

Abstract
The pathway of product release from the R state of aspartate transcarbamoylase has been determined here by solving the crystal structure of Escherichia coli aspartate transcarbamoylase (ATCase) locked in the R-quaternary structure by specific introduction of disulfide bonds. ATCase displays ordered substrate binding and product release, remaining in the R state until substrates are exhausted. The structure reported here represents ATCase in the R state bound to the final product molecule, phosphate. This structure has been difficult to obtain previously because the enzyme relaxes back to the T state after the substrates are exhausted. Hence cocrystallizing the wild-type enzyme with phosphate results in a T-state structure. In this structure of the enzyme trapped in the R state with specific disulfide bonds, we observe two phosphate molecules per active site. The position of the first phosphate corresponds to the position of the phosphate of carbamoyl phosphate and the position of the phosphonate of N-phosphonacetyl-L-aspartate. However, the second, more weakly bound phosphate, is bound in a positively charged pocket more accessible to the surface than the other phosphate. The second phosphate appears to be on the path that phosphate would have to take to exit the active site. Our results suggest that phosphate dissociation and carbamoyl phosphate binding can occur simultaneously and the dissociation of phosphate may actually promote the binding of carbamoyl phosphate for more efficient catalysis.
Keywords: protein structure-function, X-ray crystallography, allosteric transition, cooperativity, enzyme mechanism
Introduction
Escherichia coli aspartate transcarbamoylase (ATCase) is an allosteric enzyme that catalyzes the committed step of pyrimidine nucleotide biosynthesis, the carbamoylation of l-aspartate (Asp) by carbamoyl phosphate (CP) to form n-carbamoyl-l-aspartate (CA) and phosphate (Pi).1 ATCase activity is exquisitely regulated through homotropic cooperativity for Asp as well as heterotropic regulation by the allosteric effectors ATP, CTP1, and UTP in the presence of CTP.2 The quaternary structure is composed of six catalytic chains arranged as two trimeric subunits and six regulatory chains arranged as three dimeric subunits.3,4 The upper catalytic trimer consists of chains C1, C2, and C3 while the lower catalytic trimer consists of chains C4, C5, and C6 positioned directly below C1, C2, and C3 respectively. The three regulatory dimers consist of chains R1/R6, R2/R4, and R3/R5. Each regulatory chain interacts with the corresponding-numbered catalytic chain.
ATCase exists in two different structural and functional states; the low-activity, low-affinity T state, and the high-activity, high-affinity R state.5-7 The conversion of the enzyme from the T to R state occurs upon Asp binding to the holoenzyme in the presence of CP. The allosteric transition involves an 11 Å expansion and 10° rotation of the molecule along its three-fold axis as well as a 15° rotation of the regulatory dimers relative to their respective two-fold axis.8 In additon to the quaternary conformational change, the allosteric transition also involves several tertiary changes such as the reorganization of the 80s and 240's loops of the catalytic chains. Specific interchain interactions of the side chains of the 240's loop have been identified as important contributors to stabilizing the R state.9
Small-angle X-ray scattering experiments have shown that upon Asp binding to the ATCase·CP complex, the enzyme undergoes the allosteric transition from the T state to the R state. ATCase remains in the R state until all substrates are exhausted before transitioning back to the T state.10,11 These results suggest that, in the presence of additional substrates, the enzyme releases the products CA and Pi without reverting back to the T state. Therefore, in order to observe the enzyme·product complex in the active form, it requires a crystal structure of the R state bound to products, but crystallization of the wild-type enzyme with product molecules or analogs yields T-state structures.12 Stabilization of the R-state quaternary state of the holoenzyme has been made possible by means of specifically introduced interchain disulfide bonds that hold the molecule in its expanded form.13 Specifically, an Ala to Cys mutation at position 241 in the 240's loop, as well as a Cys to Ala mutation at position 47, provide the possibility to selectively form disulfide bonds between Cys241 in each upper catalytic chain to its neighbor in the lower catalytic chain, thereby locking the 240's loop into the position characteristic for the R quaternary structure.13 When the disulfide bonds are intact, the C47A/A241C holoenzyme does not exhibit cooperativity for Asp, is not influenced by the allosteric effectors, and is not activated by PALA. When the disulfide linkages are reduced, the enzyme displays wild-type activity and is activated by ATP and inhibited by CTP.13 Utilizing the disulfide linked C47A/A241C holoenzyme, a structure of the enzyme in the R state with the final product phosphate bound was obtained, which corresponds to the R-state structure of the enzyme after the dissociation of product carbamoyl aspartate from the active site.
Results and Discussion
The allosteric transition of ATCase
Over the past few years work in this laboratory has focused on obtaining structural snapshots of ATCase at each step in the chemical mechanism and the allosteric transition leading to product formation. Utilizing substrates, products, or structural analogs, significant progress has been made towards this goal. The structures of the steps leading to the allosteric transition include the T-state enzyme in the absence of ligands (1ZA1),14 and the T-state enzyme in the presence of the first substrate to bind, CP, (1ZA2).14 Following the allosteric transition to the R state, known structures include the R-state enzyme in the absence of ligands (2A0F),15 the R-state enzyme in the presence of the CP analog phosphonacetamide (2A0F),15 in the presence of phosphonacetamide and Asp (2HSE),16 and in the presence of the bisubstrate analog PALA (1D09)17. Following the chemical reaction, the R-state enzyme with products CA and Pi bound as well as immediately after CA dissociates from the active site, have yet to be determined. The only analogous structures are ATCase in the R-state with product analogs citrate and phosphate RCit_Pi (1R0B),18 and in the T state with the product molecules TCA_Pi (1R0C).12 To understand the mechanism by which ATCase releases products and binds the next round of substrates without reverting back to the T state, an R-state structure with the last product molecule, Pi, is crucial.
