

Abstract
ATP-binding cassette, sub-family G, member 2 (ABCG2) is expressed in both normal and cancer cells, and plays a crucial role in the side population (SP) formation and efflux of xenobiotics and drugs. Nrf2, a redox sensing transcription factor, upon constitutive activation in non-small-cell lung cancer cells up-regulates a wide spectrum of genes involved in redox balance, glutathione metabolism, and drug detoxification that contribute to chemoresistance and tumorigenecity. This study examined the mechanism underlying Nrf2-dependent expression of ABCG2 and its role in multidrug resistance phenotype. In silico analysis of the 5’-promoter flanking region of ABCG2 identified an antioxidant response element at -431 bp to -420 bp. A detailed promoter analysis using luciferase reporter assays demonstrated that antioxidant response element (ARE) at -431 bp to -420 bp is critical for the Nrf2-mediated expression in lung cancer cells. Electrophoresis mobility shift assays (EMSA) and chromatin-immunoprecipitation (ChIP) assays revealed that Nrf2 interacts with ABCG2 ARE element at -431 bp to -420 bp in vitro and in vivo. Disruption of Nrf2 expression in lung cancer and prostate cancer cells, by short hairpin RNA, attenuated the expression of ABCG2 transcript and protein and dramatically reduced the SP fraction in Nrf2-depleted cancer cells. Moreover, depleted levels of ABCG2 in these Nrf2-knockdown cells sensitized them to mitoxantrone and topotecan, two chemotherapy drugs detoxified mainly by ABCG2. As expected, overexpression of Nrf2 cDNA in lung epithelial cells led to an increase in ABCG2 expression and a 2-fold higher SP fraction. Thus, Nrf2-mediated regulation of ABCG2 expression maintains SP fraction and confers chemoresistance.
Keywords: Nrf2, ABCG2, lung cancer, cancer stem cells, chemo-resistance, RNAi
Introduction
Lung cancer is the leading cause of cancer-related death in both men and women in US [1]. The prognosis for lung cancer remains poor, with overall 5-year survival of 14%. The death toll caused by lung cancer alone counts more than that of breast, colorectal, and prostate cancers combined. Non-small cell lung carcinoma (NSCLC) constitutes about 85% of all lung cancers[1]. Chemotherapy is the standard treatment for advanced NSCLC patients, but chemotherapy resistance stays as an obstacle and leads to mortality.
Recent discoveries have provided clear evidence that cancers may develop from rare self-renewing stem cells, which are biologically distinct from differentiated cancer cells. The eradication of these cancer stem cells is likely a critical component of any successful anticancer strategy and this may explain why conventional cancer therapies are often effective in reducing tumor burden, but are rarely curative. Cancer stem cells have been identified in several cancerous tissues, such as acute myelogenic leukemia, neuroblastoma, lung, colon, and breast cancers [2-4]. These cancer stem cells represent only a small percentage of total cell populations, and they show distinct features, such as resistance to irradiation and chemotherapy, reconstitution of the whole populations after irradiation [3, 5]. Interestingly, cancer stem cells efficiently efflux Hoechst dye resulting in the dye-negative phenotype, also known as side population (SP) phenotype [3]. Further investigations revealed that Hoechst dye efflux and the SP formation capacity of cancer stem cells are largely attributable to ATP-binding cassette, sub-family G, member 2 (ABCG2) molecule [6-8]. ABCG2, also known as breast cancer resistance protein (BCRP), was originally cloned from multi-drug resistant breast cancer cells [9], and its up-regulation has been linked to chemo-resistance phenotype in various cancer cells [3, 6]. It was demonstrated that ABCG2 is responsible for the SP formation in lung cancer cells [10-11].
Nrf2, a cap ‘n’ collar basic leucine zipper transcription factor, protects against environmental toxicants, oxidative injury, inflammation, and apoptosis through transcriptional induction of a broad spectrum of cytoprotective genes involved in electrophile/drug detoxification function including several ATP-dependent drug efflux pumps (e.g., ATP-binding cassette, sub-family C, member 1 and ATP-binding cassette, sub-family C, member 2) [12-14]. Kelch like ECH associated protein (KEAP1) is a cytoplasmic anchor of Nrf2 and maintains steady-state levels of Nrf2 and Nrf2-dependent transcription by signaling Nrf2 for proteosomal degradation [15-16]. Somatic mutations in KEAP1 and loss of heterozygosity at KEAP1 locus result in loss of KEAP1 function in cancer cells and gain of Nrf2 function [17]. Activating mutations in Nrf2 have been recently reported in squamous cell lung carcinomas [18]. Gain of Nrf2 function in lung cancer cells up-regulates the expression of genes involved in protection against oxidative stress and thereby promotes tumorigenecity and chemo-resistance [17, 19-22].
The ABCG2 gene is highly expressed in the plasma membrane of several drug resistant cell lines, where it has been shown to transport antitumor drugs including mitoxantrone, topotecan, doxorubicin, and daunorubicin [2, 9, 23]. ABCG2 has been also identified as a protective pump against endogenous and exogenous toxic agents. Oltipraz and tert-butylhydroquinone, which are known to activate Nrf2-dependent gene, up-regulated ABCG2 expression in primary human hepatocytes and human hepatocellular carcinoma cell lines, respectively [24-25]. Because Nrf2 is a stress-inducible transcription factor, which regulates the expression of several cytoprotective genes and drug detoxification enzymes via a common antioxidant response element (ARE) located in the promoter, we decided to investigate whether Nrf2 regulates the expression of ABCG2 as well. A better understanding of the role of Nrf2 in the regulation of ABCG2 expression in cancer cells will help elucidate its role in promoting multidrug resistance phenotype in cancer cells. Here, we show that Nrf2 controls ABCG2 expression at transcriptional level and is required for maintaining of SP in A549 and H460 lung cancer cells as well as prostate cancer cells. Reduced Nrf2 expression results in enhanced sensitivity to mitoxantrone and topotecan in both A549 and H460 lung cancer cells.
