Sodium chloride (NaCl), sodium sulfate (Na2SO4) was purchased from Fisher Scientific and used as received. Synthesis of 2,5-dibromo-3-(bromomethyl)thiophene (1) was synthesized according to literature procedures. 21 Synthesis of 2,5-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl) thiophene (BEDOT-T) was synthesized according to literature.22 Indium-doped tin oxide (ITO) coated glass (dimensions 7 mm × 50 mm × 0.7 mm, Rs = 15–25 Ω, unpolished float glass) was purchased from Delta Technologies.
Instrumental
A CH Instruments 400 potentiostat and 660a potentiostat were used for electrochemical polymerizations. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker 400 FT-NMR spectrometer. 1H NMR data are reported as follows; chemical shift (multiplicity: s=singlet, d=doublet, t=triplet, q=quartet, and m=multiplet, integration). 1H and 13C chemical shifts are reported in ppm downfield from tetramethylsilane (TMS) reference. Optical properties of the polymer films were measured by both Perkin-Elmer Lambda 900 and a Varian Cary 5000 UV-Vis-NIR spectrophotometers. Polymer film thickness was measured using a Zygo New View 3-D Optical Profilometer surface profiler. Fourier transform infrared spectroscopy (FT-IR) was performed using a MAGNA-IR560. Thermal analysis was performed using TA Instruments DSC 2920 and Hi-Res TGA 2950 for Differential Scanning Calorimetry (DSC) and Thermal Gravimetric Analysis (TGA), respectively. In DSC, the samples were heated to 100°C and immediately cooled until they reach to −50°C, and then DSC analysis was performed by heating at a scan rate of 10°C/min. The polymer films on ITO glass were measured using JEOL 6335F field emission scanning electron microscope (SEM). Elemental Analysis was performed by Galbraith Laboratories.
Electrochemistry
All electrochemical experiments in organic media were performed using a three-cell configuration in 0.1 M TBAPF6/ACN with a CHI 400 potentiostat. The reference electrode was a nonaqueous Ag/Ag+ electrode consisting of a silver wire immersed in a glass capillary body fitted with a Vycor tip and filled with 0.1 M silver nitrate (AgNO3)/ACN and 0.1 M TBAPF6/ACN solutions. The Ag/Ag+ reference electrode was calibrated to be 0.4385 V vs. the normal hydrogen electrode (NHE) using a 10 mM ferrocene/ACN solution. A 1 cm2 platinum flag was used as a counter electrode in all electrochemical measurements. Aqueous experiments were carried out using saturated water solutions of NaCl on a CHI 660a potentiostat. An aqueous reference electrode (Ag/AgCl in 3 M NaCl, 0.230 V vs. NHE) was used, along with the same Pt counter. We define “conventional electrochemical polymerization” as the method by which the monomer is dissolved in electrolyte solution and a potential is applied to the working electrode by which oxidation of the monomer takes place to form corresponding polymer. After polymerization, the polymer-coated Pt working electrode was washed with ACN to remove residual monomers and oligomers. The Pt flag is routinely heated with an acetylene torch to ensure the complete removal on any residual organic components. The cyclic voltammogram of the polymer was obtained in a monomer-free solution (comprising the same electrolyte system used for polymerization minus the presence of monomer) in order to isolate the electrochemical processes of the polymer. In organic electrochemical solid-state oxidative conversions, the experimental setup was the same as in the conventional method (salt and solvent), with the only difference and outstanding advantage being that there was no need to wash the polymer and refresh the electrolyte solution due to solid-state conversion and monomer-free solution during the conversion. During aqueous electrochemical solid-state oxidative conversions, the electrolyte was saturated NaCl in DI water. Again, the bath was not changed between conversion and switching, as it was unnecessary. All spectroelectrochemical measurements for films were taken in monomer-free electrolyte systems, as described above.
Preparation of Scanning Electron Microscopy (SEM) samples
Conventional electropolymerization was employed using 10 mM monomer in a 0.1 M TBAPF6/ACN solution. For conductive networks processed from precursor polymers, 1% (w\w) precursor polymer in CH2Cl2 solution was spin-coated onto an ITO/Glass slide and placed into 0.1 M TBAPF6/ACN solution and electropolymerized.