To obtain a structure of ATCase in the R state bound to Pi, the T-state destabilized mutants such as D236A19 or E239Q20 were considered, but have been shown to display a small-angle X-ray scatter pattern intermediate between the T and R state. Instead the C47A/A241C holoenzyme was utilized as a model of the R-state. The 240's loop of ATCase has significantly different conformations in the T and R states. In the R-quaternary structure, residues from the 240's loop from the two catalytic subunits are close together, but they are far apart in the T state. Saturation with substrate completes the movement of the 240's loop into its final closed position.21 The disulfide bonds between Cys241 from C1 and C4 predispose the active site into the high-affinity, high-activity form by locking this loop into the final R-state position without substrates. Utilizing a species that is locked by disulfide bonds in the active conformation ensures that crystallization in the presence of the last remaining product, Pi, will result in an ATCase molecule in the R-quaternary structure.
R-state stabilization by disulfide-linkages
To obtain a crystal structure of the R state of ATCase with Pi bound, the C47A/A241C holoenzyme purified for crystallography must be locked in the R state. The kinetics of the purified C47A/A241C holoenzyme were compared to wild-type ATCase to ensure that the enzyme displayed characteristic R-state properties. Wild-type ATCase displays homotropic cooperativity with respect to the second substrate, Asp, due a shift from the low-activity, low-affinity T state to the high-activity high-affinity R state. Thus, the wild-type enzyme displays a sigmoidal Asp saturation curve, but ATCase locked in the R state should produce a hyperbolic saturation curve. The C47A/A241C holoenzyme with disulfides intact displayed a hyperbolic Asp saturation curve confirming the loss of homotropic cooperativity as reported previously.13
Not only does the natural substrate Asp, in the presence of a saturating concentration of CP, induce the T to R transition, but the transition can also be induced by substrate analogs such as PALA. At low concentrations of Asp, much less than the [Asp]0.5, the wild-type enzyme is essentially all in the T state, however the binding of PALA to one or more active sites shifts the entire enzyme to the R state. Thus, under these conditions PALA can activate the wild-type enzyme. The response of freshly purified disulfide-linked C47A/A241C holoenzyme to PALA was tested to demonstrate the loss of homotropic cooperativity. When the disulfide bonds were intact, the enzyme did not show PALA activation. Instead, the enzyme was inhibited by PALA at all concentrations suggesting that PALA was unable to promote the T to R transition because the enzyme was locked in the R state (Fig. 1).
Fig. 1.
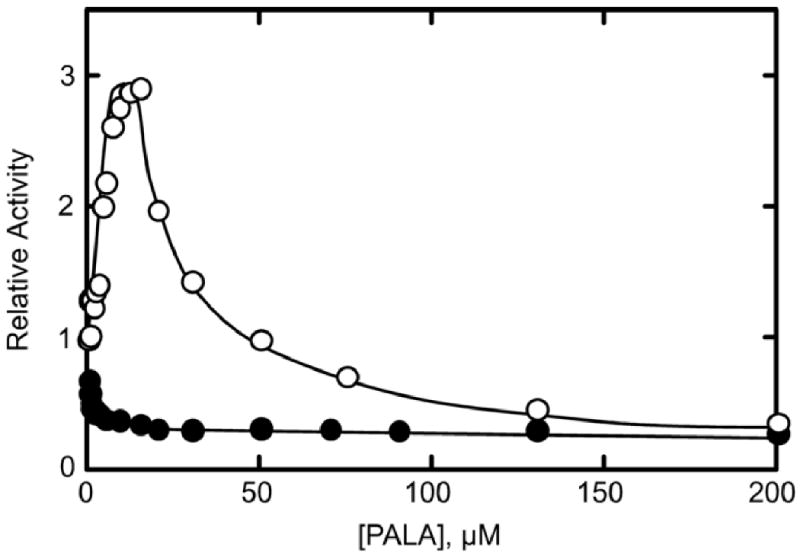
PALA saturation of the wild-type and C47A/A241C enzymes. Assays were performed in duplicate in 50 mM Tris-acetate buffer at pH 8.3 and 25° C at a saturating concentration of CP (4.8 mM) and at one-fifth the [Asp]0.5 concentration (0.5 mM for the C47A/A241C holoenzyme and 2.5 mM for wild-type ATCase). The open circles represent the wild-type ATCase which is activated then inhibited by PALA. The closed circles represent the C47A/A241C holoenzyme displaying inhibition by PALA at all concentrations.
West et al.13 reported that non-reducing SDS PAGE of the C47A/A241C holoenzyme resulted in two bands at 17 and 70 kDa. The 17 kDa band corresponds to the wild-type regulatory chain, while the 70 kDa band corresponds to two catalytic chains linked by a disulfide bond. After reducing the disulfide bonds by boiling in SDS PAGE loading dye buffer, two bands were observed at 17 and 35 kDa, identical to the bands for wild-type ATCase. After characterizing the C47A/A241C holoenzyme by reducing and non-reducing SDS PAGE to confirm that the freshly purified C47A/A241C holoenzyme displayed bands identical to those previously reported, the enzyme was concentrated for crystallography.