Materials and Methods
Cell Culture and Reagents
A549, H460, H23 and Du145 cells were purchased from ATCC and were maintained in DMEM medium with 10 % fetal bovine serum and penicillin/streptomycin. The cell lines were routinely tested for mycoplasma contamination. However, the authors did not attempt to authenticate the cell lines again. Generation of A549 and H460 cells constitutively expressing short hairpin RNA against Nrf2 or control luciferease shRNA cells with stable expression of Nrf2shRNA or control non-targeting luciferase shRNA were maintained in DMEM medium containing 1 μg/ml of puromycin (Roche, Indianapolis, IN). Human airway epithelial cell line, NuLi, derived from normal lung, was grown in a serum-free medium: BEGM (Bronchial Epithelial Growth Medium, Serum-free) from Cambrex (Cambrex, Charles city, IA) made of BEBM basal medium and SingleQuot additives supplemented with 50μg/ml G-418. Details regarding generation of A549-LuchsRNA, A549-Nrf2shRNA, H460-LucshRNA, H460-Nrf2shRNA, Du145-LucshRNA and Du145RNA cells used in this manuscript have been published (21,27). The sequence of keap1 shRNA used to downregulate Keap1 expression was: Keap1shRNA; 5’-CCGGGCCTTAATTCAGCTGAGTGTTCTCGAGAAC ACTCAGCTGAATTAAGGCTTTTTTG-3’ (Sigma-Aldrich, St. Louis, MO). Lentiviral Nrf2 expression vector (PLOHS_100067113) was purchased from Open Biosystems (Huntsville, AL). Mitoxantrone was obtained from Si gma-Aldrich (St. Louis, MO), and topotecan was purchased from LKT laboratories (St. Paul, MN).
Identification of ARE(s) in the Promoter of ABCG2
To identify the presence and location of AREs in the ABCG2 promoter, a 600 bp upstream region sequence from the transcription start site and exon-1 and exon-2 was downloaded from the NCBI database (Human Genome resources). This sequence was screened for ARE binding sites with the help of Genamics Expression 1.1 software using the primary core sequence of ARE (RTGABNNNGCR) [26] as the probe.
Plasmids and Mutagenesis
The 5’ flanking region of human ABCG2 promoter region (-496 bp to +198 bp) was PCR amplified from human genomic DNA using high-fidelity Taq polymerase (Applied Biosystems, Foster City, CA). The primers used for amplification were as follows: forward, CACTTTCTCAGAATCCCATTCAC; Reverse, GAACCTTTTGAGTGGGCACAG. The isolated PCR product was ligated to pCR2.1 vector (Invitrogen, Carlsbad, CA), and a KpnI-XhoI fragment from this construct was cloned into pGL3 basic vector (Promega, Madison, WI). A deletion construct (-310 bp to +198 bp) was generated from the full-length promoter construct. To clone the ARE enhancer sequence in pTAL vector, the ARE binding site with minimal flanking region was amplified using the following primers : forward, 5 ’-AAAAAAGGTACCATCCCATTCACCAGAAACCA; reverse primer, AAAAAACTCGAGCGAACGGAATGAACCAGAGT. Mutant ARE sequences were generated by using a site-directed mutagenesis kit from Stratagene (La Jolla, CA). Primers containing the mutant ARE sequences (GCAGCGCTTGgGcCTGGGCAACCTGTGCGTC) were used for PCR amplification of the mutant ABCG2 ARE binding site in the promoter, and PCR products were digested with DpnI for 1 h to cleave the wild-type promoter template. Sequence of each promoter construct was verified by sequencing.
DNA Transfection and Luciferase Activity
Cells were transfected at 75-85% confluency using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Briefly, cells were seeded in 24-well plates at a density of 2 × 105 cells/mL and grown overnight. After ~12 h, the medium was removed, and transfection complex containing 800 ng of plasmid DNA, 40 ng of pRL-TK plasmid (Promega, Madison, WI) at a ratio of 20:1 and transfection reagent were added to each well in the presence of fetal bovine serum. Cells were incubated for another 36 h, and then were lysed and assayed; Renilla and Firefly luciferase activities were measured using the dual luciferase assay kit (Promega, Madison, WI) with a luminometer (EG&G, Wallac, MD). Firefly luciferase activity was normalized to a Renilla luciferase activity for calculation of relative reporter activity for each construct. Results were plotted from three independent experiments with each assay conducted in triplicate.
Flow Cytometry
Analysis for the SP formation was carried out following the protocol of Goodell’s laboratory with minor modifications [27]. Briefly, cells (1 × 106/ml) were incubated at 37°C for 60 min with 5 μg/ml Hoechst 33342 (Sigma-Aldrich, St. Louis, MO), washed and re-suspended in ice-cold HBSS with 2% FCS and 2 μg/ml propidium iodide (PI) (Sigma-Aldrich, St. Louis, MO). Fumitremorgin C (FTC, 10μM), a potent and specific inhibitor of ABCG2 activity, was used as a positive control for the assay. Side population was analyzed with fluorescence-activated cell sorting (FACS) Vantage (Becton-Dickinson, Franklin Lakes, NJ).
Western Blot
Western blot was carried out using the protocol published by Singh et. al. [17]. Primary antibody incubation was carried out at 4°C for 16 h with a mouse anti-ABCG2 antibody (Sigma-Aldrich, St. Louis, MO) diluted at 1:300 in 3% bovine serum albumin (BSA), followed by incubation with horseradish peroxidase-conjugated horse anti-mouse secondary antibody (GE Healthcare, Piscataway, NJ), and developed using an ECL chemiluminescence detection system (GE Healthcare, Piscataway, NJ). Anti-α-tubulin and anti-GAPDH antibodies were used as loading control in immunoblot assays.
Cell proliferation (MTT) assays
Chemotherapy drug treatments were performed following protocols published by Singh et al [17]. The in vitro drug sensitivity experiments were carried out by using a cell proliferation assay kit (Roche, Indianapolis, IN) according to the manufacturer’s instructions.
Real-time RT-PCR
Real-time RT-PCR reactions were carried out using a protocol published by Singh, et. al. [17]. Briefly, 500ng of total RNA were reverse transcribed using high capacity cDNA synthesis kit from Applied Biosystems (Foster City, CA). An aliquot of diluted cDNA was used to measure human ABCG2 (Hs01053790_m1) and Nrf2 (Hs00232352_m1), NQO1 (Hs00168547_m1) and GCLm (Hs00157694_m1) gene expression using Taqman primer and probe mixes from Applied Biosystems (Foster City, CA). The assays were performed using the ABI 7000 TaqMan system (Applied Biosystems, Foster City, CA). β-Actin (Hs99999903_m1) was used for normalization.