Synthesis of endo- & exo- 3-(((bicyclo[2.2.1]hept-5-en-2-yl)methoxy)methyl)-2,5-dibromothiophene (2)
This material was synthesized according to literature procedure.15
Synthesis of endo- & exo- 5-(4-(((bicyclo[2.2.1]hept-5-en-2-yl)methoxy)methyl)-5-(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)thiophen-2-yl)-2,3-dihydrothieno[3,4-b][1,4]dioxine (3)
This material was synthesized according to literature procedure.15
Synthesis of poly(4)
To a vacuum-dried and nitrogen purged 50 mL three-necked round bottom flask, a solution of (3) (125 mg, 0.25 mmol) in 3 mL dry dichloromethane was injected under nitrogen. After addition of 10.3 mg of Grubb’s Gen. I alkylidene catalyst (5 mol%, 0.0125 mmol) in 2.5 mL of dichloromethane, the mixture was stirred for 1 hour, followed by quenching with 0.5 mL of ethyl vinyl ether. The polymer was precipitated in 150 mL of pentane, filtered and then dried under vacuum to yield 57 mg (0.114 mmol) of poly(4) as a gray powder. Yield: 45.6%. 1H NMR (CDCl3): δ 7.24 (1H, CH), 6.36 (1H, CH), 6.20 (1H, CH), 5.35 (2H, CH), 4.47 (2H, CH2), 4.28 (2H, CH2), 4.23-4.19 (6H, 3CH2), 3.40-3.25 (2H, CH2), 2.60-2.41 (1H, CH), 2.25 (1H, CH), 1.89-1.82 (2H, CH2), 1.67-1.44 (1H, CH), 1.37-1.23 (2H, CH2); 13C NMR (CDCl3): δ 141.83, 141.51, 138.19, 137.63, 136.69, 136.50, 135.13, 133.89, 133.76, 128.03, 124.72, 124.43, 112.06, 109.77, 99.46, 96.99, 75.66, 72.45, 67.98, 67.19, 64.98, 64.88, 64.56, 64.47, 42.62, 42.52, 39.85, 37.05, 34.15, 29.72, 26.85, 25.62, 21.30, 14.12 FTIR: 3109 cm−1 (aromatic C-H stretching); 2923 and 2852 cm−1 (aliphatic C-H stretching); 1072 cm−1 (aliphatic C-O stretching). GPC: Number-average molecular weight, Mn = 94,336 g/mol. DSC: Tg = 115°C. TGA: Tdeg = 209°C.
Synthesis of poly(6) by Condensation Polymerization
A solution of the BEDOT-T (1 eq.) in anhydrous THF under nitrogen was cooled to −78°C in a dry ice/acetone cooling bath. n-Butyllithium (2 eq.) was added drop-wise into the pre-cooled solution via syringe. After 1 hr, the reaction mixture was warmed to 0°C in ice/water bath for 15 min and then dichloro-di-n-octyl silane (1 eq.) was added drop-wise into this solution. The ice bath was removed after 30 min. The reaction solution was stirred for 3 days at room temperature. The crude polymer was precipitated out of the reaction mixture by pouring the mixture into n-pentane. The resulting precipitate was purified by Soxhlet extraction using ACN as the solvent. 1H NMR (CDCl3): δ 7.18 (s, 2H, CH); 4.43-4.25 (m, 8H, CH2); 1.44-1.16 (m, 24H, CH2); 0.9 (t, 6H, CH3); 0.15 (t, 4H, CH2); 13C NMR (CDCl3): δ 147.96, 141.89, 137.94, 133.47, 126.02, 65.01, 64.43, 33.46, 31.95, 29.70, 29.26, 23.60, 22.90, 22.71, 14.14 FTIR (KBr): 3038, 2921, 2851, 1634, 1432, 1358, 1147, 1087, 1040, 550 cm−1 GPC: Number-average molecular weight, Mn = 80,667 g/mol. DSC: Tg = 40°C. TGA: Tdeg = 290°C.
