

Abstract
Bacterial pathogens deliver multiple effector proteins into eukaryotic cells to subvert host cellular processes and an emerging theme is the cooperation between different effectors. Here, we reveal that a fine balance exists between effectors that are delivered by enteropathogenic E. coli (EPEC) which, if perturbed can have marked consequences on the outcome of the infection. We show that absence of the EPEC effector Tir confers onto the bacterium a potent ability to destroy polarized intestinal epithelia through extensive host cell detachment. This process was dependent on the EPEC effectors EspG and EspG2 through their activation of the host cysteine protease calpain. EspG and EspG2 are shown to activate calpain during EPEC infection, which increases significantly in the absence of Tir – leading to rapid host cell loss and necrosis. These findings reveal a new function for EspG and EspG2 and show that Tir, independent of its bacterial ligand Intimin, is essential for maintaining the integrity of the epithelium during EPEC infection by keeping the destructive activity of EspG and EspG2 in check.
Introduction
Many of the world's most important diseases are caused by bacterial pathogens that deliver multiple effector proteins into eukaryotic host cells. Bacterial effector proteins are an evolutionary diverse family with a wide range of functions, enabling the bacterium to modulate many host cellular processes. Typically, individual effector proteins have a modular architecture with several functional domains or motifs, which confer multiple functions onto the effector. Emerging evidence suggests that effector proteins can cooperate with each other inside the host cell (Fu and Galan, 1999; McGhie et al., 2001; Kenny et al., 2002; Dean et al., 2006; Cain et al., 2008), giving the bacterial pathogen greater versatility in its ability to control host cellular events.
Enteropathogenic E. coli (EPEC) is a bacterial pathogen that causes severe watery diarrhoea, particularly in infants, and is responsible for a large proportion of infant deaths in the developing world (Chen and Frankel, 2005). Following ingestion, EPEC binds to the surface of the human small intestine where it delivers multiple effector proteins into small intestinal cells via a bacterial-encoded type III secretion system (T3SS). The best-characterized EPEC effector proteins are encoded in a genomic pathogenicity island called the locus of enterocyte effacement (LEE) which, in addition to the T3SS genes, carries at least six effector genes (map, espF, espG, espZ, tir, espH) and the eae gene that encodes the outer membrane protein Intimin (reviewed in Dean and Kenny, 2009). At least 14 effectors located outside the LEE region have been identified (Iguchi et al., 2009) although little is known about their function. Tir is the best-studied EPEC effector and is inserted into the host plasma membrane where it acts as a receptor for Intimin, mediating intimate bacterial attachment to the host cell (Kenny et al., 1997). Tir-Intimin interaction also induces actin-polymerization to form an actin-rich ‘pedestal’ beneath the bacterium. Several other Tir-dependent signalling events have been reported that all depend on Intimin (see Dean and Kenny, 2009). Other well-studied EPEC effectors are Map and EspF, which possess several overlapping functions as both disrupt tight junctions (TJs) (McNamara et al., 2001; Dean and Kenny, 2004), target mitochondria (Kenny and Jepson, 2000; Nougayrede and Donnenberg, 2004), efface intestinal microvilli (Dean et al., 2006) and inhibit the water transporter SGLT-1 (Dean et al., 2006) with EspF, but not Map, inhibiting phagocytosis (Quitard et al., 2006). In addition, Map has been shown to induce filopodia formation (Kenny et al., 2002) and promote cell invasion (Jepson et al., 2003) with the former event regulated by Tir through its interaction with Intimin. Much less is known about the functions of the other LEE effectorsalthough EspG and its non-LEE homologue EspG2 (also known as Orf3) have been shown to play a minor role in the disruption of epithelial barrier function (Tomson et al., 2005) and cause disruption of microtubules (Matsuzawa et al., 2004; Shaw et al., 2005a). EPEC has also been shown to cause an increase in levels of the general host cysteine protease calpain (Potter et al., 2003; Hardwidge et al., 2004), although the EPEC effectors that mediate calpain upregulation have not been determined. The calpains are a family of proteases that are found in many tissue types, including the ubiquitously expressed calpain I and II. Calpain cleaves a large number of host proteins, particularly proteins involved in host focal adhesions and cytoskeletal events and indeed calpain activity has been linked to cell migration and cell detachment (Glading et al., 2002).
It has long been established that EPEC can disrupt epithelial barrier function (Canil et al., 1993) – an essential feature of the intestinal epithelium that is maintained through apically located TJs between adjacent cells (Schneeberger and Lynch, 2004). TJs create an effective barrier to the movement of small molecules across the epithelium and this can be measured as trans-epithelial electrical resistance (TER). The disruption of TJs by EPEC causes a loss of TER and is dependent on the delivery of effector proteins into host cells (Guttman and Finlay, 2009). Previous studies have shown that several EPEC effectors contribute to epithelial barrier dysfunction including Map and EspF (McNamara et al., 2001; Dean and Kenny, 2004), with EspG and EspG2 playing a minor role (Tomson et al., 2005). While the outer membrane protein Intimin is essential for EPEC-mediated barrier disruption (Dean and Kenny, 2004), the contribution of its primary receptor Tir is unclear with previous studies reporting essential (Muza-Moons et al., 2003; Miyake et al., 2005) or non-essential (Dean and Kenny, 2004) roles. Recently, we deleted the map, espF, eae and tir genes in all combinations and found that the barrier-disrupting defects of all strains missing Map, EspF and/or Intimin were reversed by deleting the tir gene (P. Dean et al., unpublished). This supported previous findings that the tir mutant does indeed disrupt barrier function and also suggested that Tir possesses a novel role to prevent undefined effectors from causing barrier dysfunction.
Here, we show that disruption of epithelial barrier function by Tir-negative mutants involves an initial lag period, which provides an explanation of why previous reports have failed to observe the barrier-disrupting capacity of the tir mutant. Moreover, we show that Tir plays a critical role in maintaining the integrity of the epithelial monolayer as its absence promotes extensive detachment of host cells. This destructive activity was shown to be mediated by the redundant functions of two effectors – EspG and EspG2 – linked to the activity of the general host protease, calpain. EspG and EspG2 are shown to activate calpain during normal EPEC infection but their damaging effects on epithelial integrity are kept in-check by the Tir effector protein. This work highlights the delicate balance that has evolved between EPEC effectors within host cells and uncovers new functions for Tir and the EspG homologues.
Results
The tir mutant causes potent disruption of epithelial barrier function, independent of Intimin
While our previous work revealed that tir–minus mutants disrupt epithelial barrier function to a level observed with wild-type (WT) EPEC, examination of the kinetics revealed a subtle difference. Thus, while EPEC infection of TC-7 polarized epithelial cells led to a gradual loss in transepithelial resistance (TER; a well-defined indicator of barrier disruption), a 1–2 h lag period consistently occurred with the tir mutant before it induced a rapid response (Fig. 1A). A similar result, whereby loss of barrier function was preceded by a lag period with the tir mutant, was also obtained with colonic T84 cells (not shown). As previously reported (Dean and Kenny, 2004), mutants missing a functional effector delivery system (espA strain) or the Intimin outer membrane protein (eae strain) are defective at causing barrier dysfunction and instead cause a progressive increase in TER values (Fig. 1A). The TER increase observed with these strains was due to increased pH of the growth medium during bacterial infection (not shown). Our studies routinely use a multiplicity of infection (moi) of 200 bacteria per host cell (Dean and Kenny, 2004; Dean et al., 2006), but much lower moi were used in studies reporting that the tir mutant does not decrease TER (Muza-Moons et al., 2003; Miyake et al., 2005). We examined whether the discrepancy in data was related to the differences in moi between the studies. A range of moi were tested (not shown) and an moi as high as 1:50 reproduced the findings of other studies (Fig. 1B), with the tir mutant, unlike the parental EPEC strain, failing to disrupt barrier function over the examined 6–7 h infection period (Muza-Moons et al., 2003; Miyake et al., 2005). By extending the infection time to 11 h, the tir mutant induced a rapid loss in barrier function after the lag, which occurred at a rate over 6 times faster (P < 0.0001) than the decrease with WT EPEC (Fig. 1C), while a TER decrease was not observed with the espA mutant over a similar 11 h infection period (Fig. 1C). Thus, these data clearly demonstrate that the tir mutant disrupts barrier function and shows that at lower bacterial doses, the lag period associated with the tir mutant persists for longer times, obscuring the strain's barrier-disrupting capacity.