Electrophoretic Mobility Shift Assays (EMSA)
Double-stranded DNA oligonucleotides corresponding to the consensus ARE sequence (5’-GCGCTTGTGACTGGGCAACCTGTGC-3’) in the ABCG2 gene promoter were synthesized commercially from Integrated DNA Technologies (Coralville, IA). The duplex was end-labeled using T4 kinase (Promega, Madison, WI) in the presence of [γ-32P] dCTP (MP Biochemicals, Pasadena, CA). The probe was purified once with NAP-5 column (GE Healthcare, Piscataway, NJ) according to the manufacturer’s instruction. The binding reactions were carried out with 5 μg of nuclear protein isolated from A549-LucshRNA cells or A549-Nrf2shRNA cells and 32P-labeled probe (5 × 105 cpm/reaction) in a 20 μl of reaction mixture containing (5 mM HEPES [pH 7.9], 100 mM NaCl, 0.25 mM EDTA, and 0.25 mM DTT). Poly (deoxyinosinic-deoxycytidylic) acid 50 μg/ml (Roche, Indianapolis, IN) was included in each reaction as nonspecific carrier DNA. The mixture was placed on ice for 30 min. Protein-DNA complexes were separated by electrophoresis on a non-denaturing 5% poly acrylamide gel electrophoresis (PAGE) gel at 200 V with a running buffer consisting of 25 mM Tris, 25 mM borate acid, and 1 mM EDTA. Gels were dried, and the radioactive bands were visualized by autoradiography. For competition reactions, either 25- or 50-fold excess of the unlabeled oligonucleotides harboring the wild type ARE sequence of the ABCG2 gene or corresponding mutant ARE (5’-GCGCTTGGGCCTGGGCAACCTGTGC-3’) was added during the preincubation period. Unlabeled oligonucleotides containing AP1 or NFκB binding sequence were used as non-specific competitors.
Chromatin Immunoprecipitation Assay (ChIP)
ChIP assays were conducted using the ChIP assay kit from Upstate Cell Signaling (Lake Placid, NY) according to the manufacturer’s instructions. Cells (~107) were harvested and chromatin was cross-linked by adding formaldehyde to the cell culture medium to a final concentration of 1% and incubating the mixture for 10 min at 37°C. Cells were washed twice with ice-cold PBS containing a protease inhibitor cocktail (Roche) and were suspended in 0.2 ml of lysis buffer (50 mM Tris-HCl pH 8.1, with 1% SDS and 5 mM EDTA). Samples were sonicated on ice to an average length of 500-1000 bp (4 pulses of 10 secs each) and were centrifuged at 10,000 × g. Solubilized chromatin was diluted 10-fold with CHIP dilution buffer (16.7 mM Tris-HCl, pH 8.1, with 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, and 167 mM NaCl) and was used for CHIP assays. An aliquot of the soluble fraction was saved for total chromatin input. Chromatin was precleared with salmon sperm DNA/protein A-agarose for 30 min and then incubated with either antibody to (Nrf2 sc-722X; Santa Cruz Biotechnology), and rabbit IgG for 18 h at 4°C with rotation. Immunoprecipitation, washing, and elution were carried out according to the manufacturer’s instructions. Cross-linked immunoprecipitates and total chromatin input were reverse cross-linked and samples were treated with proteinase-K (Sigma) and extracted with phenol-chloroform-isoamyl alcohol. DNA was precipitated and resuspended in 100 μl of TE (10 mM Tris-Cl and 0.1 mM EDTA). Then, 1 μl of DNA was used for PCR (35 cycles) with primers specific for the ABCG2 promoter. For PCR amplification, forward primer: 5’-ATCCCATTCACCAGAAACCA-3’ and reverse primer: 5’-ACTCTGGTTCATTCCGTTCG-3’ from Invitrogen (Carlsbad, CA) were used.
Statistical Analysis
Data are presented as the mean ± SD. To assess statistical significance of differences, Student’s t-test or one way ANOVA were conducted. P-values <0.05 were considered statistically significant as indicated by asterisks. IC50’s in drug-sensitization studies were obtained by using GraphPad Software (La Jolla, CA).
Results
Attenuation of Nrf2 expression in lung cancer cells leads to a decline in ABCG2 mRNA and protein level
To study the Nrf2-dependent regulation of ABCG2 expression, we selected A549 and H460 lung cancer cells as a model system, since they harbor KEAP1 gene mutation that leads to high basal level of Nrf2 pathway activity and are drug resistant [17, 22]. We have established and characterized A549 and H460 cells constitutively expressing short hairpin RNA targeting Nrf2 [17, 22]. Short-hairpin targeting luciferase shRNA expressing cells were used as non-targeting control. A549 and H460 cells stably expressing Nrf2 shRNA (A549 Nrf2shRNA, H460 Nrf2shRNA) showed significant reduction in Nrf2 mRNA expression and a parallel decrease in ABCG2 transcript levels as compared with cells expressing luciferase shRNA (A549 LucshRNA and H460 LucshRNA) (Fig. 1A). Immunoblot analysis revealed diminished levels of ABCG2 protein in Nrf2 knock-down A549 and H460 cells compared to control cells (Fig. 1B). The expression of Nrf2 and ABCG2 did not change significantly between the control cells transfected with luciferase shRNA and the untransfected parent cancer cells (Supplementary Fig. S1).
Figure 1. Nrf2-dependent ABCG2 expression in lung cancer cells.
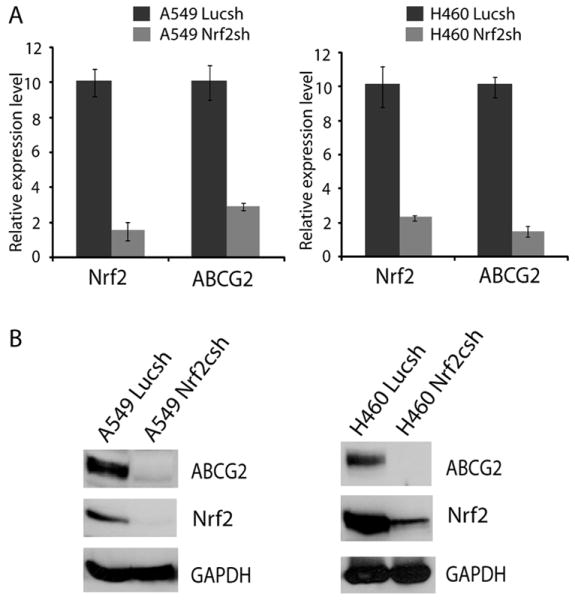
(A) Relative expression of ABCG2 in A549 and H460 cells constitutively expressing Nrf2shRNA analyzed by real-time RT-PCR. β-actin was used for normalization. Data are presented as fold change calculated using gene expression levels in control luciferase shRNA cells as baseline. (B) Immunoblot analysis of ABCG2, Nrf2, and GAPDH expression in A549 and H460 cells expressing control luciferase shRNA and Nrf2 shRNA.
