

Abstract
CONTEXT:
Hypertrophic cardiomyopathy (HCM) is known to be manifested by mutations in 12 sarcomeric genes and dilated cardiomyopathy (DCM) is known to manifest due to cytoskeletal mutations. Studies have revealed that sarcomeric mutations can also lead to DCM. Therefore, in the present study, we have made an attempt to compare and analyze the genetic variations of beta-myosin heavy chain gene (β-MYH7), which are interestingly found to be common in both HCM and DCM. The underlying pathophysiological mechanism leading to two different phenotypes has been discussed in this study. Till date, about 186 and 73 different mutations have been reported in HCM and DCM, respectively, with respect to this gene.
AIM:
The screening of β-MYH7 gene in both HCM and DCM has revealed some common genetic variations. The aim of the present study is to understand the pathophysiological mechanism underlying the manifestation of two different phenotypes.
MATERIALS AND METHODS:
100 controls, 95 HCM and 97 DCM samples were collected. Genomic DNA was extracted following rapid nonenzymatic method as described by Lahiri and Nurnberger (1991), and the extracted DNA was later subjected to polymerase chain reaction (PCR) based single stranded conformation polymorphism (SSCP) analysis to identify single nucleotide polymorphism (SNP)s/mutations associated with the diseased phenotypes.
RESULTS AND CONCLUSION:
Similar variations were observed in β-MYH7 exons 7, 12, 19 and 20 in both HCM and DCM. This could be attributed to impaired energy compromise, or to dose effect of the mutant protein, or to even environmental factors/modifier gene effects wherein an HCM could progress to a DCM phenotype affecting both right and left ventricles, leading to heart failure.
Keywords: Diastolic dysfunction, dose effect, dilated cardiomyopathy, hypertrophic cardiomyopathy, single nucleotide polymorphism, systolic dysfunction
Introduction
Cardiomyopathy is a disease of the heart muscle, associated with cardiac dysfunction. It is classified as hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM) and arrhythmogenic right ventricular dysplasia (ARVD/C). HCM is characterized by hypertrophy of the left ventricle with the predominant involvement of the interventricular septum (diastolic dysfunction). Its prevalence is found to be 1 in 500.[1,2] DCM is characterized by cardiac dilatation, predominantly of the left ventricle, and weakening of the heart muscle beyond repair, resulting in reduced contractile function and inefficient pumping of the heart (systolic dysfunction). It accounts for 70–80% of all cases of cardiomyopathy.
Till date, about 186 and 73 different mutations have been identified in HCM and DCM, respectively, with respect to β-MYH7 gene. Since most of the SNPs/ mutations have been reported in this gene, the present study includes the screening of this gene for genetic variations in both the diseases to evaluate the underlying pathophysiological mechanisms leading to either hypertrophy or dilatation.[3,4]
Common genetic variations in exons 7, 12, 19 and 20 of this gene were found in both the HCM and DCM, which emphasizes the role of genetic modifiers and environmental factors along with impaired energy compromise and dosage gene effects of the protein leading to hypertrophy or dilatation eventually resulting in heart failure.
Materials and Methods
Cardiomyopathies (HCM and DCM) were diagnosed by physical examination, echocardiogram, electrocardiogram and magnetic resonance imaging. Primary DCMs were diagnosed through coronary angiogram, wherein one can rule out the possibility of an underlying myocardial infarction/coronary artery disease, valvular disease and/or other systemic disorders. Blood was collected from 100 healthy blood donors with no history of cardiac disorders and these samples were treated as controls. Blood samples from 97 DCM cases comprising 20 juvenile, 38 primary and 39 secondary cases, and 95 HCM samples were also collected from cardiology units of CARE Hospitals, Mahavir hospitals and Niloufer hospitals, Hyderabad. Informed written consent was obtained from patients in accordance with the study protocol along with the institutional ethics committee’s approval to carry out the above study.
Genomic DNA was extracted by rapid nonenzymatic method as described by Lahiri and Nurnburger.[5] DNA was amplified based on the primer sequences available on Siedman’s database on the website http://genetics.med.harvard.edu/~seidman/cg3/genes/MYH7_exons.html.