Fig. 1.
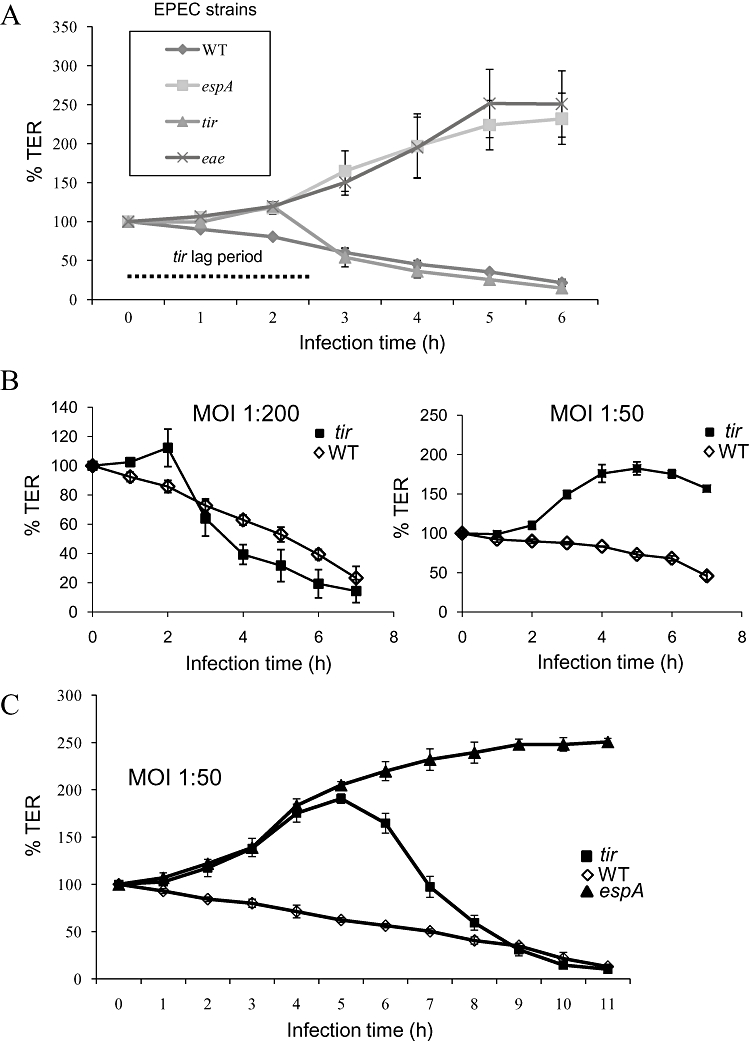
A lag phase persists in the disruption of barrier function by the tir mutant that is dictated by the multiplicity of infection (MOI). A. Polarized intestinal cells were infected with the indicated EPEC strains and the transepithelial resistance (TER) was followed. The dotted line indicates the lag period for the tir mutant where TER increases. The eae (Intimin) mutant cannot disrupt TER and was similar to that seen with the espA mutant (which cannot deliver effectors into the host cell). Points represent mean ± SEM, n = 3. B. Polarized intestinal cells were infected with wild-type (WT) EPEC or the tir mutant at the moi indicated and the transepithelial resistance (TER) was followed. The lag phase seen with the tir mutant at the moi of 1:200 (left) persisted to the end of the 7 h experiment at the moi of 1:50 (right). C. Increased infection times with the moi of 1:50 shows the tir mutant can cause a rapid TER decrease. The rate of TER decrease for the tir mutant following the lag period (35.11 h−1 ± 3.78 h−1) was significantly greater (P < 0.0001) than that of the WT (5.22 h−1 ± 1.52) while the espA mutant displayed no TER decrease. Data points show mean ± SEM, n = 3.
The absence of Tir causes destruction of the epithelium during infection
Given the different kinetics of TER decrease between the tir mutant and WT EPEC, we postulated that a different mechanism of barrier dysfunction may be involved. Microscopy analysis of infected cells revealed large voids in tir-infected monolayers by 4 h post infection that was suggestive of cell detachment (Fig. 2A) while WT EPEC-infected monolayers remained relatively intact (Fig. 2A). Indeed, by 7 h post infection, less than 20% of the monolayer remained attached following tir mutant infections, unlike ∼90% coverage for monolayers infected with WT EPEC (Fig. 2B). Supporting these data, a corresponding (∼fivefold) increase of cellular debris (measured by protein determination) was evident in the supernatant of monolayers infected with the tir versus the WT EPEC strain (Fig. 2C, P < 0.001), suggesting that the epithelium was being rapidly destroyed during tir mutant infections. Examination of host cell death prior to this extensive cell loss (4 h post infection) revealed that WT EPEC infection was linked to low levels of necrosis (assessed by trypan blue) and apoptosis (assessed by caspase-9 cleavage). However, the tir mutant infection resulted in a ∼twofold significant increase in necrosis (Fig. 2D, P < 0.002) without impacting on the low level of apoptosis (Fig. 2E, P = 0.4). These data demonstrate that the rapid disruption of epithelial barrier function observed with the tir mutant is strongly correlated to detachment of host cells and linked to a necrotic response, suggesting that the Tir effector normally suppresses these events during EPEC infection.
Fig. 2.
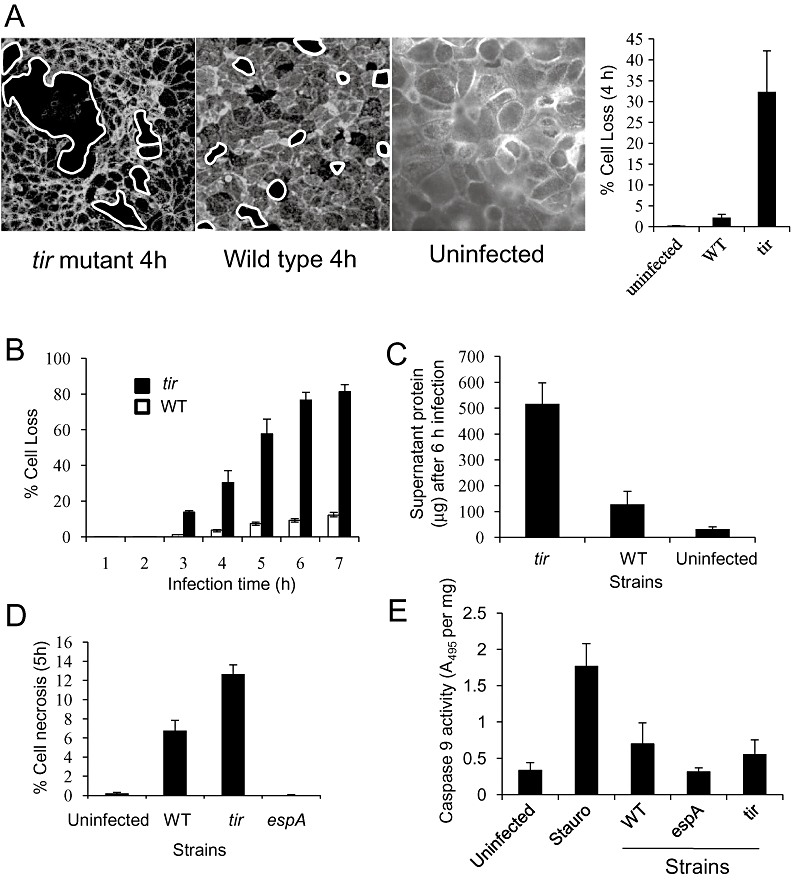
The absence of Tir causes a loss of epithelial monolayer integrity. A. Microscopy of polarized intestinal cells stained with phalloidin following a 4 h infection with the tir mutant or wild-type (WT) EPEC shows that tir-infected cells exhibit large cell-free voids in the monolayer, unlike that of WT. Percentage cell loss/unit area at 4 h post infection is shown in the right graph. B. Kinetics of cell loss during a 7 h infection with WT EPEC or the tir mutant for seven fields of view at ×63 per experiment. C. Cellular debris assessed by protein determination in the supernatant above the monolayer following a 6 h infection. D. Level of necrosis in monolayers infected with indicated strains determined by trypan blue staining (6 fields of view at ×40, n = 3). E. Levels of apoptosis in monolayers after 6 h infection with indicated EPEC strains assessed by caspase-9 cleavage activity per mg protein. Staurosporine (Stauro) was used as a positive control for apoptosis. For all graphs, data points represent mean ± SEM, n = 3.