Nrf2 directly regulates transcriptional expression of ABCG2
To investigate whether ABCG2 is a direct transcriptional target of Nrf2, we utilized promoter reporter assays to study ABCG2 promoter regulation in Nrf2-proficient (A549 control cells or parental A549 cells) and Nrf2-depleted (A549 Nrf2shRNA) lung cancer cells. Two ABCG2 promoter deletion constructs were made according to the in silico analysis of ABCG2 promoter which identified a putative ARE located at -431 bp to -420 bp upstream of the ABCG2 transcription start site (TSS). The full length reporter construct contained the putative ARE (-496 bp to +198 bp) whereas truncated ARE did not (-310 bp to +198 bp) (Fig. 2A, schematic). The two reporter constructs were transfected into A549 control cells and A549 Nrf2shRNA cells, and luciferase reporter activity was measured. As shown in Fig. 2A, promoter activity of the ABCG2 full-length construct (-496 bp to +198 bp) was significantly reduced in Nrf2-depleted A549 Nrf2shRNA cells as compared with Nrf2-proficient A549 control cells. The shorter promoter deletion construct lacking the putative ARE (-310 bp to +198 bp) exhibited similar activity in both Nrf2-depleted and control cells. To further confirm that Nrf2-dependent luciferase reporter activity was suppressed in A549Nrf2shRNA cells as compared with control cells, a Nrf2-dependent NQO1 basal promoter reporter construct [28] was included in the promoter reporter studies. The NQO1 reporter plasmid showed significantly reduced luciferase activity in A549 Nrf2 shRNA cells compared with control A549 cells (Fig. 2A). These data strongly suggest that the ARE binding site in the ABCG2 promoter region is active and functions as a transcriptional enhancer. To further confirm that the ARE sequence in ABCG2 promoter functions as an enhancer, we cloned the ABCG2 promoter ARE sequence with sufficient flanking sequence into a pTAL-Luc reporter vector containing a minimal promoter to make a ABCG2 ARE construct, and cloned ABCG2 promoter fragment with mutant ARE into a pTAL-Luc construct to make a mutant ARE construct (Fig. 2B schematic). Reporter assays results showed that mutation of the ARE core sequence significantly inhibited the Nrf2-dependent luciferase reporter activity in A549 cells (Fig. 2B). These observations suggest that the ARE element located at -431 bp to -420 bp is involved in Nrf2-dependent regulation of the ABCG2.
Figure 2. Nrf2 regulates ABCG2 transcription through an ARE element in the ABCG2 promoter.

(A) A putative ARE at -431 bp to -420 bp in the proximal promoter region of the human ABCG2 gene was identified by in silico analysis and promoter reporter assay. Left, schematic representation of the two constructs with and without putative ARE. The 5′ end of each of the constructs relative to the transcription start site (arrows) is indicated. These constructs were transfected into A549 control cells and A549 Nrf2shRNA cells, and the luciferase activity was measured. Luciferase activities were normalized to the Renilla luciferase activity of a co-transfected reporter vector. NQO1 basal promoter construct was included as a positive control for measuring Nrf2-dependent reporter expression. (B) ABCG2 promoter fragment containing the ARE core sequence was cloned upstream of heterologous promoter driving pTAL vector. Mutations in ARE core sequence (sequence shown on top) were introduced by site-directed mutagenesis. These constructs were transfected into A549 cells, and the luciferase activity was measured. ‘*’, significant when compared to wild type ARE. *, p-value< 0.05, analyzed by Student’s t test. (C) In vitro DNA binding activity of Nrf2 to ABCG2 ARE using EMSA assays. Nuclear proteins from Nrf2-depleted cells (lane 2) and control A549 cells (lane 3) were incubated with 32P-labeled oligonucleotides harboring the ARE consensus sequence of ABCG2 gene. The resulting complexes were resolved by non-denaturing PAGE and analyzed. For competition assays, a 25 and 50 -fold excess of unlabeled oligonucleotides harboring the wild type ARE (lanes 4 and 5) of the ABCG2 gene was added during the pre-incubation period. A 25–50-fold excess of unlabeled oligonucleotides with mutated ARE sequence was added (lanes 6 and 7). Nonspecific unlabeled oligonucleotides that contain AP1 and NFκB binding sequences were used as a negative control (lanes 8-11). Nuclear extract from A549 control cells were used in both specific and non-specific competitions (lanes 4 to 11). The arrow indicates ARE binding complex; FP, free probe. (D) ChIP assays were performed with A549 cells stably expressing Nrf2shRNA and the control cells expressing luciferase shRNA.
To further verify that Nrf2 binds to the ARE binding site in ABCG2 promoter in vitro and in vivo, we performed EMSA. As shown in Fig. 2C, there was a marked Nrf2-dependent complex formation when 32P-labeled ABCG2 ARE probes were incubated with nuclear extract from control A549-LucshRNA cells (doublet arrows). In contrast, the Nrf2-dependent complex formation was reduced when the 32P-labeled ARE probes were incubated with nuclear extract from Nrf2-deficient A549 cells (Fig. 2C, compare lane 3 with lane 2). The specificity of Nrf2-dependent complex formation was tested using 25X and 50X excess of unlabeled ABCG2-ARE oligonucleotides as cold competitor. The competition results demonstrated that the Nrf2-dependent complex formation was completely abolished by cold wild type ARE oligonucleotides (Fig. 2C, lanes 4 and 5), but was competed to a less extent by mutant ARE oligonucleotides (Fig. 2C, lanes 6 and 7), and was poorly competed by non-specific AP1, or NFκB oligonucleotides (Fig. 2C, lanes 8 to 11). Next, we conducted ChIP assays to examine whether Nrf2 constitutively binds to the endogenous ABCG2 promoter in cancer cells. As shown in Fig. 2D, constitutive recruitment of Nrf2 to the ARE element located in ABCG2 promoter was observed in Nrf2-proficient A549 LucshRNA control cells. As expected, in A549 Nrf2shRNA cells, Nrf2 was present at low levels and reduced amplification was detected using ABCG2 promoter primers. Immunoprecipitations with control IgG failed to enrich the ARE containing ABCG2 promoter region (Fig. 2D), suggesting that the sites were not enriched in nonspecific fashion. Overall, these results indicate that Nrf2 specifically binds to the ARE element in ABCG2 promoter. In conclusion, these promoter reporter studies, in vitro and in vivo binding assays revealed that Nrf2 regulates ABCG2 promoter activity through ARE element located at -431 bp to -420 bp from the transcription start site.