Polymerase chain reaction (PCR) was carried out in 0.2 ml tubes containing 100 ng of genomic DNA, 50 pmol each of forward and reverse primers, 0.5–1 U of Taq DNA polymerase enzyme, 200 μM of dNTPs, 1× PCR buffer and water to make up the volume to 25 μl. Initial denaturation was carried out at 95°C for 3 min, followed by a denaturation step at 95°C for 30 s. The annealing temperature varied from 54°C to 65°C based on the exon for 30 s and an extension at 72°C for 1 min. A final extension step of 72°C was for 2 min at the end of the reaction.
The amplified DNA samples were subjected to SSCP analysis on nondenaturing/polyacryalamide gels. Samples revealing any kind of band pattern variation or mobility shift were commercially sequenced on a 3730 × l DNA analyzer (Macrogen, Seoul Korea).
Results
Screening of MYH7 gene in HCM and DCM patients revealed common genetic variations in exons 7, 12, 19 and 20. Apart from these, unique SNPs/mutations in HCM (NCBI accession no. 51855930–51855950) and DCM (NCBI accession no. EF630363–EF630367 and EU091311–EU091316) were found [Reena et al. 2006,[6] Boda et al. 2009 (communicated)]. The list of SNPs/ mutations which are found to be common in both the diseased types is given in Table 1.[6]
Table 1.
Mutations/SNPs observed in both HCM and DCM
| Exon | Common variations |
Control (n) | Patient (n) | NCBI accession no. in DCM | |
|---|---|---|---|---|---|
| HCM | DCM | ||||
| 7 | Ala199Ala nt (7647) heterozygous | Ala199Ala nt (7647) homozygous | — | One patient each in both HCM and DCM | EF630363 |
| 12 | Gly354Gly (9600) heterozygous | Gly354Gly(9600) homozygous | 1 | Six in HCM and two in DCM | EF630364 |
| Lys365Lys(9633) heterozygous | Lys365Lys(9633) homozygous | — | One in HCM and three in DCM | EF630365 | |
| 19 | IVS19 + 92A/G homozygous | IVS19 + 82A/G homozygous | — | One each in HCM and DCM | EU091312 |
| 20 | IVS19 − 56A/G homozygous | IVS19 − 56A/G homozygous | — | One in HCM and two in DCM | EU091314 |
Mutational screening of MYH7 gene in HCM revealed five SNPs in exons 7, 12, 19 and 20 of which three were heterozygous and two were homozygous, [Figure 1, 2, 4] whereas the same SNPs were found to be homozygous [Figure 2] in DCM [Figure 3] sample, revealing the dose effect of the protein with the gross anatomical variations in the ventricles leading to heart failure in DCM cases.
Figure 1.
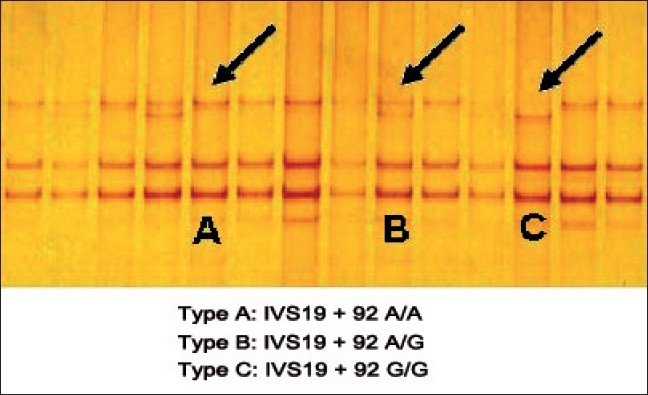
SSCP analysis of exon 19 in HCM
Figure 2.