Pre-delivery of Tir is protective against the destructive activity of tir-deficient strains
During EPEC infection, Map, EspF and Intimin (eae) collectively mediate barrier dysfunction (Dean and Kenny, 2004) and as such a map/espF/eae triple mutant is unable to disrupt TER (Fig. 3). However, we have found that a quadruple mutant (deleted for these three genes and tir) behaves like a tir mutant in that it causes rapid disruption of TER after an initial lag period that is associated with cell detachment (not shown; but see Fig. 3). This suggests that in the absence of Tir, disruption of TER does not require EPEC's prominent barrier-disrupting proteins Map, EspF or Intimin. Therefore, we utilized the map/espF/eae triple mutant to pre-deliver Tir into host cells to investigate whether Tir delivered by this strain would prevent the quadruple mutant from decreasing TER. Epithelial monolayers were infected for 1 h with the map/espF/eae triple mutant or a strain (cfm-14; TTSS-) unable to deliver Tir, after which the bacteria were killed with antibiotics. Subsequent infection by the quadruple mutant revealed that prior infection with the map/espF/eae strain prevented barrier dysfunction while cfm-14 did not, suggesting that pre-delivery of Tir was essential.
Fig. 3.
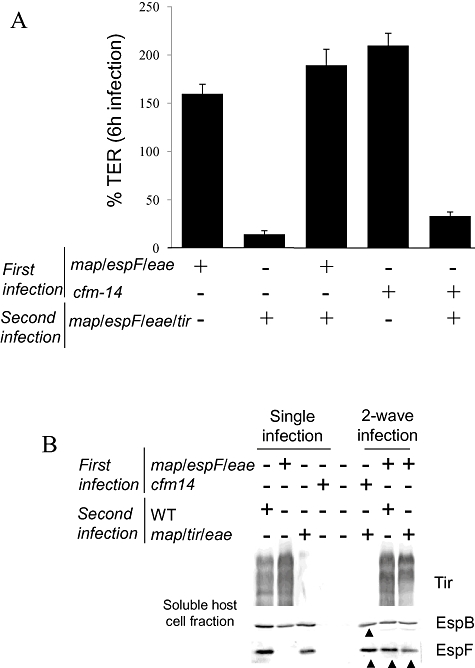
Pre-delivery of Tir into host cells protects the epithelium against the destructive activity of tir-minus mutants. A. Transepithelial resistance (TER) of polarized intestinal cells after 6 h single- or co-infection with indicated strains. For co-infections, monolayers were infected for 1 h with the triple map/espF/eae mutant (to deliver Tir into host cells) or cfm-14 (a type three secretion mutant that cannot deliver Tir), which were then killed with antibiotics. Monolayers were then infected with the quadruple map/espF/eae/tir mutant for 6 h. Data points represent mean ± SEM, n = 3. B. Western blot of infected cell lysate from cells infected with first wave and second wave infections. Black arrow heads show that the first wave of infections does not affect the delivery of effectors by the strains used in the second wave of infections. A map/tir/eae triple mutant was used in place of the map/espF/eae/tir quadruple mutant for the secondary infections to demonstrate that delivery of EspF is unaffected and was similar to single- or co-infections with WT EPEC.
As it has been reported that infection of non-polarized cells with effector-delivery competent strains inhibits the delivery of effectors by subsequent infections (Mills et al., 2008), we investigated whether our above findings were due to a defect in effector delivery by the quadruple mutant during the secondary infection. Detection of similar amounts of EspF and EspB within cells (Fig. 3B) pre-infected with the map/espF/eae followed by infection with either WT EPEC or a map/eae/tir mutant confirms that the second wave of infections are not impeded in their ability to deliver effector proteins. In addition, we found that a secondary infection with WT EPEC following pre-infection with the map/eae/tir triple mutant caused TER to decrease (not shown), further supporting the premise that effector delivery is not impeded in polarized cells. Taken together, the data show that two-wave infections work efficiently with polarized cells unlike that reported for non-polarized cells. This validates our interpretation of the above finding – that the rapid disruption of TER by the tir mutant that is linked to cell detachment and necrosis can be prevented by pre-delivery of Tir into host cells.
Barrier dysfunction by the tir mutant occurs independently of actin rearrangements, unlike that of wild-type EPEC
We previously noted that disruption of TER by EPEC can be inhibited by pre-treating monolayers with the inhibitor jasplakinolide, which stabilizes actin filaments (not shown, but see Fig. 4). As the kinetics of TER decrease for WT EPEC and the tir mutant were different and were not dependent on Map, EspF and Intimin in the latter strain, we investigated whether the cellular mechanism was different for the two strains. Pre-treatment of cells with jasplakinolide prevented WT EPEC from decreasing TER (P < 0.0001) but had little, if any impact on the ability of the tir mutant to disrupt barrier function (P = 0.472; Fig. 4). Similar results were obtained with the actin-destabilizing agent cytochalasin D (not shown but see Fig. 6A). These data suggest that barrier disruption by WT EPEC is dependent on effector-driven actin rearrangements and also demonstrates that a different cellular process, independent of actin rearrangements, is responsible for TER decrease seen with the tir mutant.
Fig. 4.
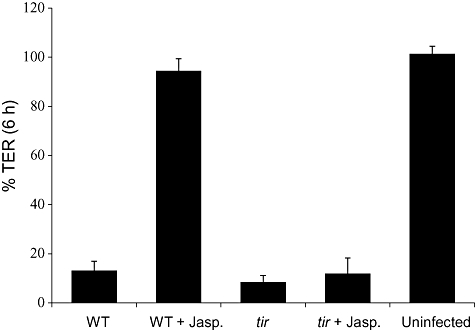
Loss of epithelial barrier function is dependent on actin rearrangements by wild-type (WT) EPEC but not the tir mutant. Transepithelial resistance (TER) of polarized intestinal cells after 6 h infection with WT EPEC or the tir mutant in the presence or absence of the actin stabilizing agent jasplakinolide. All data points are means ± SEM, n = 3. An moi of 1:200 was used for all experiments.
Fig. 6.
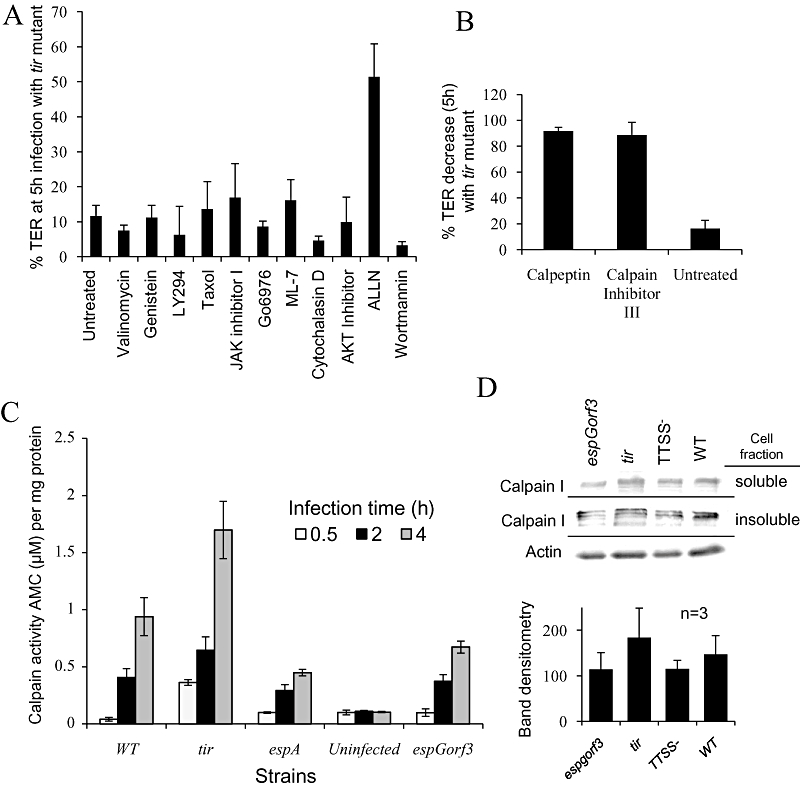
EspG and EspG2 over-activate calpain in the tir mutant. A. Inhibitor screening to identify the cellular pathway(s) involved in the potent tir mutant disruption of barrier function. Polarized intestinal cells were infected for 5 h with the tir mutant and transepithelial resistance (TER) was recorded. ALLN, a calpain inhibitor was the only inhibitor in this screen to significantly prevent the TER decrease. B. Two other calpain inhibitors calpeptin and calpain inhibitor III also prevented the TER decrease with the tir mutant. C. Calpain activity in cells infected with various EPEC strains was measured by the release of AMC from the calpain substrate Suc-LLVY-AMC per mg of cellular protein. All data points represent mean ± SEM, n = 3. D. Western blot of calpain I in host cells infected with the indicated strains. Calpain I bands were quantified by densitometry from three separate experiments. Bars show mean ± SEM.