Structure of C47A/A241C holoenzyme in the presence of Pi
The catalytic mechanism of ATCase follows an ordered-sequential mechanism with CP binding before Asp and CA leaving before Pi22,23 as outlined in Fig. 2. To understand how the final product, Pi, is released from the active site while ATCase remains in the R-quaternary state, X-ray crystallography was employed. R-state crystals were obtained with the product Pi bound by utilizing the C47A/A241C holoenzyme. Crystals were obtained with Pi bound that diffracted to a maximal resolution of 2.85 Å in the H32 space group (Table 1). The unit cell dimensions (a = b = 120.7 Å, c = 692.5 Å) were not similar to those observed for any previous ATCase crystals. Molecular replacement utilizing Phaser (CCP4) and AutoMR (PHENIX) was used to solve the structure. The asymmetric unit consisted of three catalytic-regulatory chain dimers. Two of these dimers are part of one ATCase molecule, while the other is part of an independent ATCase molecule. The RS-S_Pi structure was refined to an Rfactor/Rfree of 17.5%/24.1% using PHENIX.24
Fig. 2.
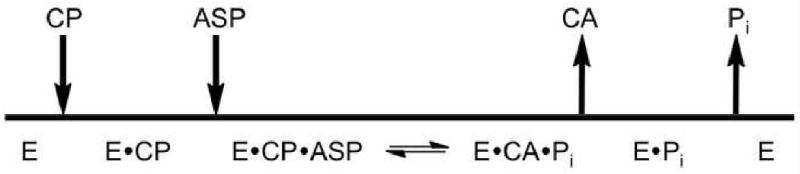
Ordered-sequential mechanism of ATCase. The figure outlines the order of substrate binding; CP binds before aspartate, and the order of product release; CA dissociates before Pi for the ATCase reaction where E·CP indicates the enzyme bound to CP.
Table 1. Data collection and refinement statistics for the RS-S_Pi ATCase structure.
| Data collection | |
|---|---|
| Space Group | H32 |
| Cell Dimensions: | |
| a, b, c (Å) | 120.7, 120.7, 692.5 |
| Wavelength (Å) | 1.0809 |
| Resolution (Å) | 50.00-2.85 |
| Rsym(%) | 0.094 (0.510) |
| Average (I/σ) | 10.0 (4.8) |
| Completeness (%) | 99.9 (100) |
| Redundancy | 13.9 (11.8) |
| Refinement | |
| Resolution (Å) | 30.00-2.85 |
| Reflections | 45,742 |
| Rwork/Rfree | 0.175/0.241 |
| Number of atoms | 11408 |
| Protein | 10596 |
| Waters | 724 |
| Phosphate | 6 |
| Occupancy | 1.0 |
| Zinc | 3 |
| RMS deviations | |
| Bond lengths (Å) | 0.011 |
| Angles (°) | 1.20 |
| Mean B value (Å2) | 46.0 |
Values in parentheses are for the highest resolution shell
Allosteric state
The allosteric transition from the T state to the R state is characterized by an 11 Å expansion along the three-fold axis. To evaluate the quaternary structure of RS-S_Pi, the vertical separation between the upper and lower catalytic subunits was computed and compared to the vertical separations of known T and R state structures: 45.6 Å25 and 56.4 Å17 respectively. The vertical separation for RS-S_Pi was determined to be 56.4 Å, indicating that the global conformation of RS-S_Pi is identical to that of the R state (Table 2).
Table 2. Comparisons of Structures.
| RS-S_Pi | RPALA | TCA,Pi | ||||
|---|---|---|---|---|---|---|
| Vertical Separation | 56.4 | 57.9 | 47.2 | |||
| (Å) Planar Angle (°) | 127.4 | 127.6 | 137.1 | |||
| Pi Interactions | Residue | Distance (Å) | Residue | Distance (Å) | Residue | Distance (Å) |
| Pi1 | Lys84 NG | 3.10 | Lys84 NG | 3.02 | ||
| Ser80 OG | 2.87 | Ser80 OG | 2.91 | |||
| Arg105 NH2 | 3.25 | Arg105 NH1 | 3.28 | |||
| Arg105 NH1 | 2.94 | Arg105 NH1 | 2.58 | |||
| Thr55 OG1 | 2.64 | Thr55 OG1 | 2.71 | |||
| Thr55 N | 2.87 | Thr55 N | 2.76 | |||
| Arg54 NH2 | 3.19 | Arg54 NH1 | 3.21 | |||
| Arg54 NE | 3.22 | |||||
| Arg54 NE | 2.88 | |||||
| Arg54 N | 3.03 | Arg54 N | 3.08 | |||
| Thr53 N | 2.62 | Thr53 N | 2.81 | |||
| Ser52 OG | 2.75 | Ser52 OG | 2.52 | |||
| Pi2 | Arg167 NH2 | 3.21 | Arg105 NH1 | 3.27 | ||
| Arg167 NE | 2.80 | Arg105 NH2 | 3.17 | |||
| His134 NE2 | 2.46 | Arg105 NH2 | 2.83 | |||
| Arg105 NH2 | 2.53 | His134 NE2 | 2.71 | |||
| Thr55 OG1 | 2.62 | |||||
In order to evaluate the differences of the RS-S_Pi active site as compared to other R-state structures, the planar angle between the CP and Asp domains26 were compared (Table 2). The planar angle is the angle formed between the centers of gravity of the CP and Asp domains and a hinge point.17 The planar angle of the active site of ATCase is different depending upon the allosteric state, with the T-state existing in a more open form with a planar angle of approximately 135° and the R-state existing in a more closed form with a planar angle of approximately 127° promoting catalysis by approximation. The average planar angle for the three catalytic chains of the RS-S_Pi structure was calculated to be 128.0 ± 0.5°, a value that is characteristic of other R-state structures (Table 2).