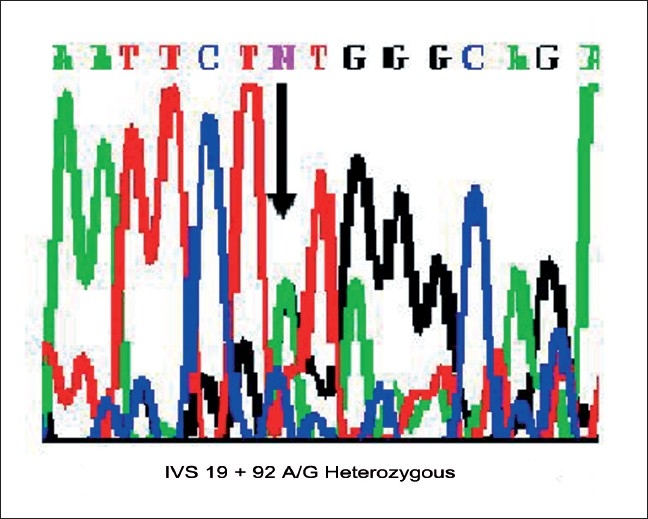
Chromatogram showing the intronic variation in Exon 19 in HCM
Figure 4.
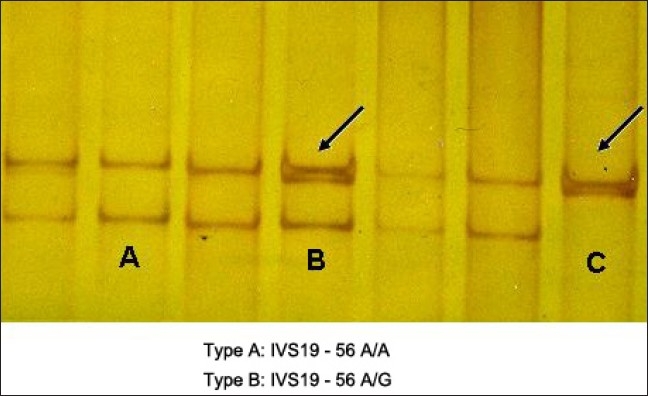
SSCP analysis of exon 20 in HCM
Figure 3.
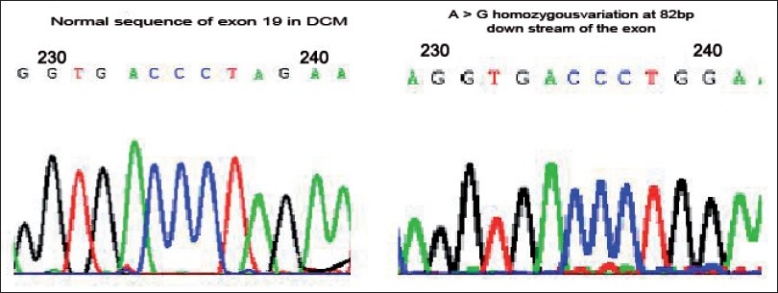
Fig 1b: Normal sequence of 82 bases downstream of the exon 19 in DCM; Fig 1c: A>G homozygous variation at 82 bases downstream of the exon 19 in DCM
Discussion
Myosin is the most conserved and a major contractile protein, which helps in force generation in the myocardium. Since the maximum number of mutations or SNPs are found to be in MYH7, we made an attempt to screen this gene in both HCM and DCM. Although we could find distinct SNPs/mutations in HCM and DCM, interestingly, certain common genetic variations were found in exons 7, 12, 19 and 20 of MYH7 gene in both the diseased phenotypes, where three of the genotypes were heterozygotes in HCM and all the genotypes were homozygotes in DCM. In the present study, we tried to hypothesize the underlying pathophysiological mechanism where the same SNPs/mutations in one gene would lead to two different diseased phenotypes.
There are various hypotheses explaining the underlying pathophysiological mechanism.