Decreasing Nrf2-dependent ABCG2 expression by shRNA results in loss of SP phenotype
ABCG2 activity is required for maintaining SP phenotype in cancer cells. To investigate whether reduced ABCG2 expression affects the SP formation in Nrf2-deficient A549 and H460 cells versus control cells, Hoechst dye exclusion assays were conducted. The results show that there are about 58% cells that belong to SP in control A549-LucshRNA cells; on the contrary, the percentage of SP drops to less than 2% in Nrf2-knockdown A549 cells. Similarly, SP content in Nrf2-knockdown H460 cells are ~0.18% compared to about 1. 3% in control H460-LucshRNA cells (Figure. 3). To confirm the specificity of the Hoechst dye efflux assay, we used Fumitremorgin C (FTC), a specific inhibitor of ABCG2. The cells were incubated with Hoechst dye in the presence of FTC followed by PI staining and flow cytometric analysis. Side population phenotype was completely blocked by FTC treatment in both A549 and H460 cells (Figure. 3 and Supplementary Figure. 2). To determine if antibiotic selection during generation of stable cell lines altered the percentage of cells in the side population, we compared the side population in A549-parent cells, A549-LucshRNA and A549-Nrf2shRNA cells (Supplementary Figure S3A). We detected about ~1.75-2 fold higher numbers of cells in the side population of A549-LucshRNA relative to A549-parent cells. Similar results were obtained with H460 parent, H460-LucshRNA and H460-Nrf2shRNA cells (Supplementary Figure S3B). However, Nrf2 depleted cells A549 and H460 had dramatically smaller fraction of cells in Side population.
Figure 3. Alteration of SP phenotype in Nrf2-depleted lung cancer cells.
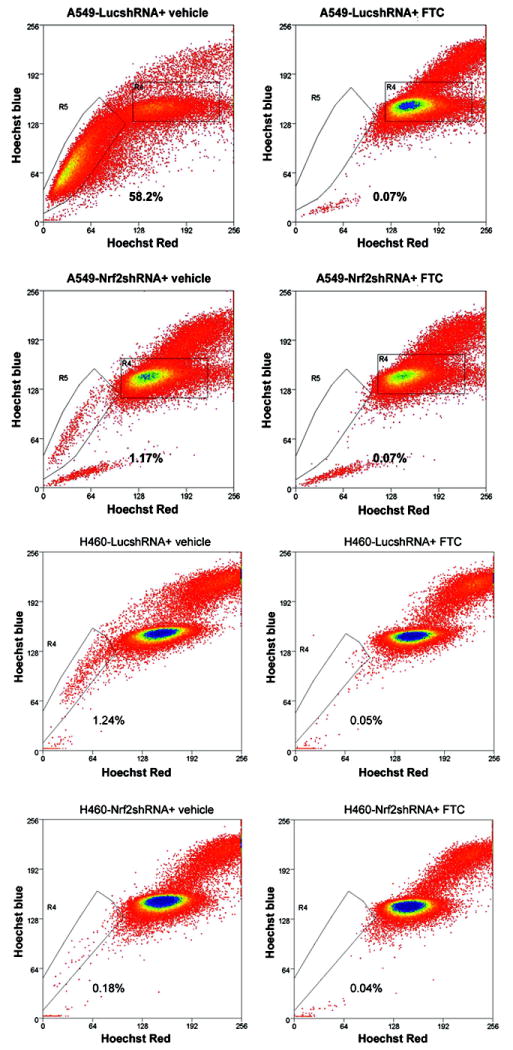
A549 and H460 cells were incubated with Hoechst 33342 dye and PI and were analyzed by flow cytometry. To demonstrate the specificity of assay, cells were incubated with Hoechst dye in the presence of Fumitremorgin C (FTC). The SP is shown as a percentage of the whole viable cell population. Reactions were done in triplicates and were repeated three times.
Inhibiting Nrf2-dependent ABCG2 expression by shRNA causes chemo-sensitization to its target anticancer drugs
ABCG2 is crucial for detoxification of several chemotherapeutic-drugs; thus, its increased expression confers chemo-resistance. It has been reported that efflux of mitoxantrone and topotecan depends on ABCG2 [9, 23]. To determine whether decreased expression of ABCG2 could sensitize A549 and H460 cells to mitoxantrone and topotecan- induced cytotoxicity, we incubated both Nrf2-depleted and control A549 and H460 cells with mitoxantrone and topotecan for 2–4 days and measured the percentage of viable cells using a MTT assay. As expected, the Nrf2-deficient A549 and H460 cells (A549 Nrf2shRNA and H460 Nrf2shRNA), with low endogenous levels of ABCG2, displayed enhanced sensitivity to mitoxantrone and topotecan in vitro (Figure. 4). The IC50 of Mitoxantrone in A549 LucshRNA and A549-Nrf2shRNA cells was 31.38 nM and 23.09 nM respectively; IC50 of Mitoxantrone in H460-LucshRNA cells and H460-Nrf2shRNA cells was 22 nM and 6 nM respectively. Similar to Mitoxantrone, Nrf2 depleted A549 and H460 cells demonstrated lower IC50 values for Topotecan (A549-LucshRNA-35.68 nM; A549-Nrf2shRNA- 11.07 nM; H460-LucshRNA-78.8 nM and H460-Nrf2shRNA-30.1nM).
Figure 4. Reversal of chemo-resistance in Nrf2-depleted lung cancer cells.