EspG and EspG2 mediate epithelial monolayer destruction in the absence of Tir
The ability of the map/espF/eae/tir quadruple mutant to cause detachment-linked TER decreases ruled out roles for Map, EspF and Intimin in the process. To investigate which effectors were involved, we examined the Δcore mutant that is missing tir, map, espF and eae (Intimin) genes and is also defective for the expression or secretion of additional effectors including EspH, EspZ, NleH, NleF and NleA (Ruchaud-Sparagano et al., 2007). The Δcore mutant mimicked the tir mutant in its ability to cause TER decrease (Fig. 5A, P = 0.7), suggesting the above described effectors were not involved. By contrast, studies with the espG/orf3/Δcore mutant (Ruchaud-Sparagano et al., 2007) implicated a critical role for the EspG homologues as this mutant was defective in causing a TER decrease (Fig. 5A). Subsequent involvement of EspG and EspG2 was supported by finding that an espG/orf3/tir triple mutant failed to decrease TER (Fig. 5B) unless complemented with plasmids encoding the espG or orf3 genes (Fig. 5B). Interestingly, the inability of the espG/orf3/tir triple mutant (which carries intact map, espF and eae genes) to disrupt barrier function implicates a critical role for Tir in the well-defined Map/EspF/Intimin-dependent barrier disruptive pathway (Dean and Kenny, 2004). This premise was supported by restoring the strain's barrier-disrupting capacity by introducing a Tir-expressing plasmid (Fig. 5B). Thus, the data show that Tir (independent of Intimin) normally suppresses an EspG/EspG2 activity that leads to loss of TER by extensive cell detachment and also demonstrates that Tir (along with Intimin) plays a key, but previously cryptic, role in regulating the epithelial barrier disrupting functions of Map and EspF.
Fig. 5.
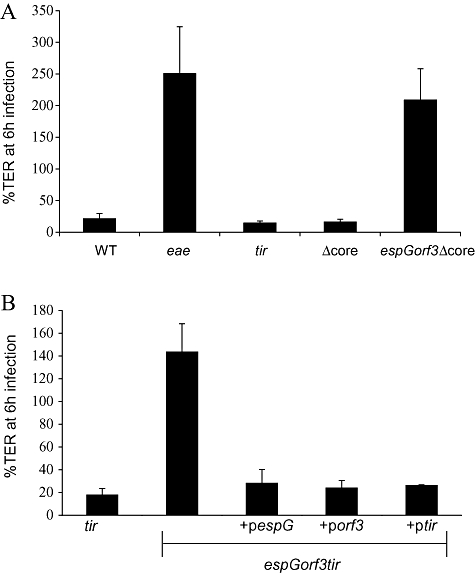
EspG and EspG2 are responsible for the loss of epithelial integrity caused by the absence of Tir. Transepithelial resistance (TER) of polarized intestinal cells infected for 6 h with indicated strains. A. Deletion of espG and orf3 (encodes EspG2), but not other effector genes prevents the TER decrease associated with the tir mutant background. Δcore is unable to express or deliver the effectors Map, EspF, Tir, EspH, EspZ, NleA, NleH1, NleH2 and also absent for Intimin. espG/orf3/Δcore is absent for the effectors EspG and EspG2 in the Δcore mutant background. B. Complementation of the espG/orf3/tir triple mutant with plasmids carrying either espG, orf3 (encodes EspG2) or tir. All infections were performed at moi of 1:200. Results show mean ± SEM, n = 3.
EspG and EspG2's destructive activity is inhibited by calpain inhibitors
A reported function of the EspG homologues is linked to their destabilization of microtubules to release the RhoA-specific GEF (GEF/H1) that activates the RhoA-Rock signalling pathway to promote stress fibre formation (Matsuzawa et al., 2004). To investigate the role of this pathway in the TER decrease caused by the tir mutant, cells were pre-treated with the ROCK inhibitor Y27632. As this treatment had no impact on the process (not shown; but see below), we attempted to identify the signalling pathway(s) mediating the EspG destructive effect. An inhibitor screen was performed using a variety of broad spectrum or specific inhibitors (Table 1) based on signalling pathways implicated in EPEC subversive events. Consistent with the finding using the ROCK inhibitor, taxol – a microtubule stabilizing agent that prevents EspG-related RhoA activation (Matsuzawa et al., 2004; Tomson et al., 2005) – did not interfere with tir mutant TER decrease (Fig. 6A, P = 0.7). Thus, this suggests that the EspG-dependent activity linked to cell detachment is not related to the destabilization of microtubules or the triggering of the GEF/H1-RhoA-Rock signalling pathway. Strikingly, only one of the 12 interrogated inhibitors (ALLN; a non-specific cysteinyl protease inhibitor) prevented tir mutant-mediated TER decreases (Fig. 6A, P < 0.003). As a target of ALLN is ubiquitously expressed calpain – a Ca2+-dependent protease that regulates cytoskeletal anchorage complexes (Lebart and Benyamin, 2006) – we investigated this linkage using two calpain inhibitors (calpeptin and calpain inhibitor III). Both inhibitors were as effective as ALLN in preventing tir mutant-associated TER decreases (Fig. 6B), suggesting that calpain activity was involved in the TER decrease linked to monolayer destruction by the tir mutant. This also suggested that EspG and EspG2 may be activating calpain to cause host cell detachment in the absence of Tir. Similar results, in which EspG/Orf3 induce host cell detachment that is mediated through a calpain dependent pathway, were also obtained with HeLa cells (P. Dean et al., unpublished).
Table 1.
Details of inhibitors used in the study.
| Inhibitor | Working concentration for polarized TC-7 cells | Inhibitor site of action |
|---|---|---|
| Jasplakinolide | 1 µM | Inhibits F-actin depolymerization |
| Cytochalasin D | 5 µM | Inhibits F-actin polymerization |
| Wortmannin | 1 µM | PI 3-kinase inhibitor |
| Valinomycin | 1 µM | Disrupts mitochondrial membrane potential |
| AKT Inhibitor I | 30 µM | AKT Inhibitor |
| Genistein | 300 µM | Protein tyrosine kinase inhibitor |
| Go6976 | 5 µM | PKC inhibitor |
| ALLN | 10 µM | Inhibits calpain I and II activity |
| ML-7 | 20 µM | Myosin light chain kinase inhibitor |
| Taxol | 50 µM | Stabilizes microtubules |
| Staurosporine | 1 µM | Broad spectrum protein kinase inhibitor, induces apoptosis |
| Calpeptin | 50 µM | Inhibits calpain I and II activity |
| Calpain Inhibitor III | 50 µM | Inhibits calpain I and II activity |
| LY294 | 25 µM | PI 3-kinase inhibitor |
| Y-27632 | 50 µM | ROCK inhibitor |
| JAK inhibitor I | 16 µM | Inhibits Janus protein tyrosine kinases |
| Cyclohexamide | 200 µg ml−1 | Inhibits protein translation |
EspG and EspG2 over-activate host calpain activity in the absence of Tir
To explore the hypothesis that the EspG homologues promote calpain activation and that Tir may regulate this event, we examined cellular calpain activity. These studies used the synthetic calpain substrate Suc-LLVY-AMC at 4 h post infection (i.e. prior to extensive cell loss; Fig. 2). Figure 6C shows limited calpain activity in uninfected cells with the espA mutant causing a near-significant increase by 4 h post infection (P = 0.07; Fig. 1). Interestingly, while WT EPEC infection significantly increased calpain activity relative to espA-infected cells (P < 0.001), there was no significant difference between activity in espG/orf3- and espA-infected cells (P = 0.3). By contrast, cells infected with the tir mutant exhibited a significant (∼twofold) increase in calpain activity compared with WT EPEC (P < 0.001; Fig. 6C). This clearly shows that EPEC induce calpain activation in polarized epithelial cells in a T3SS-dependent manner through the effectors EspG and EspG2 and that this activation is modulated by Tir.