Dose effect
Dose effect of the mutant protein plays a role, where the heterozygous condition leads to hypertrophy and homozygous [Figure 4] condition leads to DCM phenotype resulting in heart failure. This is further supported by a previous study in which heterozygous mice expressing Arg403Gln α-MHC developed left ventricular hypertrophy as commonly seen in HCM, whereas homozygous mice developed progressive DCM leading to neonatal death.[7–9]
Phenotypic plasticity
Genotype-phenotype correlations can be best explained by phenotypic plasticity where mutations/variations in the same gene and even the same mutation/variation in a different background cause disparate phenotypes.[10] In the case of cardiomyopathies, such phenotypic plasticity was best illustrated by Dr. Seidman’s group, who reported that mutations in the MYH7 and TNNT2, encoding beta-myosin heavy chain and cardiac troponin T, respectively, could cause either HCM or DCM, the opposite ends of the spectrum of phenotypic responses of the heart to injury, stress or mutations.[11]
Alterations in myofibrillar Ca2+
Same variations leading to different diseased phenotypes can also be explained by the energy compromise and alterations in myofibrillar Ca2+ sensitivity pathways. Impaired myofibrillar Ca2+ sensitivity and force transmission of cytoskeletal protein variations could result in ventricular dilatation in DCM. On the other hand, sensitization to Ca2+ is likely to promote systolic function in HCM but impaires diastolic function due to an increase in cytosolic free Ca2+.[12,13] A study on Indians by Taranjit Singh et al.[14] with a similar approach, in which MYH7 mutations were found in both HCM and DCM, further supports our data and reconfirms the fact indicating that there is a wide genetic and phenotypic heterogeneity of HCM and DCM in our population similar to that reported for other ethnic populations such as Caucasians and Japanese.[15–20]
In addition to the above discussed reasons, environmental factors or modifier gene effect could also be playing a significant role in diverse phenotypes. Therefore, our results have reinforced the fact that the progression of hypertrophy to dilatation could finally lead to heart failure.
Genotype beta–phenotype correlation
Gross abnormalities in β-MYH7 might have a little pathogenic potential in the heterozygous state and minor abnormalities may have a causative role proving dominant negative effect of the mutated protein. Polymorphisms that are associated with the disease condition could be present in the regulatory region of the gene and potentially affect the expression of the genes and hence may be relevant to disease susceptibility. Heterozygous condition is associated with mild hypertrophy (with or without obstruction), relatively good prognosis, agedependent penetrance and longer life span, whereas the homozygous state is associated with an increased risk of the disease at an early age. The heterozygous SNPs are regulatory in nature acting as susceptiblity alleles. Early onset of the disease is predicted in homozygous state with predominant left ventricle and left auricle dilatations with Ejection Fraction (EF) (>40%) along with global hypokinesia in DCM. Hypertrophy in homozygous condition may progress to DCM condition and eventually leads to heart failure.
The clinical utility of such findings can be correlated to poor prognosis and sudden cardiac deaths in DCM compared to HCM, based on the dosage effect. A report of Reena et al.[6] on genetic testing of β-MYH7 in a large kindred helped in the prediction of asymptomatic, presymptomatic and symptomatic individuals based on the mutation R870H and identified the genotype phenotype correlation being established.[6] SNPs/ mutations have been detected in other sarcomeric genes in patients not harboring β-MYH7, revealing genetic heterogeneity of the condition.
Conclusion
In the present study, common genetic variations were found in MYH7 gene in both HCM and DCM samples This could be attributed to impaired energy compromise, or to dose effect of the mutant protein, or to even environmental factors/modifier gene effects responsible for an HCM to progress to a DCM phenotype in which both right and left ventricles are affected culminating into heart failure.
Acknowledgments
The contribution of all the authors in the preparation of the manuscript is acknowledged. The present study has been funded by DST and DBT.
Footnotes
Source of Support: The present study has been funded by DST and DBT.
Conflict of Interest: None declared.