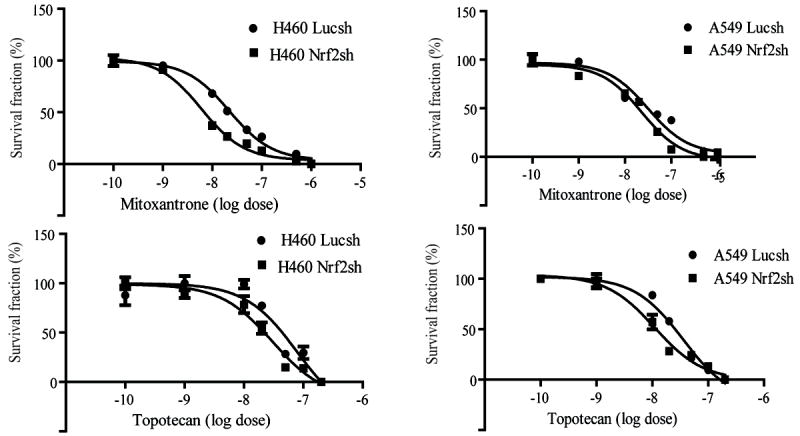
A549 cells and H460 cells were treated with mitoxantrone and topotecan for 2-4 days, and cell viability was analyzed by MTT assays. Results shown are median values obtained from three independent experiments with 8 samples for each dose. Results were analyzed by One-way ANOVA analysis, ‘*’, p-value<0.001 in all panels.
Nrf2 dependent ABCG2 expression in Prostate cancer cells
We have established and characterized Du145 cells constitutively expressing shRNA targeting Nrf2 transcript (Du145-Nrf2shRNA) and the control DU-145 cells expressing shRNA against luciferase gene (Du145-LucshRNA) [21]. Du145 cells stably expressing Nrf2shRNA (Du145-Nrf2shRNA) showed >85% reduction in Nrf2 mRNA expression and a parallel decrease in ABCG2 transcript levels as compared with cells expressing luciferase shRNA (Du145-LucshRNA) (Fig. 5A). Immunoblot analysis revealed diminished levels of Nrf2 and ABCG2 protein in Nrf2 knock-down Du145 cells compared to parent Du145 cells and Du145-LucshRNA cells (Fig. 5B-C). The expression of Nrf2 and ABCG2 in Du-145-LucshRNA cells was similar to that of the parent Du-145 cells. To determine whether Nrf2 dependent ABCG2 expression in prostate cancer cells affects the SP formation we used Hoechst dye exclusion assay. The immunoblotting data presented above shows that Du154 cells express low levels of ABCG2 protein. Analysis of side population in Du145 parent cells, Du145-LucshRNA and Du145-Nrf2shRNA cells revealed very low SP fraction in Du145 cells. We detected no cells in the SP fraction of Du145-Nrf2shRNA cells (Supplementary Figure S4). Thus, similar to Nrf2 depleted lung cancer cells, Du-145 Nrf2shRNA cells showed reduced ABCG2 mRNA, protein expression and reduced side population as compared to Nrf2 proficient cells. Collectively, these results suggest that transcriptional regulation of ABCG2 in lung and prostate cancer cells is mediated by Nrf2.
Figure 5. Attenuated expression and activity of ABCG2 in Nrf2 deficient prostate cancer cells.

(A) Relative expression of ABCG2 and Nrf2 in Du145 parent cells, Du145-LucshRNA and Du145-Nrf2shRNA cells analyzed by real-time RT-PCR. Data are presented as fold change calculated using gene expression levels in control luciferase shRNA cells as 1. (B) Immunoblot analysis of Nrf2 and ABCG2, expression in Du145 cells. (C) Densitometric quantization of Nrf2 and ABCG2 relative protein expression in Du145 parent cells, Du145-LucshRNA and Du145-Nrf2shRNA cells (arbitrary units [A.U.]).
Nrf2 Activation Induces ABCG2 Expression in Lung Epithelial Cells
Next we examined whether activation of Nrf2 induces ABCG2 expression in non-tumorigenic airway epithelial cells i.e, NuLi cells. We used a short hairpin RNA targeting Keap1 to inhibit Keap1 expression and activate Nrf2 dependent gene expression. Luciferase shRNA was used as non- targeting control shRNA. As shown in Figure 6A, reduction in Keap1 transcript levels by Keap1 shRNA resulted in a parallel increase in NQO1, GCLm and ABCG2 transcript levels. We observed an inverse correlation between Keap1 expression and Nrf2 dependent ABCG2 expression in lung epithelial cells.
Figure 6. Nrf2 activation induces ABCG2 expression in non-tumorigenic lung epithelial cells as well as tumorigenic lung epithelial cells.

(A) Up-regulation of ABCG2 expression in cells expressing Keap1 shRNA. Total RNA from NuLi cells expressing Keap1 shRNA and Luciferase shRNA was isolated and expression of Keap1 and ABCG2 was determined by real time RT-PCR. Downregulation of Keap1 expression led to an increase in Nrf2 dependent NQO1, GCLM and ABCG2 expression. ‘*’, p-value<0.05, significant when compared with LucshRNA group.
(B) Induction of ABCG2 expression in response to treatment with multiple oxidative stress inducing agents. NuLi cells were treated with TBHQ (20nM), and CS condensate (100 μg/ml) for 24h and expression of NQO1, GCLM and ABCG2 were measured by real-time RT-PCR. ‘*’, p-value<0.05, significant when compared with vehicle-treated sample.
(C) Ecotopic expression of Nrf2 in lung cancer cells upregulates Nrf2 dependent ABCG2 expression. Overexpression of Nrf2 cDNA in H23 lung cancer cells resulted in activation of Nrf2 target gene expression including ABCG2. ‘*’, p-value<0.05, significant when compared with empty vector group.
(D) Enforced expression of Nrf2 cDNA in H23 cells increases the proportion of SP cells.
To demonstrate that commonly used oxidative stress inducing agents like cigarette smoke condensate (CSC) and tert-butyl hydroquinone (tBHQ), known to upregulate Nrf2 dependent signaling, also upregulate ABCG2 expression, we treated NuLi cells with these agents and measured gene expression. The cells were exposed to cigarette smoke condensate (100μg/ml) and tBHQ (20μM) for 24h and induction of ABCG2 in response to these treatments was quantified by real-time RT-PCR. Pretreatment with Nrf2 activating agents up-regulated the expression of ABCG2 and other classical Nrf2 target genes suggesting that Nrf2 mediates inducible expression of ABCG2 (Figure 6B).
Alternatively, we overexpressed Nrf2 cDNA in a lung adenocarcinoma cell line, H23, harboring wild type Keap1 and Nrf2 activity. H23 cells were transduced with lentiviral particles containing Nrf2 cDNA or the control empty vector. Four to Six days post transduction, cells were harvested and expression of Nrf2 and its target genes including ABCG2 was measured by real-time RT-PCR and immunoblotting. As expected, Nrf2 overexpression resulted in upregulation of ABCG2 expression at mRNA and protein level (Figure 6C). Analysis of side population in H23 cells revealed that cells overexpressing Nrf2 (H23-Nrf2 cDNA) had a 2-fold higher SP fraction as compared to H23 empty vector control cells (Figure 6D). In summary, we have convincingly demonstrated that Nrf2 regulates ABCG2 gene expression and SP fraction in cancer cells.