Preparation of poly(5) and poly(7) via Organic (conventional) solid state oxidative conversion
A 1-wt % solution of the precursor polymer in CH2Cl2 was prepared. A thin film of precursor was drop-cast from this solution on a platinum button (2 mm diameter), followed by air-drying. The electrochemical conversion of precursor was then carried out at a scan rate of 100 mV/s in 0.1 M TBAPF6/ACN solution via cyclic voltammetry.
Preparation of poly(5) and poly(7) via Aqueous solid state oxidative conversion
A 10:1 mole ratio of TBAPF6 : precursor polymer solution was prepared in CH2Cl2. A thin film of precursor was drop-cast from this solution on a platinum button (2 mm diameter), followed by air-drying. Conversion was carried out in brine (saturated NaCl in deionized water) at a scan rate of 25 mV/s using cyclic voltammetry.
Preparation of poly(BEDOT-T) and poly(3) via Electrochemical Deposition
A 1 wt% solution of BEDOT-T or (3) were dissolved in TBAPF6/ACN (10 mL) solution and were polymerized via cyclic voltammetry. The resulting polymers were washed several times with ACN. After the rinsing process, the working electrode was inserted into a monomer free solution of 0.1 M TBAPF6/ACN (10 mL) and then cyclic voltammograms of the polymer were obtained at a scan rate of 100 mV/s to probe polymer redox behavior.
Results and Discussion
Monomer and Precursor Polymer Synthesis
The monomer used for the side chain, or pendant, precursor approach, (3), was prepared in a four-step procedure starting with 3-methylthiophene.21 Scheme 1 shows the reaction of 2,5-dibromo-3-(bromomethyl) thiophene (1) via Kumada coupling to form the precursor monomer. The structure was confirmed using 1H and 13C NMR and FTIR. Scheme 2 displays the variety of ways in which this monomer was used, namely the formation of a conjugated polymer by direct electrochemical polymerization and the formation and conversion of the precursor polymer. The precursor polymer was readily soluble in CHCl3 and CH2Cl2.
Scheme 1.

Synthesis of the precursor monomer, (3), used in the side chain approach.
Scheme 2.

(top) Electrochemical and chemical polymerization routes for (3); (bottom) conversion of precursor polymer, poly(4), to electrochromic polymer, poly(5).
The main chain precursor polymer, poly(6), was prepared via step-growth polymerization, as shown in Scheme 3. The formation of dilithiated monomers from n-BuLi allowed them to react with dichloro-di-n-octyl silane in order to give the corresponding polymers. Poly(6) was found to be readily soluble in common polar, aprotic organic solvents such as THF and CHCl3.
Scheme 3.
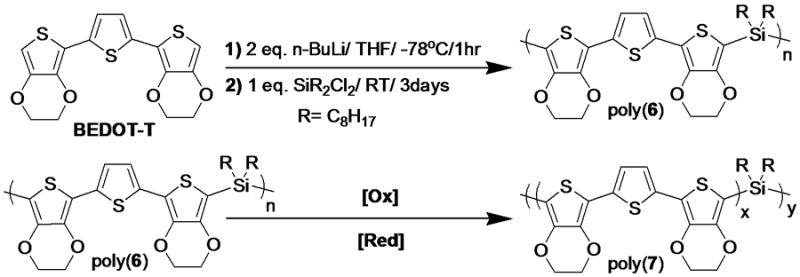
Synthesis of main chain precursor polymer, poly(6), and its conversion into a conjugated polymer, poly(7).
Poly(6) was purified by Soxhlet extraction with ACN to remove residual oligomers and low molecular weight polymers. Rigorous precision of the reactant stoichiometry was imperative to obtain a high molecular weight for the main chain precursor polymer (ca. 80 kDa).
Electrochemistry of Precursor Polymers
The aqueous electrochemical conversion of poly(4) to poly(5) was carried out using a brine solution (saturated sodium chloride in water) as the electrolyte. The films which were cast of the precursor polymer were loaded with a 10:1 mole ratio of salt to polymer. Without the addition of a salt to the polymer film matrix, aqueous electrochemical conversion of precursor polymers cannot be achieved. Typically, a water-insoluble salt is chosen for this process. Herein, we used TBAPF6 in the precursor films. In an organic electrolyte bath, the polymer films are swollen. Ion shuttling and diffusion are therefore facilitated. The salt loading allows for the presence of cavities in the film matrix, as well as proximal access to counterions during the conversion process. Thus, the films are able to convert and switch in brine.