References
- 1.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation. 1995;92:785–9. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 2.McKenna WJ, Behr ER. Hypertrophic cardiomyopathy: Management risk, stratification and prevention of sudden death. Heart. 2002;7:169–76. doi: 10.1136/heart.87.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Familial Hypertrophic Cardiomyopathy Mutation Database. Available from: http://www.angis.org.au/Databases/Heart/ [last cited on 2009 Jul 25]
- 4.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial Hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 5.Lahiri DK, Nurnberger JI., Jr A rapid non enzymatic method for preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991;19:5444. doi: 10.1093/nar/19.19.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanjore RR, Sikindlapuram AD, Calambur N, Thakkar B, Kerkar PG, Nallari P. Genotype-phenotype correlation of R870H mutation in hypertrophic cardiomyopathy. Clin Genet. 2006;69:434–6. doi: 10.1111/j.1399-0004.2006.00599.x. [DOI] [PubMed] [Google Scholar]
- 7.Fatkin D, Christe ME, Aristizabal O, McConnell BK, Srinivasan S, Schoen FJ, et al. Neonatal cardiomyopathy in mice homozygous for the Arg403Gln mutation in the alpha cardiac myosin heavy chain gene. J Clin Invest. 1999;103:147–53. doi: 10.1172/JCI4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, et al. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–4. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- 9.McConnell BK, Fatkin D, Semsarian C, Jones KA, Georgakopoulos D, Maguire CT, et al. Comparison of two murine models of familial hypertrophic ardiomyopathy. Circ Res. 2001;88:383–9. doi: 10.1161/01.res.88.4.383. [DOI] [PubMed] [Google Scholar]
- 10.Marian AJ. Phenotypic plasticity of sarcomeric protein mutations. J Am Coll Cardiol. 2007;49:25. doi: 10.1016/j.jacc.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–96. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 12.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, et al. An abnormal Ca2+ response in mutant sarcomere protein-mediated familial Hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–9. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopaties. Physiol Rev. 2002;82:945–80. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 14.Rai TS, Ahmad S, Bahl A, Ahuja M, Ahluwalia TS, Singh B, et al. Genotype phenotype correlations of cardiac beta-myosin heavy chain mutations in Indian patients with hypertrophic and dilated cardiomyopathy. Mol Cell Biochem. 2009;321:189–96. doi: 10.1007/s11010-008-9932-0. [DOI] [PubMed] [Google Scholar]
- 15.Havndrup O, Bundgaard H, Andersen PS, Larsen LA, Vuust J, Kjeldsen K, et al. The Val606Met mutation in the cardiac beta-myosin heavy chain gene in patients with familial hypertrophic cardiomyopathy is associated with a high risk of sudden death at young age. Am J Cardiol. 2001;87:1315–7. doi: 10.1016/s0002-9149(01)01532-6. [DOI] [PubMed] [Google Scholar]
- 16.Nakajima-Taniguchi C, Azuma J, Nagata S, Kishimoto T, Yamauchi-Takihara K. A missense mutation in the betamyosin heavy chain gene in a Japanese patient with hypertrophic cardiomyopathy. Jpn Circ J. 1995;59:833–7. doi: 10.1253/jcj.59.833. [DOI] [PubMed] [Google Scholar]
- 17.Mogensen J, Murphy RT, Kubo T, Bahl A, Moon JC, Klausen IC, et al. Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:2315–25. doi: 10.1016/j.jacc.2004.05.088. [DOI] [PubMed] [Google Scholar]
- 18.Liu SX, Hu SJ, Sun J, Wang J, Wang XT, Jiang Y, et al. Characteristics of the beta myosin heavy chain gene Ala26Val mutation in a Chinese family with hypertrophic cardiomyopathy. Eur J Intern Med. 2005;16:328–33. doi: 10.1016/j.ejim.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Nishi H, Kimura A, Harada H, Koga Y, Adachi K, Matsuyama K, et al. A myosin missense mutation, not a null allele, causes familial hypertrophic cardiomyopathy. Circulation. 1995;91:2911–5. doi: 10.1161/01.cir.91.12.2911. [DOI] [PubMed] [Google Scholar]
- 20.Van Driest SL, Ackerman MJ, Ommen SR, Shakur R, Will ML, Nishimura RA, et al. Prevalence and severity of “benign” mutations in the beta-myosin heavy chain, cardiac troponin T, and alpha-tropomyosin genes in hypertrophic cardiomyopathy. Circulation. 2002;106:3085–90. doi: 10.1161/01.cir.0000042675.59901.14. [DOI] [PubMed] [Google Scholar]