Discussion
In the present study, we demonstrate that Nrf2 regulates the transcriptional expression of human ABCG2 gene through an ARE element located at -431 bp to -420 bp region of the promoter, and Nrf2 is essential for maintaining ABCG2 expression and function in lung cancer and prostate cancer cells. Nrf2 depletion resulted in dramatic reduction in SP formation and reversal of chemo-resistance in non–small-cell lung cancer cell lines. Similarly, upregulation of Nrf2 activity induced the expression of ABCG2 and increased the SP fraction in lung epithelial cells.
One of the defining characteristics of cancer stem cells is their ability to transport Hoechst dye leading to SP phenotype, which is attributable to the expression of ABCG2 gene [3, 6]. A study by Scharenberg et al showed that A549 cells contain an increased fraction of SP positive cells, and high Hoechst dye efflux capacity of A549 cells correlates very strongly with ABCG2 activity [11]. Transcriptome analyses of embryonic, hematopoietic, and neural stem cells revealed a common signature of gene expression, which includes transcripts that function as cytoprotective factor against environmental and xenobiotic stress [29]. Studies showed that stem cells express high levels of drug detoxification enzymes such as P-glycoprotein, BCRP (ABCG2), glutathione S transferases, glutathione peroxidase [6, 30-31]. Interestingly, several of these cytoprotective and drug detoxification genes are classical Nrf2-dependent genes.
Nrf2, a bZIP transcription factor, protects cells from oxidative stress and xenobiotics by induction of a transcriptional program that includes major antioxidants, such as enzymes in the glutathione and thioredoxin pathways, as well as xenobiotic detoxification enzymes that include the glutathione S-transferase and UDP glycosyltransferase family, aldehyde dehydrogenase, and various drug efflux pumps-members of multidrug resistance protein family [32-34]. KEAP1 negatively regulates Nrf2 activity by targeting it for proteasomal degradation [15-16]. Nrf2-KEAP1 interactions are frequently dysfunctional in NSCLC leading to constitutive activation of Nrf2 [17]. Our previous studies have shown that point mutations in the KEAP1 gene leading to non-conservative amino acid substitutions and nonsense mutations resulting in loss of KEAP1 function in a high percentage of the NSCLC cell lines and primary tumors [17]. Recently, two Nrf2 mutation ’hot-spots’ were identified in approximately 10% of patients with squamous lung carcinoma, enabling the transcription factor to evade KEAP1-mediated repression [18, 35]. Furthermore, mutations in KEAP1 have been reported in breast cancer, gall bladder cancer and prostate cancer [17, 36-37]. High Nrf2 activity in cancer cells results in up-regulation of Phase II drug detoxification enzymes and several ATP-dependent multidrug resistant drug efflux pumps, e.g., ABCC1 and ABCC2 and promotes drug resistance [12, 38-42]. ABCG2 transcript and protein levels were dramatically reduced in Nrf2-depleted lung cancer cells suggesting that ABCG2 gene expression is Nrf2-dependent in lung cancer cells. Attenuated ABCG2 promoter reporter activity in the absence of Nrf2 activity implies that Nrf2 regulates ABCG2 transcription by modulating ABCG2 promoter activity. Loss of Nrf2 expression and a corresponding decrease in ABCG2 expression reduced the SP formation in lung cancer cells. Similar Nrf2 dependent ABCG2 expression was observed in Du145 cells, a prostate cancer cell line.
Recently, it has been reported that the expression of ABCG2 along with some other efflux pumps was decreased in hepatocytes of Nrf2 null mice compared to that of wild type mice, which indicates that Nrf2 is involved in physiological regulation of ABCG2 [43]. Tertiary butylhydroquinone, an agent known to activate Nrf2 by inducing oxidative stress, up-regulated the expression of ABCG2 in hepatocellular carcinoma cells [25]. In another study, Jigorel et al reported that Oltipraz, a small molecule activator of Nrf2, up-regulated the expression of ABCG2 in primary human hepatocytes [24]. Studies focused on regulation of the expression of ABCG2 revealed that estrogen receptor (ER)-, hypoxia-, and PPARγ-inducible cis-elements are located at 5’ promoter region of ABCG2 gene in normal SP positive cells [44-46].
Decreasing the expression of redox master regulator, Nrf2, potentially affects cellular protection against xenobiotics and drugs [17]. The attenuation in ABCG2 expression in Nrf2-deficient lung cancer cells leads to chemo-sensitization [22]. Cancer cells with reduced Nrf2 activity and a parallel decline in ABCG2 expression displayed enhanced sensitivity to mitoxantrone and topotecan. However, besides reduced ABCG2 levels, alternate mechanisms such as decrease in glutathione level, or/and reduced drug detoxification may also be involved in mitoxantrone and topotecan sensitization of Nrf2-abrogated lung cancer cells. We recently reported that Nrf2 inhibition in lung cancer cells results in enhanced intracellular accumulation carboplatin and etoposide, and thus, enhanced carboplatin and etoposide induced cell death [9, 22]. Interestingly, no significant difference in Nrf2-dependent sensitivity to another drug, methotrexate, was observed (data not shown). These findings imply that reversal of drug resistance by mitigating Nrf2-dependent drug detoxification genes is specific and it relies on different Nrf2 downstream executors in the context of different chemotherapeutic agents. One seemingly obvious advantage of targeting Nrf2 molecule, to increase sensitivity to chemotherapeutic drugs under different scenarios, is that electrophile drug detoxification pathways involved in detoxification of a wide spectrum of chemotherapy drugs are tackled at the same time, thus reducing the possibility of alternative or redundant detoxification pathway activation in targeting only one or few detoxification and/or drug pump pathways/molecules alone.
In conclusion, the loss of KEAP1 function and aberrant activation of Nrf2-dependent pathways are frequently detected in lung cancer and other solid tumors [17, 36-37]. Up-regulated Nrf2 activity in cancer cells/tissues potentially stimulates ABCG2 expression and promotes multidrug resistance phenotype. Because ABCG2 along with other cytoprotective genes are up-regulated in cancer stem cells and protect against stress, it remains to be determined whether Nrf2 and its downstream signaling is amplified in cancer stem cells and whether targeting Nrf2 activity in cancer stem cell can be effective strategy for overcoming multidrug resistance.