As a previous report has shown an increase in calpain in EPEC-infected cells (Hardwidge et al., 2004), we examined the levels of calpain in infected host cells to determine whether EspG/EspG2 induced a specific upregulation of calpain. Although the levels of the calpain I isoform were higher in the soluble and insoluble fractions of tir- and WT-infected cells as assessed by Western blot (Fig. 6D), their levels were not significantly different to the TTSS- negative control (P = 0.2 and 0.31 respectively), unlike the significant difference shown in the calpain activity assay (Fig. 6C). Similar results were obtained for calpain II (ubiquitously expressed) and calpain V (found in intestinal tissue) (not shown), suggesting that upregulation of calpain levels in the tir mutant may not significantly contribute to the increased activation of calpain. Collectively, this study demonstrates that EspG/EspG2 promotes calpain activation during normal EPEC infection, which is kept under tight control by Tir to limit the cell detachment effects mediated by calpain.
Discussion
While it is well established that bacterial effectors co-delivered into host cells can possess overlapping functions, an emerging property is their ability to cooperate with each other. Examples of this feature relate to effector-driven cytoskeletal rearrangements that are downregulated through the antagonistic action of co-delivered effectors (Fu and Galan, 1999; Kenny et al., 2002; Cain et al., 2008). However, these studies were performed on cell types that lack several key features of cells that are normally targeted by the bacteria in vivo. Here, we have used a disease-relevant small intestinal model to show that EPEC regulates the activity of the general host protease calpain through the cooperation of three of its effectors to ensure that extensive detachment of host cells is minimized during infection.
For over a decade it has been known that EPEC can disrupt epithelial barrier function by a mechanism dependent on the outer membrane protein Intimin (Canil et al., 1993). Further studies have determined that the disruptive process involves the activities of multiple effectors including prominent roles for Map and EspF and minor roles for the two EspG homologues (McNamara et al., 2001; Dean and Kenny, 2004; Tomson et al., 2005). Unexpectedly, a mutant missing the gene encoding the plasma membrane-located Tir effector, which is the main receptor for Intimin, was reported to be unable to disrupt barrier function (Muza-Moons et al., 2003; Miyake et al., 2005), while a separate study suggested otherwise (Dean and Kenny, 2004). In this study, we resolve this discrepancy by showing that although Tir has a critical role in EPEC's main barrier disrupting pathway (Map/EspF/Intimin-mediated), its contribution until now was cryptic because the absence of Tir leads to loss of barrier function by a completely separate mechanism. We show that previous studies which suggest that a tir mutant does not disrupt barrier function (Muza-Moons et al., 2003; Miyake et al., 2005) are due to modified infection protocols that resulted in low moi that unknowingly extended a lag period associated with the tir mutant, thereby obscuring the strain's barrier disrupting activity. These modified infection protocols used an moi as low as 1:3 (Miyake et al., 2005) or washed the epithelial cells after 1 h infection, thereby artificially reducing the moi (Muza-Moons et al., 2003). Our findings raise concerns on the interpretation of studies (Muza-Moons et al., 2003; Miyake et al., 2005; Tomson et al., 2005; Gill et al., 2007) with these modified infection protocols using mutants that exhibit lag periods in disrupting epithelial barrier function such as tir (this study) and espG/orf3 mutants (Matsuzawa et al., 2005; Tomson et al., 2005; P. Dean et al., unpublished).
In this study, we show that the main barrier-disrupting proteins of EPEC are dependent on both Intimin and Tir to induce barrier dysfunction. This premise was derived from the finding that the espG/orf3/tir triple mutant is completely defective at causing a loss in TER much like the eae mutant, despite the presence of EspF and Map in these strains. This was further supported by finding that the ability of the espG/orf3/tir mutant to disrupt TER is restored when the strain is complemented with a Tir-encoding plasmid. Previously, this crucial role of Tir in barrier dysfunction was obscured in the single tir mutant (Dean and Kenny, 2004) because of the unknown destructive effects of EspG/G2 in this mutant background. Taken together, this study shows that EPEC-mediated barrier disruption requires Intimin (Canil et al., 1993; Dean and Kenny, 2004), Tir (this study) and the cooperative actions of Map and EspF (McNamara et al., 2001; Dean and Kenny, 2004). The finding that cytoskeletal inhibitors prevented WT EPEC but not the tir mutant from disrupting barrier function suggested that the aforementioned barrier-disrupting proteins function by altering the actin cytoskeleton. This fits well with the recently reported role of Map as a guanine nucleotide-exchange factor for Cdc42 – a regulator of cytoskeletal rearrangements (Huang et al., 2009), EspF as a direct and indirect activator of N-WASP to induce actin nucleation (Alto et al., 2007; Cheng et al., 2008; Peralta-Ramirez et al., 2008; Sallee et al., 2008) and Tir/Intimin in inducing actin rearrangements (Kenny et al., 1997). While studies using pharmacological agents have also implicated roles for host kinases in barrier dysfunction (Guttman and Finlay, 2009), it is likely that these drugs act by preventing or counter-acting cytoskeletal rearrangements triggered by these effectors. Recently, another EPEC effector NleA was reported to disrupt epithelial barrier function (Thanabalasuriar et al., 2010) and the work presented here suggests that NleA, like Map and EspF, cannot function without Intimin or Tir being present as shown by the complete defectiveness of the eae (Intimin) or espG/orf3/tir mutants.
Tir's role as a central regulatory effector must be given serious consideration in light of past reports and the data presented here. Tir has been shown to downregulate Map-induced filopodia formation (Kenny et al., 2002), modulate EspF's ability to efface microvilli (P. Dean et al., unpublished), is implicated in NleA-mediated disruption of barrier function (as described) and is shown to suppress EspG/G2's ability to activate calpain. Thus, in addition to its crucial role in intimate attachment (Marches et al., 2000), Tir appears to play an additionally important role in regulating the functions of multiple effectors involved in several patho-physiological events.
The tir mutant disrupted barrier function up to ∼6 times faster than the parental WT strain, suggesting that it employed an alternative cellular mechanism. Although all work in this study was performed with the Caco-2 derived TC-7 cells, we found a similar result with colonic T84 cells (not shown). Morphological and inhibitor data further supported the idea that the tir-negative mutant disrupts barrier function by a distinct pathway. Thus, whereas EPEC-mediated TER decreases are linked with TJ disassembly (Simonovic et al., 2000; Dean and Kenny, 2004; Guttman and Finlay, 2009), the tir mutant was linked with extensive cell detachment with up to ∼80% of the monolayer destroyed, compared with just ∼10% for WT EPEC. In addition, we found that the TJ complexes during tir mutant infection were intact in the host cells that remaining attached (not shown), unlike that with WT EPEC (Dean and Kenny, 2004). Thus, cell detachment provided an explanation for the rapid loss of TER associated with the tir mutant as monolayer confluency is directly linked with TER. The cellular mechanism that was responsible for the destructive phenotype associated with the tir mutant was dependent on the activation of host calpains as the TER decrease caused by the tir mutant was (i) prevented by inhibitors whose primary target are the Ca2+-dependent calpains and (ii) calpain activity was significantly higher in the tir mutant infected cells. The finding that the cell-detaching effects of the tir mutant were dependent on EspG and EspG2 and that it correlated with the increase in calpain activity, which was also dependent on these two effectors, strongly supports the contention that Tir acts to suppress EspG/EspG2-induced calpain activity. Interestingly, the Tir-EspG/EspG2 calpain pathway was not exclusive to polarized intestinal cells as we found a similar finding with HeLa cells (not shown). Recent structural data suggest that EspG family members might act as a scaffold for a papain-like cysteine protease with the N-terminus noted to be topologically similar to cysteine protease inhibitors (Davis et al., 2008). This provides a mechanism whereby EspG may recruit activated calpain but with specific calpain activity regulated through EspG's N-terminal domain, dependent on an unknown function of Tir.