The aqueous conversion CV for the norbornene-based precursor, poly(4), was initiated at +0.2 V and terminated at +1.1 V. The onset for oxidative conversion occurred during the first cycle, at +0.78 V, with a shoulder appearing at +0.85 V and a peak at +1.01 V. Subsequent cycles showed increasing current growth, implying the creation of conducting polymer on the electrode surface. The redox peaks for the polymer thus obtained were broad and occurred at +0.75 V and +0.85 V when scanning anodically and cathodically, respectively. The color of the resultant material matched that of the precursor polymer that was converted under organic conditions. The CV for the organic conversion of poly(4) was initiated at −0.3 V. At a potential of +0.3 V, there was an onset for oxidation with a peak at +0.535 V. The onset oxidation potential of poly(4) was only 0.03 V lower than conventional electropolymerization of (6), but was the same as the onset potential for the electropolymerization of BEDOT-T. The oxidation potential poly(4) was higher than that of BEDOT-T by 0.14 V. The reduction potential was 0.03 V higher than the reduction potential of (4). In the second voltage scan of poly(4) in the anodic direction, only the oxidation of the conjugated poly(BEDOT-T) crosslinked units was noted, which is evidence of complete conversion. The initially yellow-brown film of poly(4) turned bluish above +0.1 V during the electrochemistry and was a reddish color in its neutral state.
The silane-based precursor system, poly(6), was converted to the conjugated polymer, poly(7), by electrochemical desilylation. In the organic case, poly(6) had an onset oxidation potential at +0.3 V and peaked at +0.6 V. Upon reversing the scan in the cathodic direction, two reduction peaks at +0.39 V and −0.05 V were observed which corresponds to the reduction of oxidized and conductive form to the neutral form of poly(7). In the second anodic scan, the disappearance of the peak at +0.6 V indicated the complete conversion of precursor poly(6) to poly(7); it displayed broad redox peaks at +0.19 V and +0.49 V. When compared to the CVs from poly(6), the polymer obtained from conventional polymerization of the monomer, BEDOT-T, resulted in similar redox peaks. It was found that the precursor polymers had an increase in the oxidation potential of approximately ~0.1 V. The conversion of poly(6) into its conductive form, poly(7), was visually confirmed by the color change from a yellow precursor film to blue (oxidized, above +0.2 V) and red (neutral, below −0.4 V) states. These colors remained stable upon exposure to atmospheric conditions for several weeks. The aqueous case for this precursor was treated the same as before, with a salt pre-loaded film. The CV was initiated at 0.0 V and terminated at +0.80 V. The onset of polymerization occurred at +0.58 V with a peak appearing at +0.75 V. Broad redox peaks appeared at +0.51 V anodically and +0.49 V. Once again, the color obtained via this process matched that of the organic case.
Spectroelectrochemistry
In order to assess the similarity of the resultant materials across polymerization methods, spectral comparisons in the visible range were made between the electropolymerization of the monomers incorporated into the precursor polymers as well as the organic and aqueous conversions of each. Figure 1 displays the oxidized and neutral states of thin films of the side chain precursor (norbornene based), as well as a film of electrochemically deposited monomer. Electropolymerization of (3) resulted in a polymer whose λmax in the neutral state was 510 nm. This material exhibited peaks at 370 nm and 743 nm in the oxidized state. Organic and aqueous converted precursor poly(4) showed neutral state peaks at 483 nm and 521 nm, respectively. The oxidized state peaks appeared at 359 nm and 353 nm, respectively. These values were in close agreement and the spectra were very similar in all cases, indicating that an equivalent electrochromic material was created from each of the three procedures. Color coordinates for the converted forms of these side chain based precursors were calculated. CIE u′v′ values are tabulated below (Table 1). Visually, the colors obtained are equivalent; the color coordinates support this observation.
Figure 1.

Neutral (solid line) and oxidized (dashed line) state spectra for (A) electropolymerized (3) [poly(3)], (B) organic converted poly(5), and (C) aqueous converted poly(5). Spectra were taken for films switched in the same electrolyte used for their respective polymerizations.