Supplementary Material
Acknowledgments
We thank Dr. Joseph B. Margolick and Mr. Hao Zhang at Department of Molecular Microbiology and Immunology, Bloomberg School of Public Health, Johns Hopkins University, for assistance with side population analysis.
Financial Support: This work was supported by NIH grants P50 CA058184, RO1 CA140492, P30ES03819, developmental grant from prostate spore P50 CA58236 (SB) and Flight Attendant Medical Research Institute (SB and AS).
Footnotes
Conflict of Interest: None
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Wulf GG, Wang RY, Kuehnle I, et al. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood. 2001;98:1166–73. doi: 10.1182/blood.v98.4.1166. [DOI] [PubMed] [Google Scholar]
- 3.Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 5.Dylla SJ, Beviglia L, Park IK, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou S, Schuetz JD, Bunting KD, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–34. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 7.Feuring-Buske M, Hogge DE. Hoechst 33342 efflux identifies a subpopulation of cytogenetically normal CD34(+)CD38(-) progenitor cells from patients with acute myeloid leukemia. Blood. 2001;97:3882–9. doi: 10.1182/blood.v97.12.3882. [DOI] [PubMed] [Google Scholar]
- 8.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A. 2004;101:781–6. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sung JM, Cho HJ, Yi H, et al. Characterization of a stem cell population in lung cancer A549 cells. Biochem Biophys Res Commun. 2008;371:163–7. doi: 10.1016/j.bbrc.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 11.Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99:507–12. doi: 10.1182/blood.v99.2.507. [DOI] [PubMed] [Google Scholar]
- 12.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 13.Rangasamy T, Cho CY, Thimmulappa RK, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–59. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morito N, Yoh K, Itoh K, et al. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene. 2003;22:9275–81. doi: 10.1038/sj.onc.1207024. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibata T, Ohta T, Tong KI, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–73. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Padmanabhan B, Tong KI, Ohta T, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 20.Ohta T, Iijima K, Miyamoto M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–9. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 21.Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, Wu Hailong, Bova S, Biswal S. Loss of Keap1 Function in Prostate Cancer Cells Causes Chemoresistance and Radioresistance and Promotes Tumor Growth. Molecular Cancer Therapeutics. 2010 doi: 10.1158/1535-7163.MCT-09-0589. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh A, Boldin-Adamsky S, Thimmulappa RK, et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008;68:7975–84. doi: 10.1158/0008-5472.CAN-08-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maliepaard M, van Gastelen MA, de Jong LA, et al. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999;59:4559–63. [PubMed] [Google Scholar]
- 24.Jigorel E, Le Vee M, Boursier-Neyret C, Parmentier Y, Fardel O. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab Dispos. 2006;34:1756–63. doi: 10.1124/dmd.106.010033. [DOI] [PubMed] [Google Scholar]
- 25.Adachi T, Nakagawa H, Chung I, et al. Nrf2-dependent and -independent induction of ABC transporters ABCC1, ABCC2, and ABCG2 in HepG2 cells under oxidative stress. J Exp Ther Oncol. 2007;6:335–48. [PubMed] [Google Scholar]
- 26.Baker AF, Landowski T, Dorr R, et al. The antitumor agent imexon activates antioxidant gene expression: evidence for an oxidative stress response. Clin Cancer Res. 2007;13:3388–94. doi: 10.1158/1078-0432.CCR-06-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harvey CJ, Thimmulappa RK, Singh A, et al. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol Med. 2009;46:443–53. doi: 10.1016/j.freeradbiomed.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanova NB, Dimos JT, Schaniel C, et al. A stem cell molecular signature. Science. 2002;298:601–4. doi: 10.1126/science.1073823. [DOI] [PubMed] [Google Scholar]
- 30.Chaudhary PM, Roninson IB. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell. 1991;66:85–94. doi: 10.1016/0092-8674(91)90141-k. [DOI] [PubMed] [Google Scholar]
- 31.Madhavan L, Ourednik V, Ourednik J. Increased “vigilance” of antioxidant mechanisms in neural stem cells potentiates their capability to resist oxidative stress. Stem Cells. 2006;24:2110–9. doi: 10.1634/stemcells.2006-0018. [DOI] [PubMed] [Google Scholar]
- 32.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 33.Ramos-Gomez M, Kwak MK, Dolan PM, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–5. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwak MK, Kensler TW, Casero RA., Jr Induction of phase 2 enzymes by serum oxidized polyamines through activation of Nrf2: effect of the polyamine metabolite acrolein. Biochem Biophys Res Commun. 2003;305:662–70. doi: 10.1016/s0006-291x(03)00834-9. [DOI] [PubMed] [Google Scholar]
- 35.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–88. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 37.Shibata T, Kokubu A, Gotoh M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–68. 68, e1–4. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 38.Hayashi A, Suzuki H, Itoh K, Yamamoto M, Sugiyama Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem Biophys Res Commun. 2003;310:824–9. doi: 10.1016/j.bbrc.2003.09.086. [DOI] [PubMed] [Google Scholar]
- 39.Kim YJ, Ahn JY, Liang P, et al. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 2007;67:546–54. doi: 10.1158/0008-5472.CAN-06-2401. [DOI] [PubMed] [Google Scholar]
- 40.Lee TD, Yang H, Whang J, Lu SC. Cloning and characterization of the human glutathione synthetase 5’-flanking region. Biochem J. 2005;390:521–8. doi: 10.1042/BJ20050439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–60. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 42.Vollrath V, Wielandt AM, Iruretagoyena M, Chianale J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem J. 2006;395:599–609. doi: 10.1042/BJ20051518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reisman SA, Yeager RL, Yamamoto M, Klaassen CD. Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol Sci. 2009;108:35–47. doi: 10.1093/toxsci/kfn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ee PL, Kamalakaran S, Tonetti D, et al. Identification of a novel estrogen response element in the breast cancer resistance protein (ABCG2) gene. Cancer Res. 2004;64:1247–51. doi: 10.1158/0008-5472.can-03-3583. [DOI] [PubMed] [Google Scholar]
- 45.Krishnamurthy P, Ross DD, Nakanishi T, et al. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24218–25. doi: 10.1074/jbc.M313599200. [DOI] [PubMed] [Google Scholar]
- 46.Szatmari I, Vamosi G, Brazda P, et al. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 2006;281:23812–23. doi: 10.1074/jbc.M604890200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.