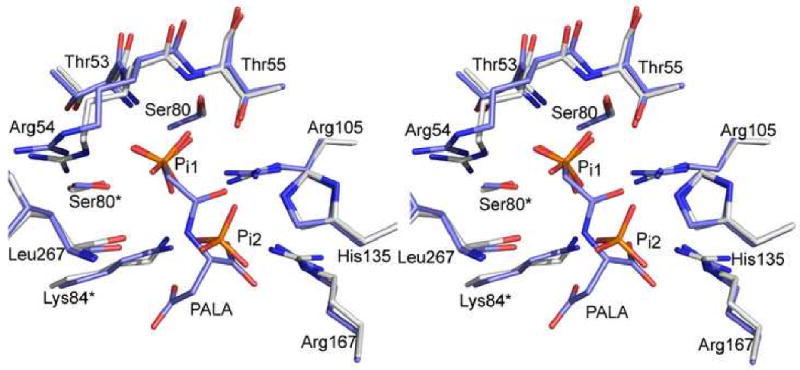
In addition to vertical separation and planar angle values consistent with typical R-state conformations, a structural alignment of the RS-S_Pi and RPALA structures was prepared. PALA is a bi-substrate analog that induces the T to R transition, so the RPALA structure is representative of the R quaternary structure. The structural alignment showed an overall RMS deviation of 2.17 Å between the two structures, and a RMS deviation of 0.84 Å for the alignment of a single catalytic chain from both structures. Fig. 3a shows the backbone RMS deviation of the holoenzyme, while Fig. 3b shows a single C1 catalytic chain between the RPALA and RS-S_Pi structures. The greatest deviation in the structures was observed in the regulatory chains, while the a comparison of a single catalytic chain of the RPALA and RS-S_Pi structures are nearly identical. The only significant shift in the backbone of the catalytic chain occurs at the N terminus shown in pink. All other residues display an RMS deviation of less than 1.0 Å.
Fig. 3.
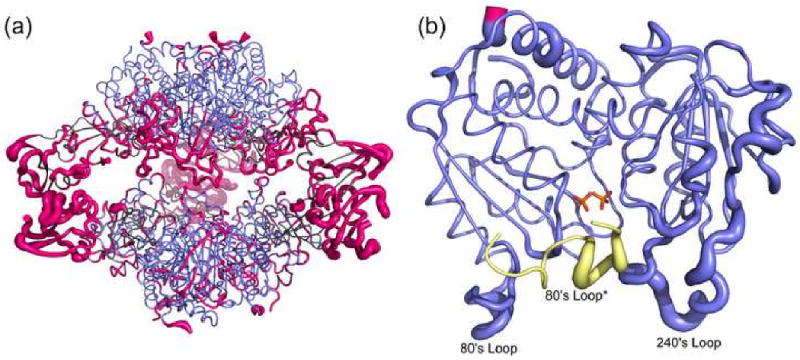
Structural alignment of the RS-S_Pi and RPALA structures. (a) Structural alignment of the c6r6 holoenzyme, with an overall RMS deviation of 2.17 Å. The catalytic chains are colored blue and the regulatory chains are colored grey. Regions with a RMS deviation greater than 1.5 Å are colored pink. (b) Structural alignment of one catalytic chain (blue, pink) of the RS-S_Pi structure and one catalytic chain of the RPALA structure, along with the 80's loop of the adjacent catalytic chain (yellow). The width of the tube is proportional to the RMS deviation between the α-carbon positions of the two structures. Regions colored in pink represent a RMS deviation greater than 1.5 Å. This figure was drawn using Pymol.39 The two phosphates bound to the active site are represented by sticks and colored by atom.
Disulfide linkages
The disulfide bonds between Cys241 of the catalytic chain from the upper and lower trimers are critical to stabilize the R state by positioning the 240's loop in its active conformation. The electron density maps confirmed the presence of disulfide linkages in the RS-S_Pi structure. Fig. 4 shows the electron density generated from a composite omit map surrounding the Cys241 of the 240's loops from C1 and C4 subunits and clearly indicates the presence of a disulfide bond. The geometry of the disulfide bond in this structure differs from ideality. The dihedral angle between the Cβ-Sγ-Sγ′-Cβ′ which is generally approximately 90° was measured for the Cys241 residues involved in the disulfide bonds, giving an average value of 160 ± 13°. The geometry of this disulfide may differ from ideality because the Cys residues involved are not native to ATCase and therefore the resulting disulfide-linked holoenzyme represents an unnatural, manipulated structure rather than one that has evolved over time to serve a specific function. The distance of the disulfide bond was 2.04 Å close to the ideal.
Fig. 4.
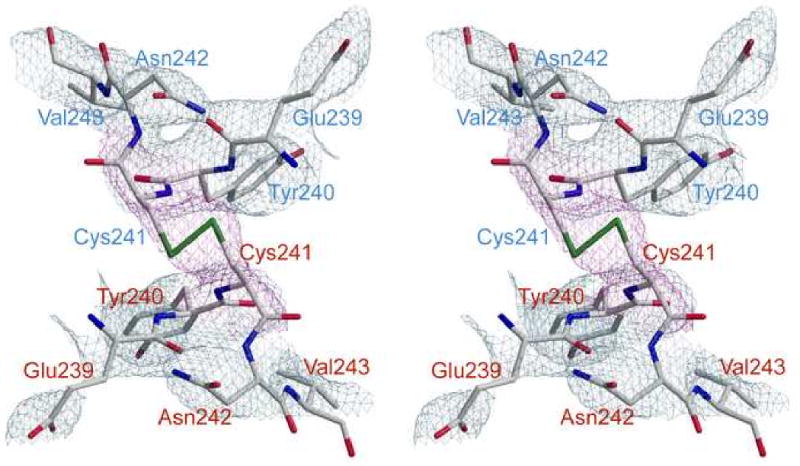
Stereoview of the 240's loops of the C1 and C4 chains of the RS-S_Pi structure with a disulfide linkage between Cys241 from the C1 and C4 chains. The refined coordinates of residues 239-240 and 242-243 of both chains are overlaid on the 2Fo–Fc electron density map (grey) contoured at 1.5σ. Cys241 from both chains is overlaid on the composite omit map (magenta). Residues from C1 are labeled in blue and those from C4 are labeled in red.