Previously, EPEC was reported to cause a threefold to fivefold increase in the levels of calpain in intestinal cells (Hardwidge et al., 2004). We were unable to detect a similar increase in protein levels for three common calpain isoforms, although we did observe subtle differences between the levels of calpain in cells infected with WT EPEC, espG/orf3 and the tir mutant. However, our finding that pre-treating host cells with cyclohexamide (which prevents protein translation) did not compromise tir mutant barrier disruption (not shown) suggests that the upregulation of calpain levels is not essential for the tir mutant to activate calpain. Further studies are underway to elucidate the mechanism by which Tir and EspG modulate host calpain activity and the contribution of the different calpain isoforms to the EPEC disease process.
Integrating the inhibitor, mutant and calpain activity data with published work offers a model whereby EPEC delivers the multi-functional EspG effectors to subvert various cellular processes including microtubule destabilization (Matsuzawa et al., 2005; Tomson et al., 2005), stress fibre formation (Matsuzawa et al., 2004), increasing paracellular permeability (Matsuzawa et al., 2005; Tomson et al., 2005), co-transporter inhibition (Gill et al., 2007) and inducing calpain activation (this study). We speculate that EPEC delivers sufficient levels of the EspG/EspG2 to modulate these cellular processes with a need for Tir to control or dampen the induction of calpain activity to prevent cell detachment. Calpain cleaves a number of host proteins, particularly those found in adhesion complexes and their cleavage results in disassembly of focal adhesions and cell rounding (Glading et al., 2002; Franco and Huttenlocher, 2005), thus the linkage between calpain and cell detachment is well established. Prevention of host cell detachment and limiting cell turnover is an important strategy of bacterial pathogens as host intestinal enterocytes are continually being replaced, preventing many harmful invaders from becoming established. Bacterial effector proteins have been identified that inhibit host cell turnover, including Cif of A/E pathogens, which blocks cell division (Marches et al., 2003), or the recently reported Shigella effector OspG, which prevents cell detachment by reinforcing focal adhesions in polarized epithelial cells (Kim et al., 2005). Our study suggests that the EPEC Tir can be included into the family of effectors that inhibit cell detachment.
What is the role of calpain activation during WT EPEC infection? Although EPEC causes detachment of non-polarized cells in a T3SS-dependent manner (Shifrin et al., 2002), this is not a prominent feature associated with EPEC-infected polarized epithelia, suggesting that the small increases in calpain activity induced by EspG/EspG2 in WT EPEC have other roles. It has been suggested that calpain plays a role in microvilli effacement by EPEC (Potter et al., 2003), but we and others find no role for EspG or EspG2 in effacement (P. Dean et al., unpublished; Shaw et al., 2005b). Equally, calpain has been implicated as a pro- and anti-apoptotic protein, but the low level of apoptotic cells induced by EPEC was similar for both the WT and the tir mutant. A more likely role relates to the cleavage of tight junctional proteins such as occludin (Zhu et al., 2006; Chun and Prince, 2009), which may explain EspG's reported role in selectively opening TJs to small molecules (Matsuzawa et al., 2005). However, the finding that Map and EspF cause a loss of the tight junctional protein occludin in infected cells (Dean and Kenny, 2004) and that an espGorf3 double mutant causes significant tight junctional damage (Matsuzawa et al., 2005; Tomson et al., 2005) implies that EPEC interaction with TJs is a complex process with overlapping effector functions. Whatever the role, it is clear that the cleavage of host proteins by calpain is not very promiscuous during EPEC infection but is under tight control as cell detachment is not a common event, suggesting that the main target of calpain, the adhesion complexes, remains relatively intact during infection.
There is a growing consensus that interplay between bacterial effector proteins is highly complex and well-regulated process. While there are only a few examples of regulatory mechanisms it is clear that the functions of effectors do not proceed without restriction but are tight controlled by the bacterium. This makes sense as most effectors are multi-functional and have subtle effects on the host cell, which if left unchecked may become too deleterious, loosening the pathogen's grip on the host cell. Future studies will determine the unanswered questions that remain including the nature of Tir suppressive function on EspG/G2 activity, the mechanism of calpain induction by EspG/G2 and the role that calpain activity plays during EPEC infection.
Experimental procedures
Bacterial strains and plasmids
Enteropathogenic E. coli strains used in this study were WT EPEC (E2348/69 O127:H6) (Levine et al., 1985), tir (Kenny et al., 1997), espA (Kenny et al., 1996), espG/orf3 (Elliott et al., 2001), cfm-14 (Donnenberg et al., 1990), the eae (Intimin) mutant CVD206 (Donnenberg and Kaper, 1991), the quadruple mutant map/espF/eae/tir (Quitard et al., 2006) and the espG/orf3/Δcore mutant (Ruchaud-Sparagano et al., 2007). The Δcore mutant was made as per the espG/orf3/Δcore mutant (Ruchaud-Sparagano et al., 2007) using EPEC rather than the espG/orf3 double mutant as a recipient for allelic exchange. The map/espF/eae triple mutant was generated via allelic exchange using the suicide vector pCVD442 (Donnenberg and Kaper, 1991) as described previously (Kenny et al., 1997). Briefly, the map mutant was used as a recipient for pCVD442-ΔespF to generate the map/espF double mutant, which was used as a recipient for pCVD442-Δeae generating the map/espF/eae triple mutant. The triple mutant espG/orf3/tir was made by allelic exchange using the espG/orf3 mutant as a recipient for the suicide vector pCVD442-Δtir. Disruption of targeted gene was verified by PCR with Western blot analysis confirming that the strain had a functional T3SS and thus could deliver EspB into host cells. The espG- (Matsuzawa et al., 2005) and tir-encoding (Kenny, 2001) plasmids were selected with carbenicilin (100 µg ml−1 final concentration) or chloramphenicol (25 µg ml−1 final concentration) respectively.
Cell culture and infection assays
Bacterial culture and infection assays were as described previously (Dean and Kenny, 2004). The TC-7 cell line (Chantret et al., 1994), a subclone of the heterogeneous intestinal cell line Caco-2, was used for all assays in this study. TC-7 cells provide a homogeneous population of enterocytes with a regular brush border and rapid doubling time (Chantret et al., 1994). This well-characterized cell line was used as it mimicked host–pathogen events better than the parent Caco-2 cell line with less inherent variability. TC-7 cells were used 12–15 days post confluence and cultured as previously described for Caco-2 cells (Dean and Kenny, 2004) in transwells (Corning) on membrane filters with 0.4 µM sized pore. Infection assays were performed as previously reported for Caco-2 (Dean and Kenny, 2004). Briefly, the OD600 of overnight bacterial cultures was measured and suspensions were diluted in Dulbecco's modified Eagle's medium (DMEM) and grown in this medium (at 37°C in 5% CO2) for 3 h. The OD600 of the suspensions was measured and diluted accordingly to the indicated moi (typically 1:200 for most experiments) and added to the apical surface of TC-7 monolayers on membrane filters. TER was measured at selected time points as previously described (Dean and Kenny, 2004) with the TER values for the TC-7 cell line consistently between 200 and 300 Ω cm−2. The term TER corresponds to the electrical resistance of the epithelium plus that of the membrane filter and solutions with TER fluctuation not providing mechanistic insight on the nature of any change.
Western blot
Infected cells were washed twice in PBS and lysed in 1% (v/v) Triton-X 100 for 5 min prior to centrifugation at 12 000 g. The ‘soluble’ supernatant containing the cytoplasmic and membrane fractions was removed and the ‘insoluble’ cell pellet was washed in PBS. Both fractions were subject to SDS-PAGE and Western blot as described (Dean and Kenny, 2004). Antibodies used were calpain-µ (Sigma), calpain-V (Biovision), calpain-I (Chemicon) and actin (Sigma). To quantify the intensity of the bands, densitometrical analysis from at three separate blots was performed using the software program Image J.
Two-wave infections
TC-7 cells (12–15 days post confluence) were left uninfected or infected 1 h with the map/espF/eae or cfm-14 mutants (moi was 1:200). After infection the cells were washed three times in DMEM and the bacteria killed (gentamycin 100 µg ml−1 and penicillin/streptomycin 100 units ml−1 final concentration in DMEM for 30 min). Cells were washed 3 times in DMEM and incubated with indicated strains using the normal infection protocol with TER monitored for a further 6 h.