Table 1.
CIE u′v′ color coordinates for electrodeposited 3 and converted precursor poly(4).
| State | Method | u′ | v′ |
|---|---|---|---|
| Neutral | Electrodeposition | 0.25 | 0.47 |
| Organic | 0.29 | 0.51 | |
| Aqueous | 0.25 | 0.46 | |
| Oxidized | Electrodeposition | 0.18 | 0.48 |
| Organic | 0.20 | 0.45 | |
| Aqueous | 0.21 | 0.48 |
Figure 2 shows films of the main chain precursor from both conversion methods, as well as electrodeposited monomer. Electrodeposited monomer BEDOT-T yielded a neutral state λmax of 538 nm. In the oxidized state, it exhibited a peak at 364 nm and a shoulder at approximately 775 nm. Organic and aqueous converted precursor poly(6) showed neutral state peaks at 524 nm and 478 nm, respectively. The aqueous converted sample also exhibited shoulders at 419 and 526 nm. The oxidized state peaks appeared at 367 nm (shoulder at 775 nm) and 368 nm (also at 692 nm), respectively. These values were again in close agreement with each other, as well as showing similarity to the side chain spectra, although the aqueous processed silane precursor did show a blue shift of 46 nm, indicating an overall shorter conjugation length. Color coordinates for the main chain approach were calculated, as well. The CIE u′v′ values are shown in Table 2. Once again, visual confirmation of matched colors was confirmed by the color coordinates.
Figure 2.

Neutral (solid line) and oxidized (dashed line) state spectra for (A) electropolymerized BEDOT-T, (B) poly(7) prepared by organic conversion and (C) poly(7) prepared by aqueous conversion. Spectra were taken for films switched in the same electrolyte used for their respective polymerizations.
Table 2.
CIE u′v′ color coordinates for BEDOT-T and converted precursor poly(6).
| State | Method | u′ | v′ |
|---|---|---|---|
| Neutral | Electrodeposition | 0.23 | 0.43 |
| Organic | 0.25 | 0.46 | |
| Aqueous | 0.25 | 0.50 | |
| Oxidized | Electrodeposition | 0.19 | 0.46 |
| Organic | 0.19 | 0.47 | |
| Aqueous | 0.19 | 0.46 |
Despite subtle differences in the spectra for each of these six different materials, the coloration consistency confirms that the material produced is an equivalent electrochromic. No significant variation in switching speeds was observed for any of the systems studied. The films produced by aqueous conversion switched states, assuring their functionality as a conjugated or conducting polymer, as well. Drug delivery applications using electrostatic interactions with molecules such as dopamine HCl (sparingly soluble in DCM) are therefore feasible. One can envision pre-loading the precursor films with such drugs, which will associate with the conducting polymer upon oxidative conversion. Subsequent redox switching would then release the drug into the medium.
SEM Analysis
Films of conjugated polymers from electrodeposition and precursor conversion were prepared and analyzed by SEM to compare the morphologies of the conductive films after processing. As shown in Figure 3, the film obtained from conversion of a spun-coated sample of the main chain precursor polymer (left) showed a smooth, thin film. However, the film generated via conventional electrochemical deposition of BEDOT-T (right) is a rough, grainy film. These morphological changes are due to the difference in mechanism of polymerization in each case. Smooth films are unlikely to result from nucleation and growth mechanisms, such as electrodeposition. It should be noted that side-chain conversions result in crosslinked films, which also have rough morphologies. The main chain precursors, on the other hand, yield the observed smooth topography.
Figure 3.

Comparison SEM pictures of films of conducting polymers achieved by (top) main chain precursor conversion [poly(7)] and (bottom) electropolymerization [PBEDOT-T].