The disulfide bonds in this structure predispose the active site into the high-affinity form characteristic of the R state. This is clear from the tube diagram in Fig. 3b, which compares the catalytic chains of the RS-S_Pi and RPALA structures. When substrates, or the substrate analog PALA, are bound to ATCase, the 240's loop is in its final closed conformation. The tube diagram in Fig. 3 indicates that the 240's loop of the PALA-liganded structure is nearly identical to the position of the 240's loop in the disulfide-linked structure (less than 1.0 Å RMS deviation).
Active site ligands
In the H32 space group, the asymmetric unit contains three catalytic chains and three regulatory chains. The C47A/A241C holoenzyme was crystallized in the presence of 100 mM KH2PO4 and two molecules of phosphate were observed in each active site at 100% occupancy. In the catalytic cycle, phosphate is the final product to dissociate from the active site prior to the beginning of a new catalytic cycle with the binding of CP followed by Asp. While only one Pi molecule is produced per catalytic cycle, it is not surprising to observe two Pi molecules in the active site in the absence of additional ligands, because the R-state active site consists of many positively charged residues resulting in high affinity for its negatively-charged substrates. There are approximately 12 hydrogen-bonding interactions between the enzyme and the first phosphate molecule (Pi1) and approximately four hydrogen-bonding interactions between the enzyme and the second phosphate molecule (Pi2). These interactions are outlined in Table 2 and shown in Fig. 5.
Fig. 5.
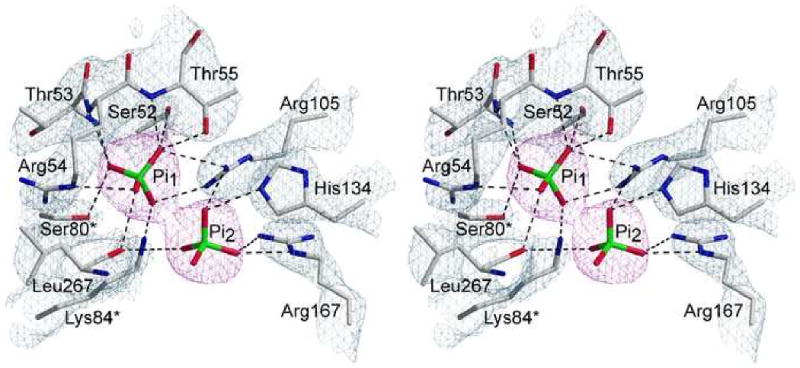
Stereoview of the active site in the C47A/A241C holoenzyme complexed with Pi. The refined coordinates of the side-chains and backbone are overlaid on the 2Fo–Fc electron density map (grey) shown contoured at 1.5σ. The two Pi molecules are overlaid on the composite omit map (magenta) shown contoured at 1.5σ.
The crystal structure of ATCase has been solved in the presence of a number substrates and analogs that contain either a phosphate or phosphonate functional group such as RPALA,17 RPAM,15 RPAM_MAL,27 RCit_Pi,18 and TCP.14 This negatively charged group binds specifically into a positively charged pocket of the active site and shares a common position and similar hydrogen-binding interactions among these structures. For instance when the active sites of the above-mentioned structures are overlaid, the phosphate or phosphonate moiety binds in a common position. Notably, the position of Pi1 in the RS-S_Pi structure binds in the same position and makes similar hydrogen-bonding interactions as the previously solved structures. When the active site from RPALA is overlaid with the RS-S_Pi structure, the phosphonate group of PALA is in the identical position as Pi1 and displays identical hydrogen bonding interactions with active site residues. Fig. 6 shows the overlay of active sites from the RPALA and RS-S_Pi structures to clearly indicate the similar positions of Pi1 and the phosphonate group of PALA.17
Fig. 6.
Stereoview of the active site superposition of the RS-S_Pi structure complexed with Pi1 and Pi2 (white carbons) and the RPALA structure complexed with PALA (blue carbons). This figure was drawn using Pymol.39
Pi2 is bound approximately 4.3 Å away from Pi1 in the active site and thus does not occupy the common position described above. There are far less hydrogen-bonding interactions of Pi2 with active site residues than Pi1 (Table 2) indicating that this Pi is not as tightly bound, but Pi2 is bound in a positively charged pocket more accessible to the surface. The data suggest that Pi2 may occupy a stabilized position in the pathway that the product molecule follows as it is guided out of the active site. This notion is further supported by the fact that the Pi1 cannot leave the active site pocket without moving towards the Pi2 site. This is not the first instance where two identical ligand molecules have been observed at the active site of an ATCase structure. The TCP structure contains two identical substrate molecules at the active site, whereas the RS-S_Pi structure contains two identical product molecules at the active site. In the TCP structure, one CP molecule is bound in the position comparable to the analogous moiety of PALA from the RPALA structure, and forms many hydrogen-bonding interactions with active site residues. The other CP molecule is far from the PALA position, approximately 9 Å from the first CP molecule, and makes fewer interactions with active site residues suggesting that this position is on the path that CP takes when binding to the active site. Similarly, Pi1 from the RS-S_Pi structure is bound in the identical position of the phosphate of the tightly bound CP from the TCP structure and makes many hydrogen-binding interactions with active site residues. Pi2 is positioned 4.3 Å from Pi1 and makes very few interactions with active site residues, suggesting that this position is on the path that Pi makes when dissociating from the active site. Furthermore, when the RS-S_Pi and TCP active sites are overlaid, Pi1 is bound in the identical position of the phosphate of the tightly bound CP, and Pi2 is positioned between the two CP molecules without any clashes. If CP follows the same path as it binds to the active site in T and R states, then this may indicate that the active site can accommodate Pi dissociation while leaving enough space for CP to bind simultaneously and Pi dissociation my help lead CP into the active site.