Cell death and detachment assays
The level of infection-induced cell detachment was determined by centrifuging host cells within the supernatant at 500 g for 10 min, with two separate PBS washes. This isolated the majority of host cells away from the bacteria, which remained in the supernatant. The cell pellet was lysed with 0.1 M NaOH in PBS and the protein content determined using Bradford reagent (Sigma) assaying at 495 nm. The level of cell necrosis of infected monolayers was determined by incubating with 0.2% (w/v) trypan blue in PBS for 5 min, washing in PBS and visualizing cells using a conventional light microscope. The number of blue cells was recorded per field of view using a ×40 objective lens examining six fields of view per experiment. The level of apoptosis was determined by assaying caspase-9 cleavage using the substrate LEHD-pNA. The liberation of pNA was detected spectrophotometry at 405 nm according to manufacturer's instructions (Calbiochem 218824). As a positive control, cells were incubated in DMEM containing staurosporine (1 µM) to induce apoptosis (Bertrand et al., 1994).
Confocal microscopy
At select times post infection, the TC-7 monolayers were washed gently in PBS and fixed in 2.5% (w/v) paraformaldehyde for 15 min. Cells were then washed and incubated in a solution of phalloidin to stain F-actin as previously described (Dean and Kenny, 2004). Cells were visualized using a Leica SP2 confocal microscope by making a series of optical sections along the z-axis and reconstructing the monolayer as a composite projection. The level of cell detachment was determined using Leica confocal software by calculating the area of cell-free voids as regions of interests in the field of view and expressing as a percentage of the total field.
Inhibitor assays
All inhibitors were added to the apical and basal wells of polarized TC-7 cells (12–15 days post confluent) for 2 h prior to infection then removed from the apical well (2 washes in DMEM) prior to the addition of bacteria. Bacterial infection and TER monitoring were performed as above. Although the TC-7 cells were washed prior to infection, each inhibitor was assessed for its effect on bacterial growth at the concentration used. No inhibitor used in this study affected bacterial growth in DMEM over the time-course of the experiment (not shown). Working concentrations of inhibitors used in this study and their mode of action is given in Table 1. All inhibitors were purchased from Calbiochem except valinomycin and wortmannin, which were from Sigma.
Calpain activity
Measurement of calpain activity was performed based on the catalytic cleavage of the synthetic calpain substrate Suc-LLVY-AMC in the presence of Ca2+ and the reducing agent TCEP. The liberation of AMC during the reaction was detected at a wavelength of 460 nm. AMC (µM) released was determined using an AMC standard curve. Following infection, TC-7 cells were harvested and washed with PBS, lysed in Cytobuster (Calbiochem) and incubated on ice for 30 min. The lysate was centrifuged at 4°C at 12 000 g and the protein content was determined using BCA reagent. Calpain activity was determined using Suc-LLVY-AMC with the calpain activity kit according to the manufacturer's instructions (Calbiochem).
Statistical analysis
Significance levels within data sets were determined using a one-way anova with a post hoc Tukey test indicating significances between individual data points.
Acknowledgments
We are indebted to Dr Trevor Booth (Bioimaging, Newcastle) for help with confocal microscopy, Drs Nicolas Garmy, Marc Maresca and Mr Jian Yang for helping set up assays and Dr Akio Abe for the espG plasmids. This work was funded by a Wellcome Trust senior fellowship awarded to BK (ref: WT064409MA).
References
- Alto NM, Weflen AW, Rardin MJ, Yarar D, Lazar CS, Tonikian R, et al. The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. J Cell Biol. 2007;178:1265–1278. doi: 10.1083/jcb.200705021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand R, Solary E, O’Connor P, Kohn KW, Pommier Y. Induction of a common pathway of apoptosis by staurosporine. Exp Cell Res. 1994;211:314–321. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- Cain RJ, Hayward RD, Koronakis V. Deciphering interplay between Salmonella invasion effectors. PLoS Pathog. 2008;4:e1000037. doi: 10.1371/journal.ppat.1000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canil C, Rosenshine I, Ruschkowski S, Donnenberg MS, Kaper JB, Finlay BB. Enteropathogenic Escherichia coli decreases the transepithelial electrical resistance of polarized epithelial monolayers. Infect Immun. 1993;61:2755–2762. doi: 10.1128/iai.61.7.2755-2762.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantret I, Rodolosse A, Barbat A, Dussaulx E, Brot-Laroche E, Zweibaum A, Rousset M. Differential expression of sucrase-isomaltase in clones isolated from early and late passages of the cell line Caco-2: evidence for glucose-dependent negative regulation. J Cell Sci. 1994;107:213–225. doi: 10.1242/jcs.107.1.213. [DOI] [PubMed] [Google Scholar]
- Chen HD, Frankel G. Enteropathogenic Escherichia coli: unravelling pathogenesis. FEMS Microbiol Rev. 2005;29:83–98. doi: 10.1016/j.femsre.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Skehan BM, Campellone KG, Leong JM, Rosen MK. Structural mechanism of WASP activation by the enterohaemorrhagic E. coli effector EspF(U) Nature. 2008;454:1009–1013. doi: 10.1038/nature07160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Prince A. TLR2-induced calpain cleavage of epithelial junctional proteins facilitates leukocyte transmigration. Cell Host Microbe. 2009;5:47–58. doi: 10.1016/j.chom.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Wang J, Tropea JE, Zhang D, Dauter Z, Waugh DS, Wlodawer A. Novel fold of VirA, a type III secretion system effector protein from Shigella flexneri. Protein Sci. 2008;17:2167–2173. doi: 10.1110/ps.037978.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean P, Kenny B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol Microbiol. 2004;54:665–675. doi: 10.1111/j.1365-2958.2004.04308.x. [DOI] [PubMed] [Google Scholar]
- Dean P, Kenny B. The effector repertoire of enteropathogenic E. coli: ganging up on the host cell. Curr Opin Microbiol. 2009;12:101–109. doi: 10.1016/j.mib.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean P, Maresca M, Schuller S, Phillips AD, Kenny B. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proc Natl Acad Sci USA. 2006;103:1876–1881. doi: 10.1073/pnas.0509451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991;59:4310–4317. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Calderwood SB, Donohue-Rolfe A, Keusch GT, Kaper JB. Construction and analysis of TnphoA mutants of enteropathogenic Escherichia coli unable to invade HEp-2 cells. Infect Immun. 1990;58:1565–1571. doi: 10.1128/iai.58.6.1565-1571.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SJ, Krejany EO, Mellies JL, Robins-Browne RM, Sasakawa C, Kaper JB. EspG, a novel type III system-secreted protein from enteropathogenic Escherichia coli with similarities to VirA of Shigella flexneri. Infect Immun. 2001;69:4027–4033. doi: 10.1128/IAI.69.6.4027-4033.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. 2005;118:3829–3838. doi: 10.1242/jcs.02562. [DOI] [PubMed] [Google Scholar]
- Fu Y, Galan JE. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature. 1999;401:293–297. doi: 10.1038/45829. [DOI] [PubMed] [Google Scholar]
- Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, et al. Mechanism underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenic E. coli. J Clin Invest. 2007;117:428–437. doi: 10.1172/JCI29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glading A, Lauffenburger DA, Wells A. Cutting to the chase: calpain proteases in cell motility. Trends Cell Biol. 2002;12:46–54. doi: 10.1016/s0962-8924(01)02179-1. [DOI] [PubMed] [Google Scholar]
- Guttman JA, Finlay BB. Tight junctions as targets of infectious agents. Biochim Biophys Acta. 2009;1788:832–841. doi: 10.1016/j.bbamem.2008.10.028. [DOI] [PubMed] [Google Scholar]
- Hardwidge PR, Rodriguez-Escudero I, Goode D, Donohoe S, Eng J, Goodlett DR, et al. Proteomic analysis of the intestinal epithelial cell response to enteropathogenic Escherichia coli. J Biol Chem. 2004;279:20127–20136. doi: 10.1074/jbc.M401228200. [DOI] [PubMed] [Google Scholar]