Conclusion
Conjugated polymers were prepared in water. The resultant polymers were redox active, switched states in aqueous electrolyte, exhibited electrochromism, and matched the coloration of materials prepared via organic processing and electrochemical deposition. This expands the versatility of the precursor method as well as creates an inexpensive and environmentally sound processing step. High yields were achieved and maintained through these conversions, in agreement with prior solid state experiments. We speculate that any precursor which is created via the main or side chain approach may be converted to corresponding conducting polymers using aqueous solid state oxidative conversion, provided its oxidation potential does not exceed the electrochemical window of the electrolyte. The importance of this work is the use of a “greener” processing technique which also represents a large reduction in cost over organic salts and solvents. Further, aqueous-converted precursors could be employed in a variety of medical and biological applications of conducting polymers without affecting the morphology of the film.
Supplementary Material
Acknowledgments
The authors thank the NIEHS/NIH (PHS Grant ES013557) for financial support of this work.
References and Notes
- 1.a) Lerch K, Jonas F, Linke MJ. Chem Phys Phy-Chem Biol. 1998;95:1506. [Google Scholar]; b) Sotzing GA, Briglin S, Grubbs RH, Lewis NS. Anal Chem. 2000;72:3181. doi: 10.1021/ac991079x. [DOI] [PubMed] [Google Scholar]; c) Allcock HR, Dodge JA, Van Dyke LS, Martin CR. Chem Mater. 1992;4:780–788. [Google Scholar]; d) Trchova M, Stejskal J, Prokes J. Synth Met. 1999;101:840–841. [Google Scholar]; e) Sapp SA, Sotzing GA, Reynolds JR. Chem Mater. 1998;10:2101–2108. [Google Scholar]; f) Yu G, Heeger AJ. Synth Met. 1997;85:1183. [Google Scholar]; g) Friend RH, Gymer RW, Holmes AB, Burroughes JH, Marks RN, Taliani C, Bradley DDC, Dos Santos DA, Bredas JL, Logdlund M, Salaneck WR. Nature (London) 1999;397:121–128. [Google Scholar]; h) Sirringhaus H, Tessler N, Friend RH. Science. 1998;280:1741–1744. doi: 10.1126/science.280.5370.1741. [DOI] [PubMed] [Google Scholar]; i) Torsi L, Dodabalapur A, Rothberg LJ, Fung AWP, Katz HE. Science. 1996;272:1462–1464. doi: 10.1126/science.272.5267.1462. [DOI] [PubMed] [Google Scholar]; j) McQuade DT, Pullen AE, Swager TM. Chem Rev. 2000;100:2537–2574. doi: 10.1021/cr9801014. [DOI] [PubMed] [Google Scholar]; k) Roncali J, Garnier F. J Phys Chem. 1988;92:833–840. [Google Scholar]; l) Evanoff DD, Jr, Carroll JB, Roeder RD, Hunt ZJ, Lawrence JR, Foulger SH. J Polym Sci Part A: Polym Chem. 2008;46:7882–7897. [Google Scholar]; m) Udum YA, Yildiz E, Gunbas G, Toppare L. J Polym Sci Part A: Polym Chem. 2008;46:3723–3731. [Google Scholar]; n) Turkarslan O, Ak M, Tanyeli C, Akhmedov IM, Toppare L. J Polym Sci Part A: Polym Chem. 2007;45:4496–4503. [Google Scholar]; o) Chang HW, Lin KH, Chueh CC, Liou GS, Chen WC. J Polym Sci Part A: Polym Chem. 2009;47:4037–4050. [Google Scholar]
- 2.Taranekar P, Park J, Patton D, Fulghum T, Ramon G, Bittner E, Advincula R. Adv Mater. 2006;18:2461–2465. [Google Scholar]
- 3.Advincula R. J Coat Tech. 2008;5:34–37. [Google Scholar]