By locking ATCase in the active conformation through the introduction of specific disulfide bonds, crystallizing the enzyme and determining its structure, an R-state model of the enzyme has been generated that can be used to complete the X-ray crystallographic snapshots of each step in the catalytic cycle. Thus far we have observed the R-state active site with the final product molecule Pi bound; a structure that could not be obtained utilizing the wild-type enzyme. Furthermore, no mutations near the active site were necessary, so all the important protein-ligand interactions are maintained. We look forward to utilizing ATCase locked in the R state to solve the structures of the R state of ATCase at each of the remaining steps of the catalytic cycle.
Materials and Methods
Materials
Chemicals
l-aspartate, n-carbamoyl-l-aspartate, potassium dihydrogen phosphate, and uracil were obtained from Sigma. PALA was obtained from the NCI, National Institutes of Health. Sodium dodecyl sulfate and the protein assay dye were purchased from Bio-Rad. Carbamoyl phosphate dilithium salt obtained from Sigma was purified before use by precipitation from 50% (v/v) ethanol and was stored desiccated at -20 °C.1 50 μl dialysis buttons were obtained from Hampton Research. Dialysis tubing was obtained from BioDesign Inc. of New York.
Methods
Overexpression and purification of the mutant enzyme
The mutant holoenzyme was overexpressed from plasmid pEK613 and purified to homogeneity as described previously.28 After concentration, the purity of the enzyme was checked by SDS-PAGE,29 non-reducing SDS-PAGE (in which 2-mercaptoethanol is omitted from the sample buffer), and nondenaturing PAGE.30,31 The concentration of the mutant enzyme was determined by the Bio-Rad version of the Bradford dye binding assay.32
Aspartate transcarbamoylase assay
The aspartate transcarbamoylase activity was measured at 25 °C by the colorimetric method.33 Aspartate saturation curves were performed in duplicate. Assays were performed in 50 mM Tris-acetate buffer, pH 8.3, in the presence of saturating carbamoyl phosphate (4.8 mM). Data analysis of the steady-state kinetics was carried out as described previously.34 Fitting of the experimental data to theoretical equations was accomplished by non-linear regression. The data were analyzed using an extension of the Hill equation that included a term for substrate inhibition. If the fit to the Hill equation gave a Hill coefficient of 1 or less, the experimental data were fit to the Michaelis-Menten equation with an additional term for substrate inhibition.35
Crystallization and data collection
The C47A/A241C holoenzyme was crystallized by microdialysis, using 50 μl wells (Hampton Research). To crystallize in the presence of phosphate the enzyme solution, at 10 mg/ml, was dialyzed against a solution of 100 mM potassium dihydrogen phosphate, 3 mM sodium azide (pH 5.9). Crystals grew to average dimensions of 0.8 mm × 0.6 mm × 0.5 mm within one week. Crystals were transferred into a freezing solution containing 20% PEG 400 in crystallization buffer for ∼1 min prior to freezing in liquid nitrogen.
The X-ray data were collected at Brookhaven National Synchrotron Light Source (Brookhaven National Laboratory, New York), beamline X29. The C47A/A241C holoenzyme crystallized in space group H32 with unit cell dimensions a = b = 120.7, c = 692.5 (Å), α = β = 90, γ = 120 (°) and diffracted to 2.85 Å resolution.
Molecular replacement and structural refinement
The initial model for the structure was derived from the coordinates of the wild-type structure of E. coli ATCase crystallized in the presence of PALA (PDB code 1D09) with all of the water molecules and ligands removed.17 A Matthews coefficient of 2.43 suggested four catalytic-regulatory dimers in the asymmetric unit. Molecular replacement was performed using Phaser within the CCP4 Suite36 in addition to molecular replacement by AutoMR within PHENIX.24 Both programs solved the structure with three catalytic-regulatory dimers in the asymmetric unit, which gives a Matthews coefficient of 3.24 and 62% solvent.
The coordinates from the molecular replacement were subjected to rigid-body refinement and simulated annealing within PHENIX, reducing the Rfactor/Rfree to 0.260/0.275. Further structure refinement was performed using PHENIX24 bringing the final Rfactor/Rfree to 0.175/0.241. The N terminus of the regulatory chain was disordered; for this reason, the first twelve amino acid residues were removed from the model. Two molecules of phosphate were fit on the basis of a composite omit map generated by PHENIX.24 The initial model contained Ala at position 241, but the disulfide linkages between Cys241 of catalytic subunits C1 and C4 could clearly be observed in the Fo–Fc electron density maps as negative density. COOT37 was used to mutate and auto-fit Cys residues into the negative density at positions 241 of all catalytic chains. Following refinement in PHENIX24 each Cys241 fit into the density appropriately. Water molecules were added to the structure using PHENIX24 on the basis of Fo–Fc electron density maps at or above the 3.0σ level. The model was checked for errors using PROCHECK.38 The details of data processing and refinement statistics are given in Table I.
Protein data bank deposition
The coordinate and structure factors for the C47A/A241C holoenzyme complex with phosphate have been deposited in the RCSB Protein Data Bank under accession code 3MPU.
Acknowledgments
This work was supported in part by Grant GM26237 from the National Institutes of Heath. Data for the crystals with Pi bound were measured at Beamline X29 of the National Synchrotron Light Source. We thank Howard Robinson of Brookhaven National Laboratory for data collection and assistance with data processing. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886.