- Huang Z, Sutton SE, Wallenfang AJ, Orchard RC, Wu X, Feng Y, et al. Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat Struct Mol Biol. 2009;16:853–860. doi: 10.1038/nsmb.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi A, Thomson NR, Ogura Y, Saunders D, Ooka T, Henderson IR, et al. Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J Bacteriol. 2009;191:347–354. doi: 10.1128/JB.01238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson MA, Pellegrin S, Peto L, Banbury DN, Leard AD, Mellor H, Kenny B. Synergistic roles for the Map and Tir effector molecules in mediating uptake of enteropathogenic Escherichia coli (EPEC) into non-phagocytic cells. Cell Microbiol. 2003;5:773–783. doi: 10.1046/j.1462-5822.2003.00315.x. [DOI] [PubMed] [Google Scholar]
- Kenny B. The enterohaemorrhagic Escherichia coli (serotype O157:H7) Tir molecule is not functionally interchangeable for its enteropathogenic E. coli (serotype O127:H6) homologue. Cell Microbiol. 2001;3:499–510. doi: 10.1046/j.1462-5822.2001.00133.x. [DOI] [PubMed] [Google Scholar]
- Kenny B, Jepson M. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cell Microbiol. 2000;2:579–590. doi: 10.1046/j.1462-5822.2000.00082.x. [DOI] [PubMed] [Google Scholar]
- Kenny B, Lai LC, Finlay BB, Donnenberg MS. EspA, a protein secreted by enteropathogenic Escherichia coli, is required to induce signals in epithelial cells. Mol Microbiol. 1996;20:313–323. doi: 10.1111/j.1365-2958.1996.tb02619.x. [DOI] [PubMed] [Google Scholar]
- Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell. 1997;91:511–520. doi: 10.1016/s0092-8674(00)80437-7. [DOI] [PubMed] [Google Scholar]
- Kenny B, Ellis S, Leard AD, Warawa J, Mellor H, Jepson MA. Co-ordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol Microbiol. 2002;44:1095–1107. doi: 10.1046/j.1365-2958.2002.02952.x. [DOI] [PubMed] [Google Scholar]
- Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci USA. 2005;102:14046–14051. doi: 10.1073/pnas.0504466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebart MC, Benyamin Y. Calpain involvement in the remodeling of cytoskeletal anchorage complexes. FEBS J. 2006;273:3415–3426. doi: 10.1111/j.1742-4658.2006.05350.x. [DOI] [PubMed] [Google Scholar]
- Levine MM, Nataro JP, Karch H, Baldini MM, Kaper JB, Black RE, et al. The diarrheal response of humans to some classic serotypes of enteropathogenic Escherichia coli is dependent on a plasmid encoding an enteroadhesiveness factor. J Infect Dis. 1985;152:550–559. doi: 10.1093/infdis/152.3.550. [DOI] [PubMed] [Google Scholar]
- McGhie EJ, Hayward RD, Koronakis V. Cooperation between actin-binding proteins of invasive Salmonella: SipA potentiates SipC nucleation and bundling of actin. EMBO J. 2001;20:2131–2139. doi: 10.1093/emboj/20.9.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara BP, Koutsouris A, O’Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest. 2001;107:621–629. doi: 10.1172/JCI11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marches O, Nougayrede JP, Boullier S, Mainil J, Charlier G, Raymond I, et al. Role of tir and intimin in the virulence of rabbit enteropathogenic Escherichia coli serotype O103:H2. Infect Immun. 2000;68:2171–2182. doi: 10.1128/iai.68.4.2171-2182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marches O, Ledger TN, Boury M, Ohara M, Tu X, Goffaux F, et al. Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol Microbiol. 2003;50:1553–1567. doi: 10.1046/j.1365-2958.2003.03821.x. [DOI] [PubMed] [Google Scholar]
- Matsuzawa T, Kuwae A, Yoshida S, Sasakawa C, Abe A. Enteropathogenic Escherichia coli activates the RhoA signaling pathway via the stimulation of GEF-H1. EMBO J. 2004;23:3570–3582. doi: 10.1038/sj.emboj.7600359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa T, Kuwae A, Abe A. Enteropathogenic Escherichia coli type III effectors EspG and EspG2 alter epithelial paracellular permeability. Infect Immun. 2005;73:6283–6289. doi: 10.1128/IAI.73.10.6283-6289.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E, Baruch K, Charpentier X, Kobi S, Rosenshine I. Real-time analysis of effector translocation by the type III secretion system of enteropathogenic Escherichia coli. Cell Host Microbe. 2008;3:104–113. doi: 10.1016/j.chom.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Miyake M, Hanajima M, Matsuzawa T, Kobayashi C, Minami M, Abe A, Horiguchi Y. Binding of intimin with Tir on the bacterial surface is prerequisite for the barrier disruption induced by enteropathogenic Escherichia coli. Biochem Biophys Res Commun. 2005;337:922–927. doi: 10.1016/j.bbrc.2005.09.130. [DOI] [PubMed] [Google Scholar]
- Muza-Moons MM, Koutsouris A, Hecht G. Disruption of cell polarity by enteropathogenic Escherichia coli enables basolateral membrane proteins to migrate apically and to potentiate physiological consequences. Infect Immun. 2003;71:7069–7078. doi: 10.1128/IAI.71.12.7069-7078.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nougayrede JP, Donnenberg MS. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell Microbiol. 2004;6:1097–1111. doi: 10.1111/j.1462-5822.2004.00421.x. [DOI] [PubMed] [Google Scholar]
- Peralta-Ramirez J, Hernandez JM, Manning-Cela R, Luna-Munoz J, Garcia-Tovar C, Nougayrede JP, et al. EspF Interacts with nucleation-promoting factors to recruit junctional proteins into pedestals for pedestal maturation and disruption of paracellular permeability. Infect Immun. 2008;76:3854–3868. doi: 10.1128/IAI.00072-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter DA, Srirangam A, Fiacco KA, Brocks D, Hawes J, Herndon C, et al. Calpain regulates enterocyte brush border actin assembly and pathogenic Escherichia coli-mediated effacement. J Biol Chem. 2003;278:30403–30412. doi: 10.1074/jbc.M304616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitard S, Dean P, Maresca M, Kenny B. The enteropathogenic Escherichia coli EspF effector molecule inhibits PI-3 kinase-mediated uptake independently of mitochondrial targeting. Cell Microbiol. 2006;8:972–981. doi: 10.1111/j.1462-5822.2005.00680.x. [DOI] [PubMed] [Google Scholar]
- Ruchaud-Sparagano MH, Maresca M, Kenny B. Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell Microbiol. 2007;9:1909–1921. doi: 10.1111/j.1462-5822.2007.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallee NA, Rivera GM, Dueber JE, Vasilescu D, Mullins RD, Mayer BJ, Lim WA. The pathogen protein EspF(U) hijacks actin polymerization using mimicry and multivalency. Nature. 2008;454:1005–1008. doi: 10.1038/nature07170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- Shaw RK, Smollett K, Cleary J, Garmendia J, Straatman-Iwanowska A, Frankel G, Knutton S. Enteropathogenic Escherichia coli type III effectors EspG and EspG2 disrupt the microtubule network of intestinal epithelial cells. Infect Immun. 2005a;73:4385–4390. doi: 10.1128/IAI.73.7.4385-4390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RK, Cleary J, Murphy MS, Frankel G, Knutton S. Interaction of enteropathogenic Escherichia coli with human intestinal mucosa: role of effector proteins in brush border remodeling and formation of attaching and effacing lesions. Infect Immun. 2005b;73:1243–1251. doi: 10.1128/IAI.73.2.1243-1251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrin Y, Kirschner J, Geiger B, Rosenshine I. Enteropathogenic Escherichia coli induces modification of the focal adhesions of infected host cells. Cell Microbiol. 2002;4:235–243. doi: 10.1046/j.1462-5822.2002.00188.x. [DOI] [PubMed] [Google Scholar]
- Simonovic I, Rosenberg J, Koutsouris A, Hecht G. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol. 2000;2:305–315. doi: 10.1046/j.1462-5822.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- Thanabalasuriar A, Koutsouris A, Weflen A, Mimee M, Hecht G, Gruenheid S. The bacterial virulence factor NleA is required for the disruption of intestinal tight junctions by enteropathogenic Escherichia coli. Cell Microbiol. 2010;12:31–41. doi: 10.1111/j.1462-5822.2009.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomson FL, Viswanathan VK, Kanack KJ, Kanteti RP, Straub KV, Menet M, et al. Enteropathogenic Escherichia coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Mol Microbiol. 2005;56:447–464. doi: 10.1111/j.1365-2958.2005.04571.x. [DOI] [PubMed] [Google Scholar]
- Zhu L, Li X, Zeng R, Gorodeski GI. Changes in tight junctional resistance of the cervical epithelium are associated with modulation of content and phosphorylation of occludin 65-kilodalton and 50-kilodalton forms. Endocrinology. 2006;147:977–989. doi: 10.1210/en.2005-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]