- 4.Katzenmeyer AM, Bayam Y, Logeeswaran VJ, Pitcher MW, Nur Y, Seyyidocglu S, Toppare LK, Talin AA, Han H, Davis CE, Islam MA. Journal of Nanomaterials. 2009:4. doi: 10.1155/2009/832327. Article ID 832327. [DOI] [Google Scholar]
- 5.Taranekar P, Abdulbaki M, Krishnamoorti R, Krishnamoorti S, Waenkaew P, Patton D, Fulghum T, Advincula R. Macromolecules. 2006;39:3848–3854. [Google Scholar]; b) Kiskan B, Yagci Y, Sahmetlioglu E, Toppare L. J Polym Sci Part A: Polym Chem. 2007;45:999–1006. [Google Scholar]
- 6.a) Watson KJ, Wolfe PS, Nguyen ST, Zhu J, Mirkin CA. Macromolecules. 2000;13:4628–4633. [Google Scholar]; b) Jagadesan S, Advincula R, Valiyaveettil S. Adv Mater. 2005;17:1282–1285. [Google Scholar]
- 7.Jang S, Sotzing GA, Marquez M. Macromolecules. 2002;35:7293–7300. [Google Scholar]
- 8.Jang S, Sotzing GA, Marquez M. Macromolecules. 2004;37:4351–4359. [Google Scholar]
- 9.Kang HA, Bronstein HE, Swager TM. Macromolecules. 2008;41:5540–5547. [Google Scholar]
- 10.Zhao C, Zhang Y, Pan S, Rothberg L, Ng MK. Macromolecules. 2007;40:1816–1823. [Google Scholar]
- 11.Jang S, Seshadri V, Khil M, Kumar A, Marquez M, Mather PT, Sotzing GA. Adv Mater. 2005;17(18):2177–2180. [Google Scholar]
- 12.Jang S, Marquez M, Sotzing GA. J Am Chem Soc. 2004;126:9476–9477. doi: 10.1021/ja047766+. [DOI] [PubMed] [Google Scholar]
- 13.Chahma M, Hicks RG. Can J Chem. 2004;82:1629–1633. [Google Scholar]
- 14.Bokria JG, Kumar A, Seshadri V, Tran A, Sotzing GA. Adv Mater. 2008;20:1175–1178. [Google Scholar]
- 15.Yavuz MS, Jensen GC, Penaloza DP, Seery TAP, Pendergraph SA, Rusling JF, Sotzing GA. Langmuir. 2009;25:13120–13124. doi: 10.1021/la901779k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Invernale MA, Ding Y, Mamangun DMD, Yavuz MS, Sotzing GA. Adv Mater. 2010 doi: 10.1002/adma.200902975. (in press) [DOI] [PubMed] [Google Scholar]
- 17.a) Darmanin T, Nicolas M, Guittard F. Langmuir. 2008;24:9739–9746. doi: 10.1021/la801040z. [DOI] [PubMed] [Google Scholar]; b) Lin P, Yan F, Chan HLW. Langmuir. 2009;25:7465–7470. doi: 10.1021/la900387m. [DOI] [PubMed] [Google Scholar]
- 18.Mortimer RJ, Graham KR, Grenier CRG, Reynolds JR. ACS Applied Materials & Interfaces ACS ASAP, (Web) 2009 October 8; doi: 10.1021/am900431z. [DOI] [PubMed] [Google Scholar]
- 19.a) George PM, LaVan DA, Burdick JA, Chen C-Y, Liang E, Langer R. Adv Mater. 2006;18:577–581. [Google Scholar]; b) Niamlang S, Sirivat A. International Journal of Pharmaceutics. 2009;371:126–133. doi: 10.1016/j.ijpharm.2008.12.032. [DOI] [PubMed] [Google Scholar]
- 20.a) Luo SC, Ali EM, Tansil NC, Yu H, Gao S, Kantchev EAB, Ying JY. Langmuir. 2008;24:8071–8077. doi: 10.1021/la800333g. [DOI] [PubMed] [Google Scholar]; b) Salto C, Saindon E, Bolin M, Kanciurzewska A, Fahlman M, Jager EWH, Tengvall P, Arenas E, Berggren M. Langmuir. 2008;24:14133–14138. doi: 10.1021/la8028337. [DOI] [PubMed] [Google Scholar]; c) Ludwig KA, Uram JD, Yang J, Martin DC, Kipke DR. J Neural Eng. 2006;3:59–70. doi: 10.1088/1741-2560/3/1/007. [DOI] [PubMed] [Google Scholar]
- 21.a) Yassar A, Garnier F. Macromolecules. 1995;28:4548–4553. [Google Scholar]; b) Mandal SS, Chakraborty J, De A. J Chem Soc. 1999;18:2639–2644. [Google Scholar]
- 22.Sotzing GA, Reynolds JR, Steel PJ. Chem Mater. 1996;8:882–889. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.