Abbreviations used
- ATCase
aspartate transcarbamoylase (EC 2.1.3.2, aspartate carbamoyltransferase)
- CA
carbamoyl aspartate
- CP
carbamoyl phosphate
- PALA
N- phosphonacetyl-L-aspartate
- [Asp]0.5
the aspartate concentration at one-half of the maximal observed specific activity
- 80's loop
a loop in the catalytic chain of ATCase comprised of residues 73-88
- 240's loop
a loop in the catalytic chain of ATCase comprised of residues 230-245
- RS-S_Pi
the X-ray structure of the C47A/A241C disulfide-linked ATCase holoenzyme in the presence of Pi
- Pi1 and Pi2
the two phosphate molecules bound at the active site of the RS-S_Pi structure
- RPALA
the X-ray structure of wild-type ATCase in the presence of PALA (PDB code 1D09)
- TCA_Pi
the X-ray structure of wild-type ATCase in the presence of CA and Pi (PDB code 1R0C)
- TCP
the X-ray structure of wild-type ATCase in the presence of two molecules of CP (PDB code 1ZA2)
- RPAM_MAL
the X-ray structure of wild-type ATCase in the presence of phosphonoacetamide and malonate (PDB code 1AT1)
- RCit_Pi
the X-ray structure of wild-type ATCase in the presence of citrate and phosphate (PDB code 1R0B)
- RPAM
the X-ray structure of D236A ATCase in the presence of phosphonoacetamide (PDB code 2A0F)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gerhart JC, Pardee AB. Enzymology of control by feedback inhibition. J Biol Chem. 1962;237:891–896. [PubMed] [Google Scholar]
- 2.Wild JR, Loughrey-Chen SJ, Corder TS. In the presence of CTP, UTP becomes an allosteric inhibitor of aspartate transcarbamylase. Proc Natl Acad Sci USA. 1989;86:46–50. doi: 10.1073/pnas.86.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber KK. New Structural Model of E. coli Aspartate Transcarbamylase and the Amino-acid Sequence of the Regulatory Polypeptide Chain. Nature. 1968;218:1116–1119. doi: 10.1038/2181116a0. [DOI] [PubMed] [Google Scholar]
- 4.Wiley DC, Lipscomb WN. Crystallographic Determination of Symmetry of Aspartate Transcarbamylase. Nature. 1968;218:1119–1121. doi: 10.1038/2181119a0. [DOI] [PubMed] [Google Scholar]
- 5.Gerhart JC, Schachman HK. Allosteric interactions in aspartate transcarbamylase II. Evidence for different conformational states of the protein in the presence and absence of specific ligands. Biochemistry. 1968;7:538–552. doi: 10.1021/bi00842a600. [DOI] [PubMed] [Google Scholar]
- 6.Howlett GJ, Blackburn MN, Compton JG, Schachman HK. Allosteric regulation of aspartate transcarbamoylase. Analysis of the structural and functional behavior in terms of a two-state model. Biochemistry. 1977;16:5091–5099. doi: 10.1021/bi00642a023. [DOI] [PubMed] [Google Scholar]
- 7.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamylase: The relations between structure and function. Science. 1988;241:669–674. doi: 10.1126/science.3041592. [DOI] [PubMed] [Google Scholar]
- 8.Ke HM, Lipscomb WN, Cho Y, Honzatko RB. Complex of N-phosphonacetyl-L-aspartate with aspartate carbamoyltransferase: X-ray refinement, analysis of conformational changes and catalytic and allosteric mechanisms. J Mol Biol. 1988;204:725–747. doi: 10.1016/0022-2836(88)90365-8. [DOI] [PubMed] [Google Scholar]
- 9.Ladjimi MM, Kantrowitz ER. A possible model for the concerted allosteric transition in Escherichia coli aspartate transcarbamylase as deduced from site-directed mutagenesis studies. Biochemistry. 1988;27:276–283. doi: 10.1021/bi00401a042. [DOI] [PubMed] [Google Scholar]
- 10.Tsuruta H, Sano T, Vachette P, Tauc P, Moody MF, Wakabayashi K, Amemiya Y, Kimura K, Kihara H. Structural kinetics of the allosteric transition of aspartate transcarbamoylase produced by physiological substrates. FEBS Lett. 1990;263:66–68. doi: 10.1016/0014-5793(90)80706-o. [DOI] [PubMed] [Google Scholar]
- 11.Tsuruta H, Vachette P, Sano T, Moody MF, Tauc P, Amemiya Y, Wakabayashi K, Kihara H. Kinetics of the quaternary structure change of aspartate transcarbamoylase triggered by succinate, a competitive inhibitor. Biochemistry. 1994;33:10007–10012. doi: 10.1021/bi00199a026. [DOI] [PubMed] [Google Scholar]
- 12.Huang J, Lipscomb WN. Products in the T-state of aspartate transcarbamylase: crystal structure of the phosphate and N-carbamyl-L-aspartate ligated enzyme. Biochemistry. 2004;43:6422–6426. doi: 10.1021/bi0302144. [DOI] [PubMed] [Google Scholar]
- 13.West JM, Tsuruta H, Kantrowitz ER. Stabilization of the R allosteric structure of E. coli aspartate transcarbamoylase by disulfide bond formation. J Biol Chem. 2002;277:47300–47304. doi: 10.1074/jbc.M209913200. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Stieglitz KA, Cardia JP, Kantrowitz ER. Structural basis for ordered substrate binding and cooperativity in aspartate transcarbamoylase. Proc Natl Acad Sci U S A. 2005;102:8881–8886. doi: 10.1073/pnas.0503742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stieglitz KA, Dusinberre KJ, Cardia JP, Tsuruta H, Kantrowitz ER. Structure of the E. coli aspartate transcarbamoylase trapped in the middle of the catalytic cycle. J Mol Biol. 2005;352:478–486. doi: 10.1016/j.jmb.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Eldo J, Kantrowitz ER. Structural Model of the R state of Escherichia coli aspartate transcarbamoylase with substrates bound. J Mol Biol. 2007;371:1261–1273. doi: 10.1016/j.jmb.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin L, Stec B, Lipscomb WN, Kantrowitz ER. Insights into the mechanism of catalysis and heterotropic regulation of E. coli aspartate transcarbamoylase based upon a structure of enzyme complexed with the bisubstrate analog N-phosphonacetyl-L-aspartate at 2.1 Å. Proteins: Struct Funct Genet. 1999;37:729–742. [PubMed] [Google Scholar]
- 18.Huang J, Lipscomb WN. Aspartate transcarbamylase (ATCase) of Escherichia coli: a new crystalline R-state bound to PALA, or to product analogues citrate and phosphate. Biochemistry. 2004;43:6415–21. doi: 10.1021/bi030213b. [DOI] [PubMed] [Google Scholar]
- 19.Chan RS, Sakash JB, Macol CP, West JM, Tsuruta H, Kantrowitz ER. The role of intersubunit interactions for the stabilization of the T State of Escherichia coli aspartate transcarbamoylase. J Biol Chem. 2002;277:49755–49760. doi: 10.1074/jbc.M208919200. [DOI] [PubMed] [Google Scholar]
- 20.Tauc P, Vachette P, Middleton SA, Kantrowitz ER. The structural consequences of a single amino acid mutation on aspartate transcarbamylase from Escherichia coli. J Mol Biol. 1990;214:327–335. doi: 10.1016/0022-2836(90)90164-H. [DOI] [PubMed] [Google Scholar]
- 21.Fetler L, Vachette P, Hervé G, Ladjimi MM. Unlike the quaternary structure transition, the tertiary structure change of the 240s loop in allosteric aspartate transcarbamylase requires active site saturation by substrate for completion. Biochemistry. 1995;34:15654–15660. doi: 10.1021/bi00048a008. [DOI] [PubMed] [Google Scholar]
- 22.Hsuanyu Y, Wedler FC. Kinetic mechanism of native Escherichia coli aspartate transcarbamylase. Arch Biochem Biophys. 1987;259:316–330. doi: 10.1016/0003-9861(87)90498-x. [DOI] [PubMed] [Google Scholar]
- 23.Wedler FC, Gasser FJ. Ordered substrate binding and evidence for a thermally induced change in mechanism for E. coli aspartate transcarbamylase. Arch Biochem Biophys. 1974;163:57–68. doi: 10.1016/0003-9861(74)90454-8. [DOI] [PubMed] [Google Scholar]
- 24.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 25.Kosman RP, Gouaux JE, Lipscomb WN. Crystal structure of CTP-ligated T state aspartate transcarbamoylase at 2.5 Å resolution: Implications for aspartate transcarbamoylase mutants and the mechanism of negative cooperativity. Proteins: Struct Funct Genet. 1993;15:147–176. doi: 10.1002/prot.340150206. [DOI] [PubMed] [Google Scholar]
- 26.Stieglitz K, Stec B, Baker DP, Kantrowitz ER. Monitoring the transition from the T to the R state in E. coli aspartate transcarbamoylase by X-ray crystallography: Crystal structures of the E50A mutant in four distinct allosteric states. J Mol Biol. 2004;341:853–868. doi: 10.1016/j.jmb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Gouaux JE, Stevens RC, Lipscomb WN. Crystal structures of aspartate carbamoyltransferase ligated with phosphonoacetamide, malonate and CTP or ATP at 2.8 Å resolution and neutral pH. Biochemistry. 1990;29:7702–7715. doi: 10.1021/bi00485a020. [DOI] [PubMed] [Google Scholar]
- 28.Nowlan SF, Kantrowitz ER. Superproduction and rapid purification of E. coli aspartate transcarbamoylase and its catalytic subunit under extreme derepression of the pyrimidine pathway. J Biol Chem. 1985;260:14712–14716. [PubMed] [Google Scholar]
- 29.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Davis BJ. Disc electrophoresis-II Method and application to human serum proteins. Ann N Y Acad Sci. 1964;121:404–427. doi: 10.1111/j.1749-6632.1964.tb14213.x. [DOI] [PubMed] [Google Scholar]
- 31.Ornstein L. Disc electrophoresis. I. Background and theory. Ann N Y Acad Sci. 1964;121:321–349. doi: 10.1111/j.1749-6632.1964.tb14207.x. [DOI] [PubMed] [Google Scholar]
- 32.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 33.Pastra-Landis SC, Foote J, Kantrowitz ER. An improved colorimetric assay for aspartate and ornithine transcarbamylases. Anal Biochem. 1981;118:358–363. doi: 10.1016/0003-2697(81)90594-7. [DOI] [PubMed] [Google Scholar]
- 34.Silver RS, Daigneault JP, Teague PD, Kantrowitz ER. Analysis of two purified mutants of Escherichia coli aspartate transcarbamylase with single amino acid substitutions. J Mol Biol. 1983;168:729–745. doi: 10.1016/s0022-2836(83)80072-2. [DOI] [PubMed] [Google Scholar]
- 35.Pastra-Landis SC, Evans DR, Lipscomb WN. The effect of pH on the cooperative behavior of aspartate transcarbamylase from Escherichia coli. J Biol Chem. 1978;253:4624–4630. [PubMed] [Google Scholar]
- 36.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 39.DeLano WL. The PyMol molecular graphics system. DeLano Scientific LLC; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]